
MicroRNA Expression during Bovine Oocyte Maturation and Fertilization

Abstract
:1. Introduction
2. Results
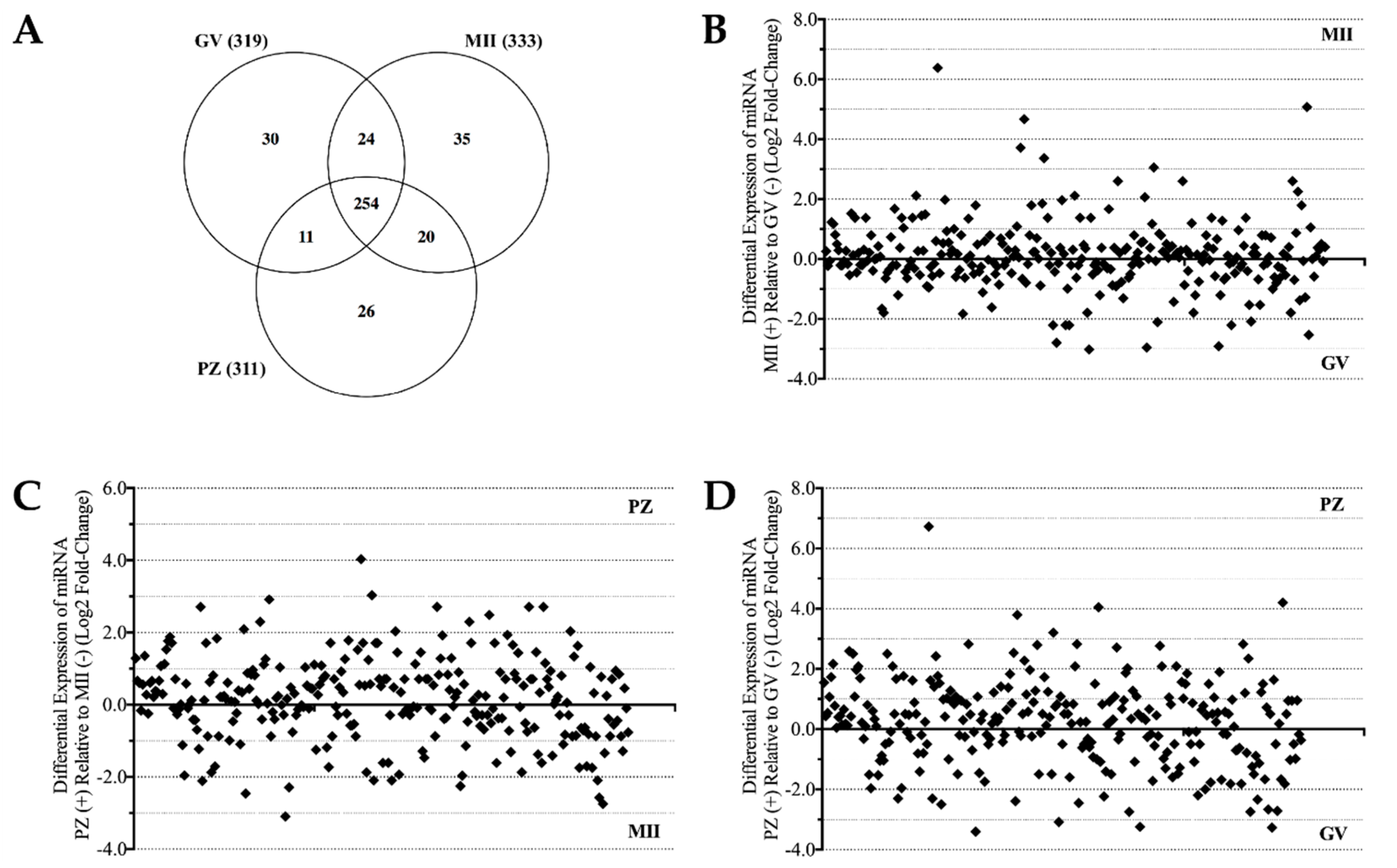
2.1. MicroRNA Detection and Differential Expression Analysis
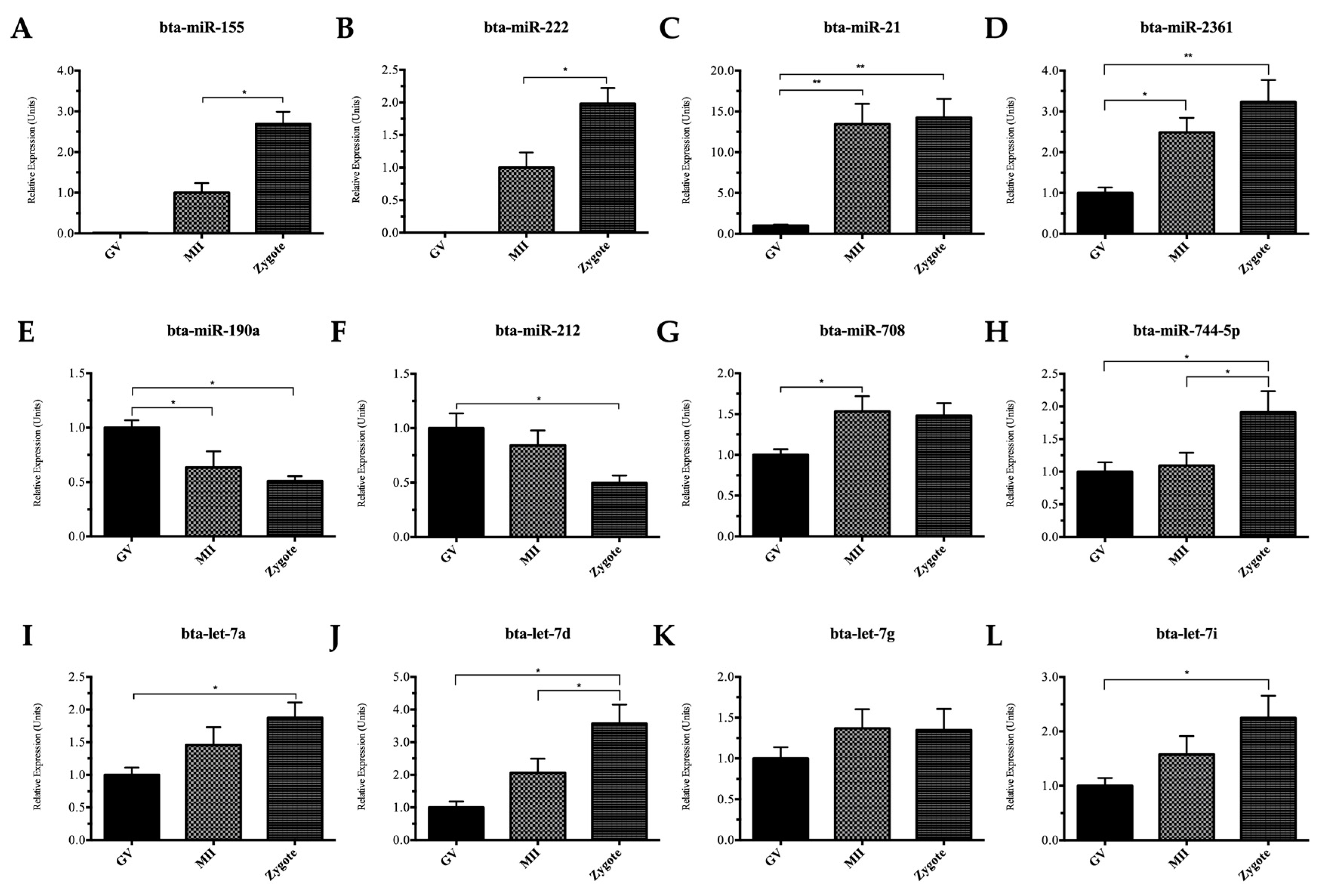
2.2. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR) Validation of miRNAs
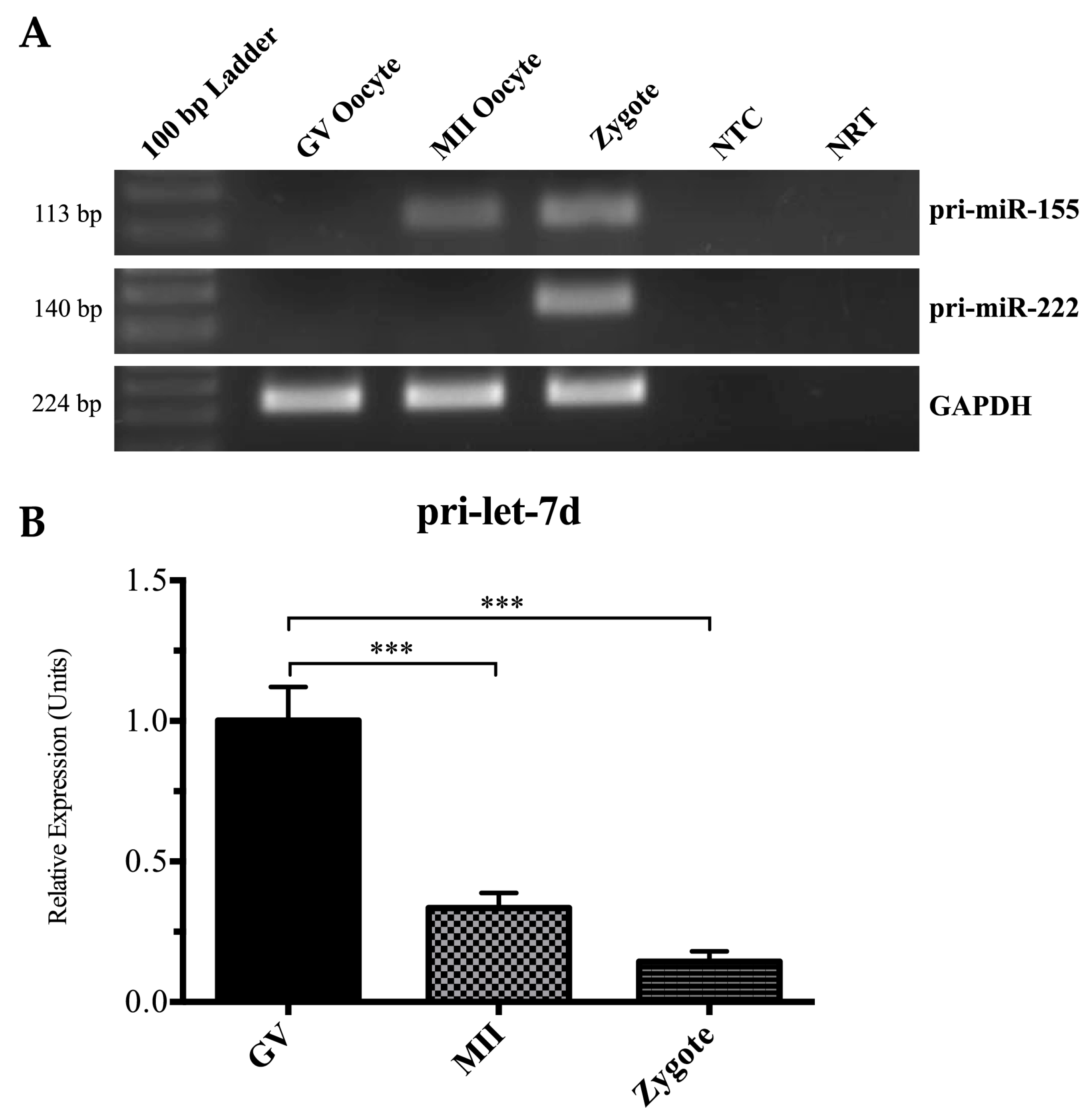
2.3. Detection of Pri-miRNAs
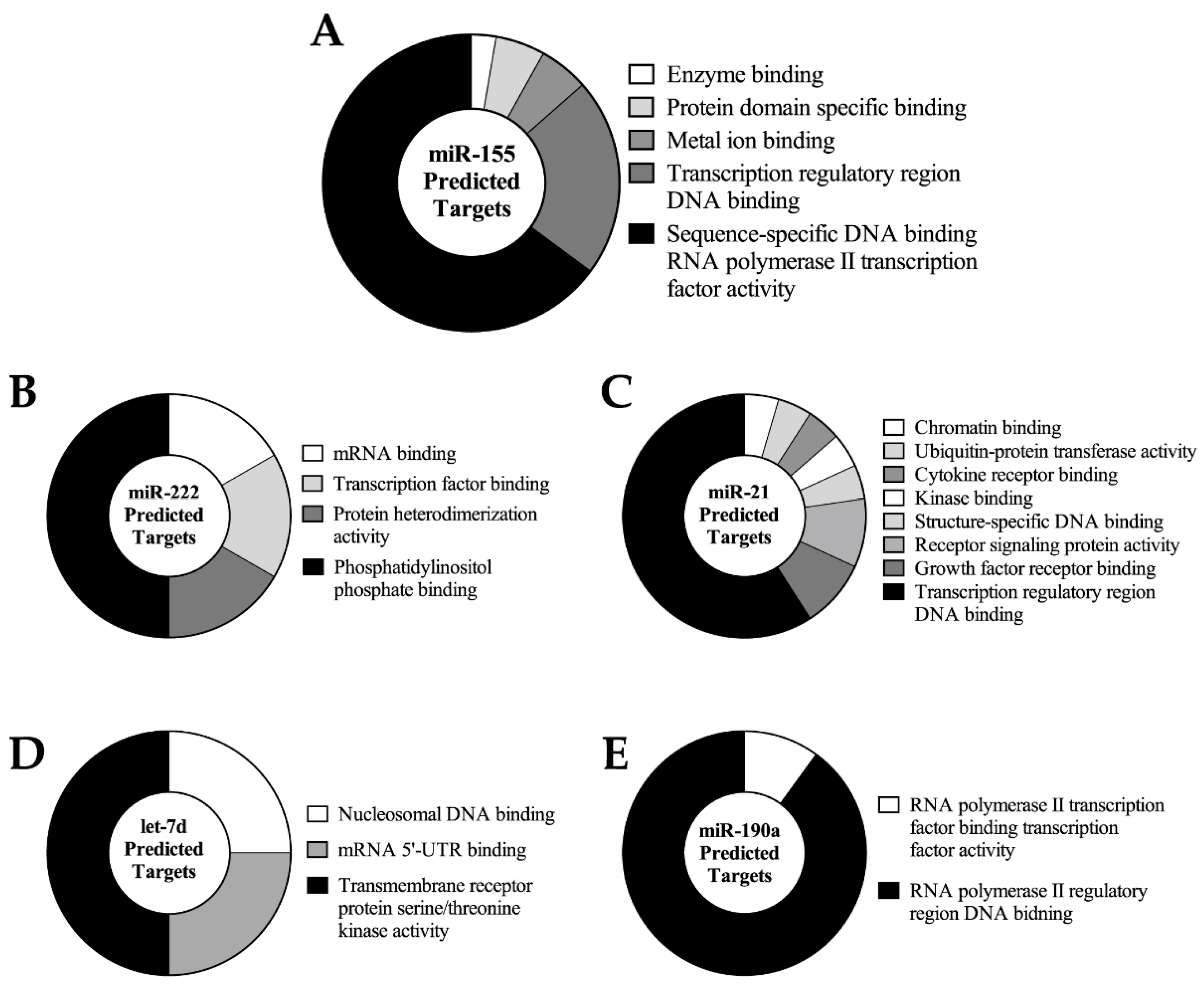
2.4. Predicted miRNA Target Cluster Analysis
2.5. Correlation of Predicted Targets with the Bovine Oocyte and Embryo Proteome
3. Discussion
4. Materials and Methods
4.1. In Vitro Oocyte Maturation, Fertilization and Collection
4.2. RNA Isolation, cDNA Synthesis, and PCR
4.3. Small RNA Library Prep and Next-Generation Sequencing
4.4. Bioinformatics Analysis
4.5. Statistical Analysis
4.6. Predicted Target Cluster Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| GV | germinal vesicle |
| GVBD | germinal vesicle breakdown |
| MII | metaphase II |
| PZ | presumptive zygote |
| miRNA | microRNA |
| ncRNA | non-coding RNA |
| pri-miRNA | primary-microRNA |
| pre-miRNA | precursor-microRNA |
| AGO | Argonaute |
| miRISC | miRNA-induced silencing complex |
| UTR | untranslated region |
| MET | maternal-to-embryonic transition |
| RNA-Seq | RNA sequencing |
| NGS | next-generation sequencing |
| qRT-PCR | quantitative reverse transcription PCR |
| GO | gene ontology |
| piRNA | piwi-interacting RNA |
References
- Hyttel, P.; Xu, K.P.; Smith, S.; Callesen, H.; Greve, T. Ultrastructure of the final nuclear maturation of bovine oocytes in vitro. Anat. Embryol. 1987, 176, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Callesen, H.; Greve, T.; Hyttel, P. Preovulatory endocrinology and oocyte maturation in superovulated cattle. Theriogenology 1986, 25, 71–86. [Google Scholar] [CrossRef]
- Song, J.L.; Wessel, G.M. How to make an egg: Transcriptional regulation in oocytes. Differentiation 2005, 73, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Paynton, B.V.; Rempel, R.; Bachvarova, R. Changes in state of adenylation and time course of degradation of maternal mRNAs during oocyte maturation and early embryonic development in the mouse. Dev. Biol. 1988, 129, 304–314. [Google Scholar] [CrossRef]
- Assou, S.; Al-edani, T.; Haouzi, D.; Philippe, N.; Lecellier, C.H.; Piquemal, D.; Commes, T.; Ait-Ahmed, O.; Dechaud, H.; Hamamah, S. MicroRNAs: New candidates for the regulation of the human cumulus-oocyte complex. Hum. Reprod. 2013, 28, 3038–3049. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, A.D.; Gilbert, I.; Caballero, J.; Barreto, R.; Fournier, E.; Tossou, P.; Sirard, M.-A.; Clarke, H.J.; Khandjian, É.W.; Richard, F.J.; et al. The gametic synapse: RNA transfer to the bovine oocyte. Biol. Reprod. 2014, 91, 90. [Google Scholar] [CrossRef] [PubMed]
- Fair, T.; Carter, F.; Park, S.; Evans, A.C.O.; Lonergan, P. Global gene expression analysis during bovine oocyte in vitro maturation. Theriogenology 2007, 68, S91–S97. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Kaneda, M.; O’Carroll, D.; Hajkova, P.; Barton, S.C.; Sun, Y.A.; Lee, C.; Tarakhovsky, A.; Lao, K.; Surani, M.A. Maternal microRNAs are essential for mouse zygotic development. Genes Dev. 2007, 21, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Tesfaye, D.; Worku, D.; Rings, F.; Phatsara, C.; Tholen, E.; Schellander, K.; Hoelker, M. Identification and expression profiling of microRNAs during bovine oocyte maturation using heterologous approach. Mol. Reprod. Dev. 2009, 76, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans. Science 2001, 294, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Hutvágner, G.; Zamore, P.D. RNAi: Nature abhors a double-strand. Curr. Opin. Genet. Dev. 2002, 12, 225–232. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004, 14, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, Y.; Yeom, K.-H.; Kim, Y.-K.; Jin, H.; Kim, V.N. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar] [CrossRef] [PubMed]
- Bohnsack, M.T.; Czaplinski, K.; GÖRLICH, D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 2004, 10, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.; Güttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear export of microRNA precursors. Science 2004, 303, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.I.; Chendrimada, T.P.; Cooch, N.; Shiekhattar, R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 2005, 123, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. MicroRNAs: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Truesdell, S.S.; Mortensen, R.D.; Seo, M.; Schroeder, J.C.; Lee, J.H.; LeTonqueze, O.; Vasudevan, S. MicroRNA-mediated mRNA translation activation in quiescent cells and oocytes involves recruitment of a nuclear microRNP. Sci. Rep. 2012, 2, 842. [Google Scholar] [CrossRef] [PubMed]
- Ørom, U.A.; Nielsen, F.C.; Lund, A.H. MicroRNA-10a binds the 5′UTR of ribosomal protein mRNAs and enhances their translation. Mol. Cell 2008, 30, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.N.; Habermacher, R.; Martine, U.; Closs, E.I.; Filipowicz, W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 2006, 125, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.C. MicroRNAs in embryonic stem cells and early embryonic development. J. Cell. Mol. Med. 2008, 12, 2181–2188. [Google Scholar] [CrossRef] [PubMed]
- Giraldez, A.J.; Mishima, Y.; Rihel, J.; Grocock, R.J.; van Dongen, S.; Inoue, K.; Enright, A.J.; Schier, A.F. Zebrafish miR-430 promotes deadenylation and clearance of maternal mRNAs. Science 2006, 312, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, Y.; Totoki, Y.; Toyoda, A.; Watanabe, T.; Yamamoto, Y.; Tokunaga, K.; Sakaki, Y.; Sasaki, H.; Hohjoh, H. Small RNA class transition from siRNA/piRNA to miRNA during pre-implantation mouse development. Nucleic Acids Res. 2010, 38, 5141–5151. [Google Scholar] [CrossRef] [PubMed]
- Mondou, E.; Dufort, I.; Gohin, M.; Fournier, E.; Sirard, M.A. Analysis of microRNAs and their precursors in bovine early embryonic development. Mol. Hum. Reprod. 2012, 18, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Sun, J.; Dong, H.; Luo, O.; Zheng, X.; Obergfell, C.; Tang, Y.; Bi, J.; O’Neill, R.; Ruan, Y.; et al. Transcriptional profiles of bovine in vivo pre-implantation development. BMC Genomics 2014, 15, 756. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.M.; Chitwood, J.L.; Ross, P.J. RNA-Seq profiling of single bovine oocyte transcript abundance and its modulation by cytoplasmic polyadenylation. Mol. Reprod. Dev. 2015, 82, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, D.R.; Fröhlich, T.; Otte, K.A.; Beck, A.; Habermann, F.A.; Wolf, E.; Arnold, G.J. Stage-specific proteome signatures in early bovine embryo development. J. Proteome Res. 2014, 13, 4363–4376. [Google Scholar] [CrossRef] [PubMed]
- Roovers, E.F.; Rosenkranz, D.; Mahdipour, M.; Han, C.-T.; He, N.; Chuva de Sousa Lopes, S.M.; van der Westerlaken, L.A.J.; Zischler, H.; Butter, F.; Roelen, B.A.J.; et al. Piwi proteins and piRNAs in mammalian oocytes and early embryos. Cell Rep. 2015, 10, 2069–2082. [Google Scholar] [CrossRef] [PubMed]
- Lodde, V.; Modina, S.; Maddox-Hyttel, P.; Franciosi, F.; Lauria, A.; Luciano, A.M. Oocyte morphology and transcriptional silencing in relation to chromatin remodeling during the final phases of bovine oocyte growth. Mol. Reprod. Dev. 2008, 75, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Barnetova, I.; Morovic, M.; Strejcek, F.; Østrup, O.; Hyttel, P.; Niemann, H.; Laurincik, J.; Fulka, J.; Fulka, H. RNA polymerase II transcriptional silencing in growing and fully grown germinal vesicle oocytes isolated from gonadotropin-stimulated and non-stimulated gilts. Mol. Reprod. Dev. 2012, 79, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Landys, N.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, M.; Gross, B.; et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007, 2, 2366–2382. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.-C.; Clarkin, K.C.; Wahl, G.M. Sensitivity and selectivity of the DNA damage sensor responsible for activating p53-dependent G1 arrest. Proc. Natl. Acad. Sci. USA 1996, 93, 4827–4832. [Google Scholar] [CrossRef] [PubMed]
- Matwee, C.; Betts, D.H.; King, W.A. Apoptosis in the early bovine embryo. Zygote 2000, 8, 57–68. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Chaudhuri, A.A.; Rao, D.S.; Baltimore, D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc. Natl. Acad. Sci. USA 2009, 106, 7113–7118. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, Y.; Tagawa, H.; Takahashi, N.; Watanabe, A.; Guo, Y.-M.; Iwamoto, K.; Yamashita, J.; Saitoh, H.; Kameoka, Y.; Shimizu, N.; et al. Aberrant overexpression of microRNAs activate AKT signaling via down-regulation of tumor suppressors in natural killer-cell lymphoma/leukemia. Blood 2009, 114, 3265–3275. [Google Scholar] [CrossRef] [PubMed]
- Tomek, W.; Smiljakovic, T. Activation of Akt (protein kinase B) stimulates metaphase I to metaphase II transition in bovine oocytes. Reproduction 2005, 130, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Muggenhumer, D.; Vesely, C.; Nimpf, S.; Tian, N.; Yongfeng, J.; Jantsch, M.F. Drosha protein levels are translationally regulated during Xenopus oocyte maturation. Mol. Biol. Cell 2014, 25, 2094–2104. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, N.; Wani, S.; Xu, Q.; Gu, J.; Lea, K.; Heater, S.; Barbacioru, C.; Steptoe, A.L.; Martin, H.C.; Nourbakhsh, E.; et al. MicroRNAs and their isomiRs function cooperatively to target common biological pathways. Genome Biol. 2011, 12, R126. [Google Scholar] [CrossRef] [PubMed]
- Neilsen, C.T.; Goodall, G.J.; Bracken, C.P. IsomiRs—The overlooked repertoire in the dynamic microRNAome. Trends Genet. 2012, 28, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.C.; Chan, E.; Molnar, A.; Sarkar, R.; Alexieva, D.; Isa, I.M.; Robinson, S.; Zhang, S.; Ellis, P.; Langford, C.F.; et al. 5′ isomiR variation is of functional and evolutionary importance. Nucleic Acids Res. 2014, 42, 9424–9435. [Google Scholar] [CrossRef] [PubMed]
- Flemr, M.; Ma, J.; Schultz, R.M.; Svoboda, P. P-body loss is concomitant with formation of a messenger RNA storage domain in mouse oocytes. Biol. Reprod. 2010, 82, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Flemr, M.; Moravec, M.; Libova, V.; Sedlacek, R.; Svoboda, P. Lin28a is dormant, functional, and dispensable during mouse oocyte-to-embryo transition. Biol. Reprod. 2014, 90, 131. [Google Scholar] [CrossRef] [PubMed]
- Flemr, M.; Malik, R.; Franke, V.; Nejepinska, J.; Sedlacek, R.; Vlahovicek, K.; Svoboda, P. A retrotransposon-driven dicer isoform directs endogenous small interfering RNA production in mouse oocytes. Cell 2013, 155, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, G.C.; Kurjanowicz, P.; Mereilles, F.V.; King, W.A.; LaMarre, J. Telomere length and telomerase activity in bovine pre-implantation embryos in vitro. Reprod. Domest. Anim. 2015, 50, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Nebreda, A.R.; Ferby, I. Regulation of the meiotic cell cycle in oocytes. Curr. Opin. Cell Biol. 2000, 12, 666–675. [Google Scholar] [CrossRef]
- Huang, X.; Shen, Y.; Liu, M.; Bi, C.; Jiang, C.; Iqbal, J.; McKeithan, T.W.; Chan, W.C.; Ding, S.-J.; Fu, K. Quantitative proteomics reveals that miR-155 regulates the PI3K-AKT pathway in diffuse large B-cell lymphoma. Am. J. Pathol. 2012, 181, 26–33. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stage | Raw Reads (Million) | Post-Filter Reads (Million) | miRNA (%) | RefSeq Gene Reads (%) | RefSeq Coding Reads (%) | rRNA (%) | Unannotated (%) |
|---|---|---|---|---|---|---|---|
| GV | 62.6 | 44.4 | 1.43 | 27.81 | 1.19 | 0.10 | 69.47 |
| MII | 57.0 | 39.6 | 1.64 | 27.78 | 1.17 | 0.10 | 69.31 |
| PZ | 55.2 | 33.5 | 1.53 | 27.77 | 1.56 | 0.09 | 69.05 |
| miRNA of Interest | MII vs. PZ miRNA Change | Number of Predicted miRNA Targets in Proteome | MII vs. PZ Common Protein Change |
|---|---|---|---|
| miR-155 | Up | 14 | 67% Down |
| miR-222 | Up | 15 | 56% Down |
| let-7d | Up | 37 | 73% Down |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gilchrist, G.C.; Tscherner, A.; Nalpathamkalam, T.; Merico, D.; LaMarre, J. MicroRNA Expression during Bovine Oocyte Maturation and Fertilization. Int. J. Mol. Sci. 2016, 17, 396. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030396
Gilchrist GC, Tscherner A, Nalpathamkalam T, Merico D, LaMarre J. MicroRNA Expression during Bovine Oocyte Maturation and Fertilization. International Journal of Molecular Sciences. 2016; 17(3):396. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030396
Chicago/Turabian StyleGilchrist, Graham C., Allison Tscherner, Thomas Nalpathamkalam, Daniele Merico, and Jonathan LaMarre. 2016. "MicroRNA Expression during Bovine Oocyte Maturation and Fertilization" International Journal of Molecular Sciences 17, no. 3: 396. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030396