
Transcriptome-Based Discovery of Fusarium graminearum Stress Responses to FgHV1 Infection

Abstract
:
1. Introduction
2. Results
2.1. FgHV1 Was Transmitted to All F. graminearum Asexual Spores
2.2. Illumina RNA-Seq and Overall Transcriptional Profiles in F. graminearum in the Presence of FgHV1
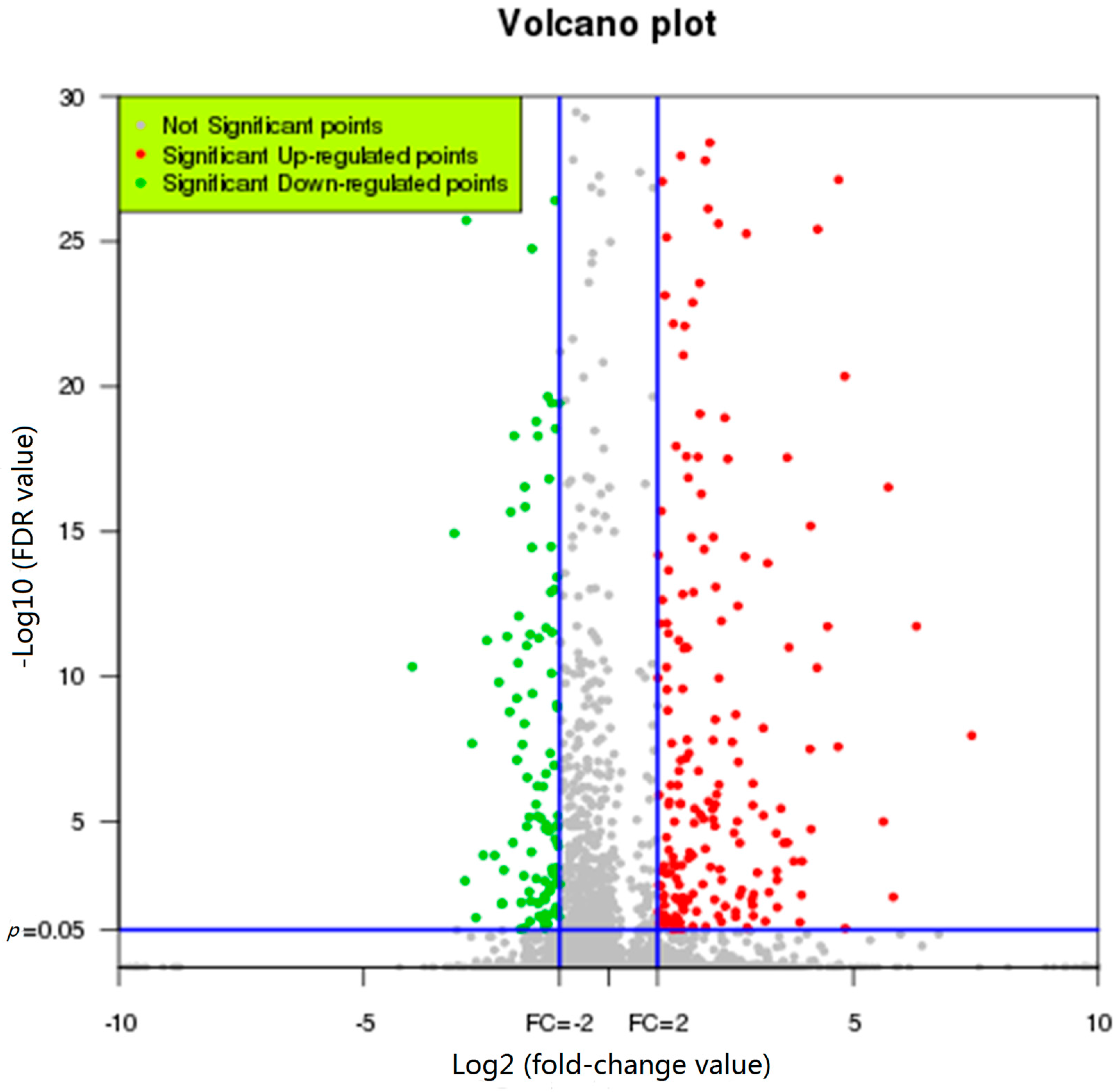
2.3. Representative Transcripts with Significant Changes in Expression in Response to FgHV1 Infection
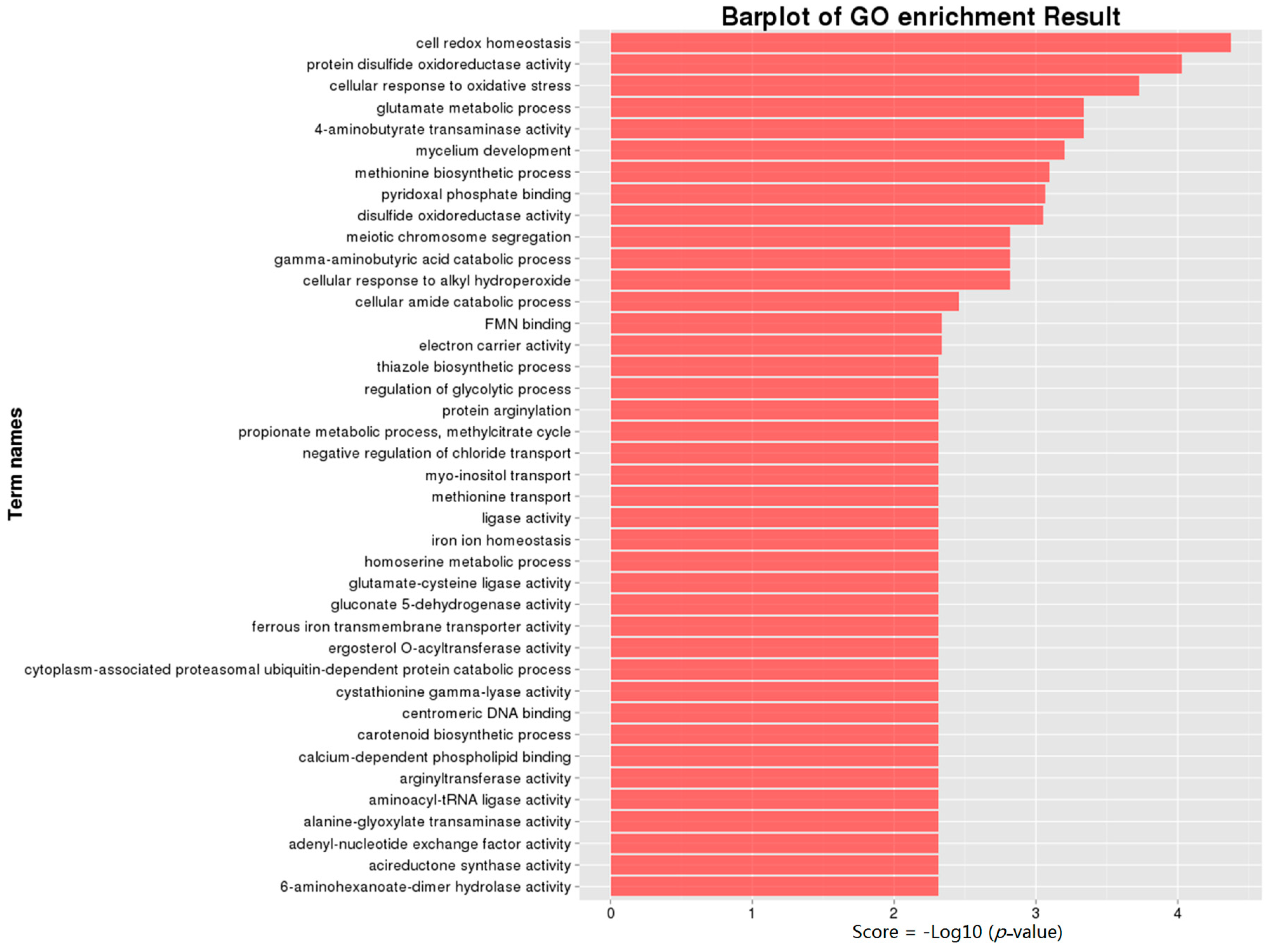
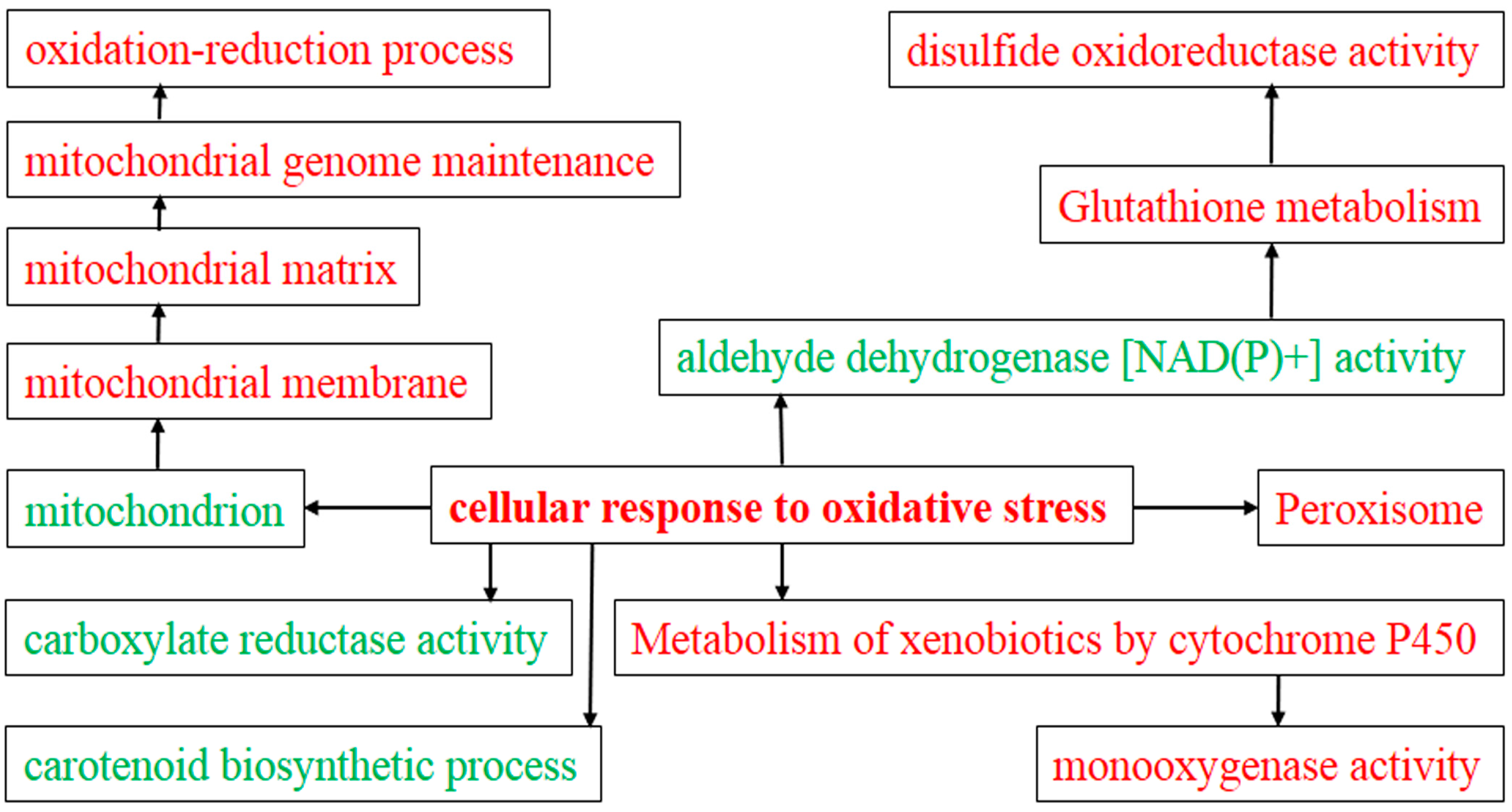
2.4. Gene Ontology (GO) Analysis of DEGs
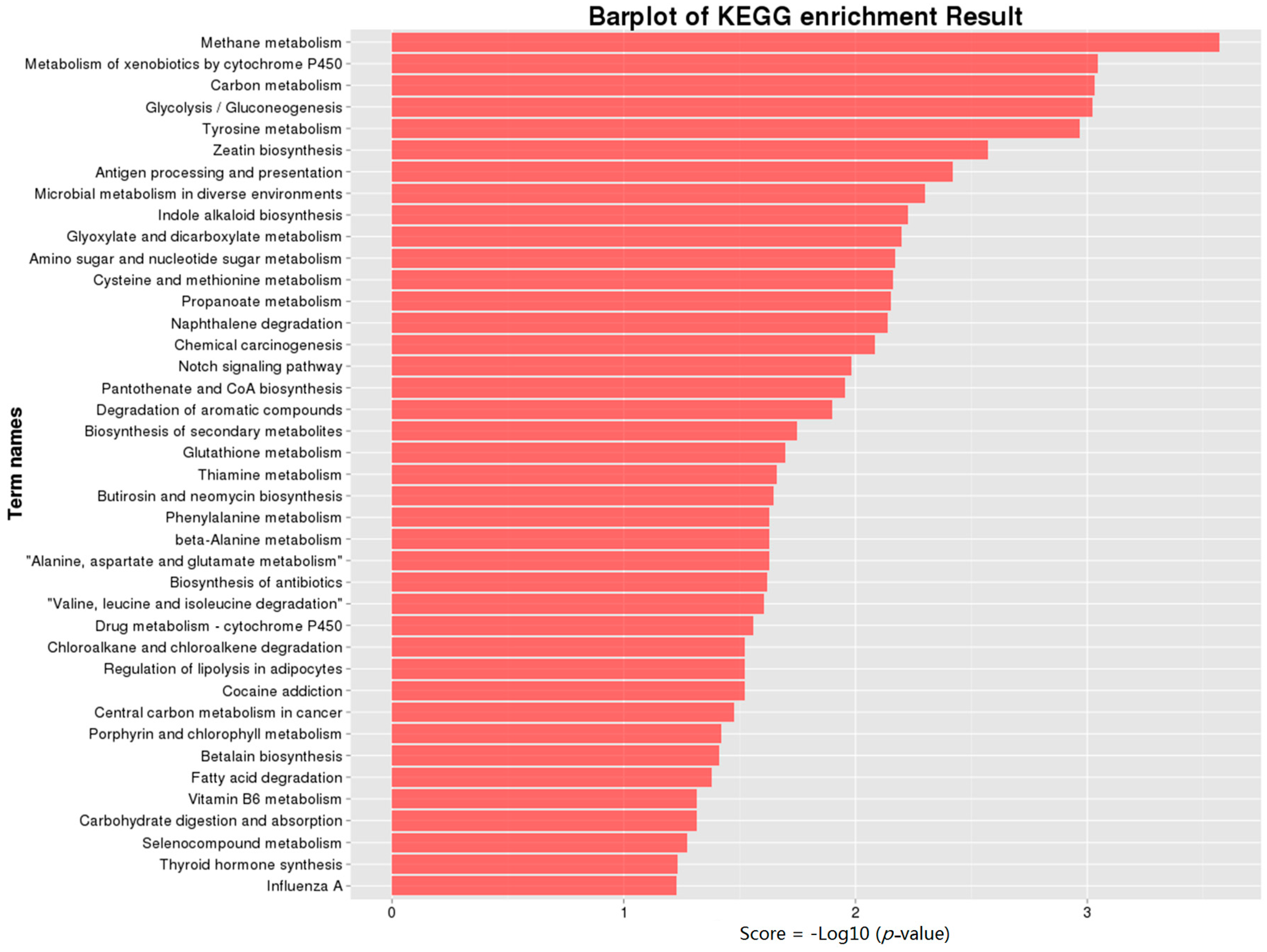
2.5. Kyoto Encyclopedia of Genes and Genomes Pathway (KEGG) Annotation of DEGs
2.6. Induction of H2O2 Accumulation and Hypersensitive Response by FgHV1
2.7. Quantitative Real-Time Reverse Transcription PCR Validation of the RNA-Seq Data
3. Discussion
4. Materials and Methods
4.1. Fungal Strains and Culture Conditions
4.2. Vertical Transmission Rate via Asexual Spores
4.3. RNA Extraction and cDNA Library Construction and Sequencing
4.4. Identification of Differentially Expressed Genes
4.5. GO Enrichment and KEGG Analysis
4.6. Detection of FgHV1 Induced H2O2 Accumulation and Hypersensitive Response
4.7. Real-Time RT-PCR
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Márquez, L.M.; Redman, R.S.; Rodriguez, R.J.; Roossinck, M.J. A virus in a fungus in a plant: Three-way symbiosis required for thermal tolerance. Science 2007, 315, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Shapira, R.; Choi, G.H.; Nuss, D.L. Virus-like genetic organization and expression strategy for a double-stranded RNA genetic element associated with biological control of chestnut blight. EMBO J. 1991, 10, 731. [Google Scholar] [PubMed]
- Chu, Y.-M.; Jeon, J.-J.; Yea, S.-J.; Kim, Y.-H.; Yun, S.-H.; Lee, Y.-W.; Kim, K.-H. Double-stranded RNA mycovirus from Fusarium graminearum. Appl. Environ. Microbiol. 2002, 68, 2529–2534. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, A.; Proctor, R. Molecular biology of Fusarium mycotoxins. Int. J. Food Microbiol. 2007, 119, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, C.A.; Güldener, U.; Xu, J.-R.; Trail, F.; Turgeon, B.G.; di Pietro, A.; Walton, J.D.; Ma, L.-J.; Baker, S.E.; Rep, M. The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 2007, 317, 1400–1402. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Kwon, S.-J.; Lee, K.-M.; Son, M.; Kim, K.-H. Complete nucleotide sequence of double-stranded RNA viruses from Fusarium graminearum strain DK3. Arch. Virol. 2009, 154, 1855–1858. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kondo, H.; Liu, L.; Guo, L.; Qiu, D. A novel virus in the family Hypoviridae from the plant pathogenic fungus Fusarium graminearum. Virus Res. 2013, 174, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, H.; Chen, X.; Qiu, D.; Guo, L. Molecular characterization of a novel hypovirus from the plant pathogenic fungus Fusarium graminearum. Virology 2015, 481, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Lin, Y.; Zhang, H.; Wang, S.; Qiu, D.; Guo, L. Molecular characterization of a novel mycovirus of the family Tymoviridae isolated from the plant pathogenic fungus Fusarium graminearum. Virology 2016, 489, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, J.; Zhang, H.; Qiu, D.; Guo, L. Two novel relative double-stranded RNA mycoviruses infecting Fusarium poae strain SX63. Int. J. Mol. Sci. 2016, 17, 641. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; He, H.; Yang, X.; Zeng, H.; Qiu, D.; Guo, L. The complete genome sequence of a novel Fusarium graminearum. Arch. Virol. 2016, 161, 1–5. [Google Scholar]
- Xie, J.; Jiang, D. New insights into mycoviruses and exploration for the biological control of crop fungal diseases. Annu. Rev. Phytopathol. 2014, 52, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Hillman, B.I.; Halpern, B.T.; Brown, M.P. A viral dsRNA element of the chestnut blight fungus with a distinct genetic organization. Virology 1994, 201, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Gao, S.; Choi, G.H.; Nuss, D.L. Extensive alteration of fungal gene transcript accumulation and elevation of G-protein-regulated cAMP levels by a virulence-attenuating hypovirus. Proc. Natl. Acad. Sci. USA 1996, 93, 7996–8000. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.D.; Nuss, D.L. Specific and common alterations in host gene transcript accumulation following infection of the chestnut blight fungus by mild and severe hypoviruses. J. Virol. 2004, 78, 4145–4155. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-M.; Cho, W.K.; Yu, J.; Son, M.; Choi, H.; Min, K.; Lee, Y.-W.; Kim, K.-H. A comparison of transcriptional patterns and mycological phenotypes following infection of Fusarium graminearum by four mycoviruses. PLoS ONE 2014, 9, e100989. [Google Scholar] [CrossRef] [PubMed]
- Turcu, F.E.R.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363. [Google Scholar] [CrossRef] [PubMed]
- Shelest, E. Transcription factors in fungi. FEMS Microbiol. Lett. 2008, 286, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Park, J.; Jang, S.; Kim, S.; Kong, S.; Choi, J.; Ahn, K.; Kim, J.; Lee, S.; Kim, S. FTFD: An informatics pipeline supporting phylogenomic analysis of fungal transcription factors. Bioinformatics 2008, 24, 1024–1025. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Jin, J.; Kim, H.; Kim, J.-C.; Yun, S.-H.; Lee, Y.-W. Gip2, a putative transcription factor that regulates the aurofusarin biosynthetic gene cluster in Gibberella zeae. Appl. Environ. Microbiol. 2006, 72, 1645–1652. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Waalwijk, C.; de Wit, P.J.; van der Lee, T.; Tang, D. EBR1, a novel Zn2Cys6 transcription factor, affects virulence and apical dominance of the hyphal tip in Fusarium graminearum. Mol. Plant-Microbe Interact. 2011, 24, 1407–1418. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Nuss, D.L. A host dicer is required for defective viral RNA production and recombinant virus vector RNA instability for a positive sense RNA virus. Proc. Natl. Acad. Sci. USA 2008, 105, 16749–16754. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Choi, G.H.; Nuss, D.L. A single Argonaute gene is required for induction of RNA silencing antiviral defense and promotes viral RNA recombination. Proc. Natl. Acad. Sci. USA 2009, 106, 17927–17932. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gao, Q.; Huang, M.; Liu, Y.; Liu, Z.; Liu, X.; Ma, Z. Characterization of RNA silencing components in the plant pathogenic fungus Fusarium graminearum. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, P.; Zhang, J.; Qiu, D.; Guo, L. Generation of a high resolution map of sRNAs from Fusarium graminearum and analysis of responses to viral infection. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Chapman, E.J.; Prokhnevsky, A.I.; Gopinath, K.; Dolja, V.V.; Carrington, J.C. Viral RNA silencing suppressors inhibit the microRNA pathway at an intermediate step. Genes Dev. 2004, 18, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, L.; Csorba, T.; Pantaleo, V.; Chapman, E.J.; Carrington, J.C.; Liu, Y.P.; Dolja, V.V.; Calvino, L.F.; López-Moya, J.J.; Burgyán, J. Small RNA binding is a common strategy to suppress RNA silencing by several viral suppressors. EMBO J. 2006, 25, 2768–2780. [Google Scholar] [CrossRef] [PubMed]
- Yaegashi, H.; Yoshikawa, N.; Ito, T.; Kanematsu, S. A mycoreovirus suppresses RNA silencing in the white root rot fungus, rosellinia necatrix. Virology 2013, 444, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Segers, G.C.; van Wezel, R.; Zhang, X.; Hong, Y.; Nuss, D.L. Hypovirus papain-like protease p29 suppresses RNA silencing in the natural fungal host and in a heterologous plant system. Eukaryot. Cell 2006, 5, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, C.; Li, Y.; Lam, T.-W.; Yiu, S.-M.; Kristiansen, K.; Wang, J. Soap2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Clean Ratio (%) b | rRNA Ratio (%) c | Genome Mapping Reads | Genome Mapping Ratio (%) d |
|---|---|---|---|---|---|---|
| HN101 a | 47,553,250 | 45,550,250 | 95.79 | 0.31 | 42,645,244 | 89.68 |
| HN102 a | 67,251,960 | 61,857,850 | 91.98 | 2.71 | 54,383,992 | 80.87 |
| HN103 a | 59,989,576 | 55,086,452 | 91.82 | 1.78 | 48,820,879 | 81.38 |
| HN10-11F1 a | 47,513,984 | 45,490,462 | 95.74 | 0.14 | 43,087,301 | 90.68 |
| HN10-11F2 a | 41,710,160 | 39,258,412 | 94.12 | 1.20 | 33,089,576 | 79.33 |
| HN10-11F3 a | 57,445,044 | 52,402,112 | 91.22 | 2.33 | 45,621,588 | 79.42 |
| Gene Locus | Fold_Change (Group 1/Group 2) | p-Value | Gene Function |
|---|---|---|---|
| Representative down-regulated genes in FgHV1-infected strain | |||
| FGSG_00226 | 0.062191 | 9.03 × 10−13 | Domain: Fungal trichothecene efflux pump (TRI12) |
| FGSG_13962 | 0.112587 | 1.77 × 10−17 | Alcohol dehydrogenase |
| FGSG_02138 | 0.131142 | 4.73 × 10−5 | Cytochrome P450 oxidoreductase |
| FGSG_10802 | 0.133304 | 1.99 × 10−28 | Domain: LrgB-like family |
| FGSG_04468 | 0.144817 | 4.92 × 10−10 | Domain: Transmembrane amino acid transporter protein |
| FGSG_06127 | 0.146473 | 6.70 × 10−85 | Formate dehydrogenase |
| FGSG_07582 | 0.152292 | 0 | MFS transporter |
| FGSG_03984 | 0.152931 | 1.161 × 10−3 | l-lactate dehydrogenase |
| FGSG_01450 | 0.157498 | 6.18 × 10−57 | Domain: Calcipressin |
| FGSG_11272 | 0.178327 | 1.05 × 10−13 | ABC transporter |
| FGSG_07598 | 0.183762 | 2.45 × 10−32 | Domain: Alcohol acetyltransferase |
| FGSG_07509 | 0.199109 | 5.39 × 10−6 | Domain: Major Facilitator Superfamily |
| FGSG_09697 | 0.202726 | 5.33 × 10−226 | Domain: multidrug resistance protein |
| FGSG_10920 | 0.211498 | 3.28 × 10−12 | Domain: Zinc-binding dehydrogenase |
| FGSG_11271 | 0.222594 | 3.45 × 10−4 | Domain: Fungal specific transcription factor |
| Representative up-regulated genes in FgHV1-infected strain | |||
| FGSG_11202 | 19.12455 | 9.96 × 10−13 | Domain: Guanylate kinase |
| FGSG_01766 | 31.36052 | 2.99 × 10−41 | Branched-chain amino acid aminotransferase |
| FGSG_02809 | 39.84652 | 6.84 × 10−182 | Domain: SRR1 |
| FGSG_11457 | 48.10646 | 9.17 × 10−137 | Endoribonuclease L-PSP |
| FGSG_04616 | 56.20855 | 1.91 × 10−4 | Glutathione-dependent formaldehyde-activating enzyme |
| Gene Locus | Fold_Change (Group 1/Group 2) | p-Value | Transcription Factor Family |
|---|---|---|---|
| FGSG_11271 | 0.222594 | 3.44 × 10−4 | Zn2Cys6 |
| FGSG_03292 | 0.290465 | 3.15 × 10−4 | Zn2Cys6 |
| FGSG_04554 | 0.295730 | 0 | Nucleic acid-binding, OB-fold |
| FGSG_06503 | 0.324989 | 1.20 × 10−4 | Zn2Cys6 |
| FGSG_09001 | 0.335331 | 4.02 × 10−6 | bZIP |
| FGSG_07583 | 0.367170 | 3.21 × 10−4 | Negative transcriptional regulator |
| FGSG_07192 | 0.396762 | 1.71 × 10−8 | Zn2Cys6 |
| FGSG_04626 | 0.405624 | 4.77 × 10−5 | GATA type zinc finger |
| FGSG_04288 | 2.109185 | 2.43 × 10−42 | C2H2 zinc finger |
| FGSG_12742 | 2.715052 | 6.74 × 10−5 | Zn2Cys6 |
| FGSG_02516 | 2.825014 | 9.17 × 10−306 | bHLH |
| FGSG_10360 | 3.148453 | 4.11 × 10−6 | C2H2 zinc finger |
| FGSG_09991 | 3.360416 | 3.57 × 10−7 | Nucleic acid-binding, OB-fold |
| FGSG_05567 | 3.933166 | 1.58 × 10−30 | bHLH |
| FGSG_00764 | 4.940559 | 1.712 × 10−3 | C2H2 zinc finger |
| FGSG_09832 | 5.399239 | 4.23 × 10−20 | bZIP |
| FGSG_04209 | 7.590574 | 2.44 × 10−4 | Negative transcriptional regulator |
| FGSG_03881 | 7.804580 | 1.49 × 10−4 | C2H2 zinc finger |
| FGSG_08246 | 28.29656 | 5.34 × 10−23 | C2H2 zinc finger |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Zhang, J.; Li, P.; Qiu, D.; Guo, L. Transcriptome-Based Discovery of Fusarium graminearum Stress Responses to FgHV1 Infection. Int. J. Mol. Sci. 2016, 17, 1922. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111922
Wang S, Zhang J, Li P, Qiu D, Guo L. Transcriptome-Based Discovery of Fusarium graminearum Stress Responses to FgHV1 Infection. International Journal of Molecular Sciences. 2016; 17(11):1922. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111922
Chicago/Turabian StyleWang, Shuangchao, Jingze Zhang, Pengfei Li, Dewen Qiu, and Lihua Guo. 2016. "Transcriptome-Based Discovery of Fusarium graminearum Stress Responses to FgHV1 Infection" International Journal of Molecular Sciences 17, no. 11: 1922. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111922