
Is There Room for Second-Generation Antipsychotics in the Pharmacotherapy of Panic Disorder? A Systematic Review Based on PRISMA Guidelines



Abstract
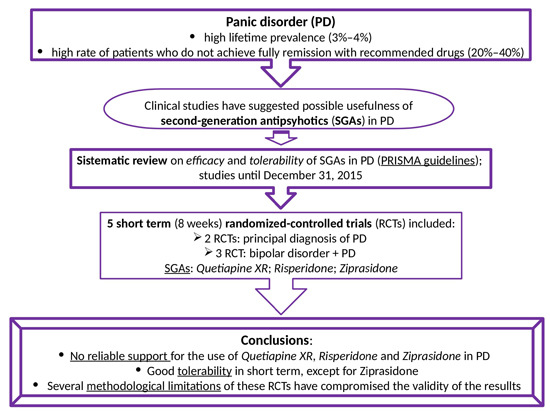
:
1. Introduction
2. Results
Risk of Bias in Individual Studies
3. Discussion
3.1. Efficacy in Patients with a Principal Diagnosis of PD
3.2. Efficacy in Patients with BD with Co-Occurring PD
3.3. Tolerability
3.4. Limitations and Future Research
4. Materials and Methods
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kessler, R.C.; Petukhova, M.; Sampson, N.A.; Zaslavsky, A.M.; Wittchen, H.U. Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int. J. Methods Psychiatr. Res. 2012, 21, 169–184. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Perna, G.; Guerriero, G.; Caldirola, D. Emerging drugs for panic disorder. Expert Opin. Emerg. Drugs 2011, 16, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, D.S.; Anderson, I.M.; Nutt, D.J.; Allgulander, C.; Bandelow, B.; den Boer, J.A.; Christmas, D.M.; Davies, S.; Fineberg, N.; Lidbetter, N.; et al. Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: A revision of the 2005 guidelines from the British Association for Psychopharmacology. J. Psychopharmacol. 2014, 28, 403–439. [Google Scholar] [CrossRef] [PubMed]
- Freire, R.C.; Zugliani, M.M.; Garcia, R.F.; Nardi, A.E. Treatment-resistant panic disorder: A systematic review. Expert Opin. Pharmacother. 2015, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Van Apeldoorn, F.J.; van Hout, W.J.; Mersch, P.P.; Huisman, M.; Slaap, B.R.; Hale, W.W., 3rd; Visser, S.; van Dyck, R.; den Boer, J.A. Is a combined therapy more effective than either CBT or SSRI alone? Results of a multicenter trial on panic disorder with or without agoraphobia. Acta Psychiatr. Scand. 2008, 117, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Perna, G.; Schruers, K.; Alciati, A.; Caldirola, D. Novel investigational therapeutics for panic disorder. Expert Opin. Investig. Drugs 2015, 24, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; He, W.; Hu, G.; Li, M. Anxiolytic-like property of risperidone and olanzapine as examined in multiple measures of fear in rats. Pharmacol. Biochem. Behav. 2010, 95, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Mead, A.; Li, M.; Kapur, S. Clozapine and olanzapine exhibit an intrinsic anxiolytic property in two conditioned fear paradigms: Contrast with haloperidol and chlordiazepoxide. Pharmacol. Biochem. Behav. 2008, 90, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Tsuchiya, K.; Koyama, T. Effects of typical and atypical antipsychotic drugs on freezing behavior induced by conditioned fear. Pharmacol. Biochem. Behav. 1996, 55, 195–201. [Google Scholar] [CrossRef]
- Lissek, S.; Rabin, S.J.; McDowell, D.J.; Dvir, S.; Bradford, D.E.; Geraci, M.; Pine, D.S.; Grillon, C. Impaired discriminative fear-conditioning resulting from elevated fear responding to learned safety cues among individuals with panic disorder. Behav. Res. Ther. 2009, 47, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Lissek, S.; Rabin, S.; Heller, R.E.; Lukenbaugh, D.; Geraci, M.; Pine, D.S.; Grillon, C. Overgeneralization of conditioned fear as a pathogenic marker of panic disorder. Am. J. Psychiatry 2010, 167, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Perna, G. Understanding anxiety disorders: The psychology and the psychopathology of defence mechanisms against threats. Riv. Psichiatr. 2013, 48, 73–75. [Google Scholar] [PubMed]
- Perna, G.; Guerriero, G.; Brambilla, P.; Caldirola, D. Panic and the brainstem: Clues from neuroimaging studies. CNS Neurol. Disord. Drug Targets 2014, 13, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Stahl, S.M. Stahl’s Essential Psychopharmacology: Neuroscientific Basis and Practical Applications, 3rd ed.; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Blier, P.; Szabo, S.T. Potential mechanisms of action of atypical antipsychotic medications in treatment-resistant depression and anxiety. J. Clin. Psychiatry 2005, 66 (Suppl. S8), 30–40. [Google Scholar] [PubMed]
- Marek, G.J.; Carpenter, L.L.; McDougle, C.J.; Price, L.H. Synergistic action of 5-HT2A antagonists and selective serotonin reuptake inhibitors in neuropsychiatric disorders. Neuropsychopharmacol.: Off. Publ. Am. Coll. Neuropsychopharmacol. 2003, 28, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.E.; VanDoren, M.J.; Duncan, G.E.; Lieberman, J.A.; Morrow, A.L. Olanzapine and clozapine increase the GABAergic neuroactive steroid allopregnanolone in rodents. Neuropsychopharmacol.: Off. Publ. Am. Coll. Neuropsychopharmacol. 2003, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Majewska, M.D. Steroid regulation of the GABAA receptor: Ligand binding, chloride transport and behaviour. Ciba Found. Symp. 1990, 153, 83–97; discussion 97–106. [Google Scholar] [PubMed]
- Akwa, Y.; Purdy, R.H.; Koob, G.F.; Britton, K.T. The amygdala mediates the anxiolytic-like effect of the neurosteroid allopregnanolone in rat. Behav. Brain Res. 1999, 106, 119–125. [Google Scholar] [CrossRef]
- Perna, G.A.A.; Riva, A.; Micieli, W.; Caldirola, D. Long-term pharmacological treatments of anxiety disorders. An updated systematic review. Curr. Psychiatry Rep. 2015, 18, 23. [Google Scholar] [CrossRef] [PubMed]
- LaLonde, C.D.; van Lieshout, R.J. Treating generalized anxiety disorder with second generation antipsychotics: A systematic review and meta-analysis. J. Clin. Psychopharmacol. 2011, 31, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Hershenberg, R.; Gros, D.F.; Brawman-Mintzer, O. Role of atypical antipsychotics in the treatment of generalized anxiety disorder. CNS Drugs 2014, 28, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.R.; Woo, Y.S.; Bahk, W.M. The potential role of atypical antipsychotics in the treatment of panic disorder. Hum. Psychopharmacol. 2014, 29, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gotzsche, P.C.; Ioannidis, J.P.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: Explanation and elaboration. J. Clin. Epidemiol. 2009, 62, e1–e34. [Google Scholar] [CrossRef] [PubMed]
- Goddard, A.W.; Mahmud, W.; Medlock, C.; Shin, Y.W.; Shekhar, A. A controlled trial of quetiapine XR coadministration treatment of SSRI-resistant panic disorder. Ann. Gen. Psychiatry 2015, 14, 26. [Google Scholar] [CrossRef] [PubMed]
- Prosser, J.M.; Yard, S.; Steele, A.; Cohen, L.J.; Galynker, I.I. A comparison of low-dose risperidone to paroxetine in the treatment of panic attacks: A randomized, single-blind study. BMC Psychiatry 2009, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Suppes, T.; McElroy, S.L.; Sheehan, D.V.; Hidalgo, R.B.; Cosgrove, V.E.; Gwizdowski, I.S.; Feldman, N.S. A randomized, double-blind, placebo-controlled study of ziprasidone monotherapy in bipolar disorder with co-occurring lifetime panic or generalized anxiety disorder. J. Clin. Psychiatry 2014, 75, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, D.V.; McElroy, S.L.; Harnett-Sheehan, K.; Keck, P.E., Jr.; Janavs, J.; Rogers, J.; Gonzalez, R.; Shivakumar, G.; Suppes, T. Randomized, placebo-controlled trial of risperidone for acute treatment of bipolar anxiety. J. Affect. Disord. 2009, 115, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, D.V.; Harnett-Sheehan, K.; Hidalgo, R.B.; Janavs, J.; McElroy, S.L.; Amado, D.; Suppes, T. Randomized, placebo-controlled trial of quetiapine XR and divalproex ER monotherapies in the treatment of the anxious bipolar patient. J. Affect. Disord. 2013, 145, 83–94. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Practice Guidelines for the Treatment of Patients with Panic Disorder, 2nd ed.; American Psychiatric Association: Arlington, VA, USA, 2009. [Google Scholar]
- Perna, G.; Gabriele, A.; Caldirola, D.; Bellodi, L. Hypersensitivity to inhalation of carbon dioxide and panic attacks. Psychiatry Res. 1995, 57, 267–273. [Google Scholar] [CrossRef]
- Perna, G.; Caldirola, D.; Arancio, C.; Bellodi, L. Panic attacks: A twin study. Psychiatry Res. 1997, 66, 69–71. [Google Scholar] [CrossRef]
- Perna, G.; Bussi, R.; Allevi, L.; Bellodi, L. Sensitivity to 35% carbon dioxide in patients with generalized anxiety disorder. J. Clin. Psychiatry 1999, 60, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Newcomer, J.W. Second-generation (atypical) antipsychotics and metabolic effects: A comprehensive literature review. CNS Drugs 2005, 19 (Suppl. S1), 1–93. [Google Scholar] [CrossRef] [PubMed]
- Coplan, J.; Gugger, J.J.; Tasleem, H. Tardive dyskinesia from atypical antipsychotic agents in patients with mood disorders in a clinical setting. J. Affect. Disord. 2013, 150, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Bandelow, B.; Lichte, T.; Rudolf, S.; Wiltink, J.; Beutel, M.E. The diagnosis of and treatment recommendations for anxiety disorders. Dtsch. Arzteblatt Int. 2014, 111, 473–480. [Google Scholar]
- Donovan, M.R.; Glue, P.; Kolluri, S.; Emir, B. Comparative efficacy of antidepressants in preventing relapse in anxiety disorders—A meta-analysis. J. Affect. Disord. 2010, 123, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Dilsaver, S.C. Comorbidity of panic disorder in bipolar illness: Evidence from the Epidemiologic Catchment Area Survey. Am. J. Psychiatry 1995, 152, 280–282. [Google Scholar] [PubMed]
- MacKinnon, D.F.; Xu, J.; McMahon, F.J.; Simpson, S.G.; Stine, O.C.; McInnis, M.G.; DePaulo, J.R. Bipolar disorder and panic disorder in families: An analysis of chromosome 18 data. Am. J. Psychiatry 1998, 155, 829–831. [Google Scholar] [PubMed]
- Logue, M.W.; Durner, M.; Heiman, G.A.; Hodge, S.E.; Hamilton, S.P.; Knowles, J.A.; Fyer, A.J.; Weissman, M.M. A linkage search for joint panic disorder/bipolar genes. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2009, 150B, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Kilbane, E.J.; Gokbayrak, N.S.; Galynker, I.; Cohen, L.; Tross, S. A review of panic and suicide in bipolar disorder: Does comorbidity increase risk? J. Affect. Disord. 2009, 115, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Forty, L.; Smith, D.; Jones, L.; Jones, I.; Caesar, S.; Cooper, C.; Fraser, C.; Gordon-Smith, K.; Hyde, S.; Farmer, A.; et al. Clinical characteristics of unipolar disorder and bipolar disorder according to the lifetime presence of recurrent panic attacks. Bipolar Disord. 2009, 11, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Feske, U.; Frank, E.; Mallinger, A.G.; Houck, P.R.; Fagiolini, A.; Shear, M.K.; Grochocinski, V.J.; Kupfer, D.J. Anxiety as a correlate of response to the acute treatment of bipolar I disorder. Am. J. Psychiatry 2000, 157, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Frank, E.; Shear, M.K.; Rucci, P.; Cyranowski, J.M.; Endicott, J.; Fagiolini, A.; Grochocinski, V.J.; Houck, P.; Kupfer, D.J.; Maser, J.D.; et al. Influence of panic-agoraphobic spectrum symptoms on treatment response in patients with recurrent major depression. Am. J. Psychiatry 2000, 157, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Tondo, L.; Vazquez, G.; Baldessarini, R.J. Mania associated with antidepressant treatment: Comprehensive meta-analytic review. Acta Psychiatr. Scand. 2010, 121, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Ketter, T.A.; Miller, S.; Dell’Osso, B.; Wang, P.W. Treatment of bipolar disorder: Review of evidence regarding quetiapine and lithium. J. Affect. Disord. 2016, 191, 256–273. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.F. False suffocation alarms, spontaneous panics, and related conditions. An integrative hypothesis. Arch. Gen. Psychiatry 1993, 50, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Perna, G.C.D.; Bellodi, L. Panic disorder: From respiration to the homeostatic brain. Acta Neuropsychiatr. 2004, 16, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Vidyasagar, M. Identifying predictive features in drug response using machine learning: Opportunities and challenges. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Maron, E.; Nutt, D. Biological predictors of pharmacological therapy in anxiety disorders. Dialogues Clin. Neurosci. 2015, 17, 305–317. [Google Scholar] [PubMed]
- Caldirola, D.; Perna, G. Is there a role for pharmacogenetics in the treatment of panic disorder? Pharmacogenomics 2015, 16, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Perna, G.; Bertani, A.; Caldirola, D.; Gabriele, A.; Cocchi, S.; Bellodi, L. Antipanic drug modulation of 35% CO2 hyperreactivity and short-term treatment outcome. J. Clin. Psychopharmacol. 2002, 22, 300–308. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders DSM-III; Amer Psychiatric Pub: Washington, DC, USA, 1980. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders DSM-III-R; Amer Psychiatric PuB: Washington, DC, USA, 1987. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders DSM-IV; Amer Psychiatric Pub: Washington, DC, USA, 1994. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders DSM-IV-TR Fourth Edition (Text Revision), 4th ed.; Amer Psychiatric Pub: Washington, DC, USA, 2000. [Google Scholar]
- Centers for Disease Control and Prevention International Classification of Diseases, Ninth Revision (ICD-9). Available online: http://www.cdc.gov/nchs/icd/icd9.htm (accessed on January 2016).
- Centers for Disease Control and Prevention International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9-CM). Available online: http://www.cdc.gov/nchs/icd/icd9cm.htm (accessed on January 2016).
- Centers for Disease Control and Prevention International Classification of Diseases, Tenth Revision (ICD-10). Available online: http://www.cdc.gov/nchs/icd/icd10.htm (accessed on January 2016).
- Higgins, J.P.; Altman, D.G.; Gøtzsche, P.C.; Jüni, P.; Moher, D.; Oxman, A.D.; Savovic, J.; Schulz, K.F.; Weeks, L.; Sterne, J.A.; et al. The Cochrane Collaboration's tool for assessing risk of bias in randomised trials. BMJ 2011, 343, d5928. [Google Scholar] [CrossRef] [PubMed] [Green Version]


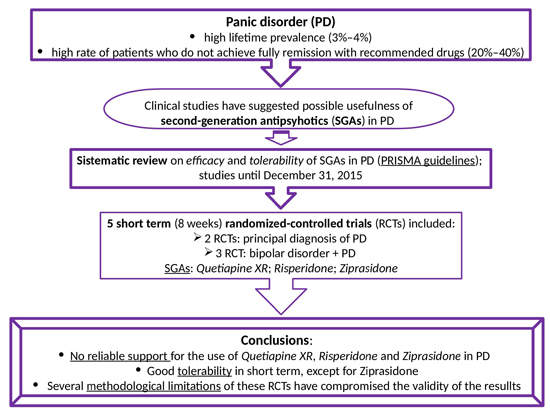{kind=link}
{kind=link}
| Authors, Year [Reference] | Study Design | Duration | Recruitment | Participants | Other Psychiatric Diagnoses (Number of Participants) | Psychiatric Assessment Instruments |
| Goddard et al., 2015 [26] | Single-site, double-blind, placebo-controlled, randomized, quetiapine XR (flexible-dose) coadministration trial | 8 weeks | Mixed strategies (referrals from local clinicians, flyers in the university hospital, on-line bulletins, ads in local newspapers) | Patients with primary, current PD with or without AG, SSRI-/SNRI-resistant | In the ITT patients: GAD (n = 8), PTSD (n = 3), MDD (n = 8), depression NOS (n = 2), dysthymia (n = 1), ADD (n = 1), bulimia (n = 1) | Mini-International Neuropsychiatric Interview (MINI Plus) for DSM-IV. Qualification of interviewer: not reported |
| Prosser et al., 2009 [27] | Single-site, single (i.e., rater)-blind, medication-controlled, flexible doses, randomized | 8 weeks | Mixed strategies (recruitment from inpatients psychiatric units, a psychiatric outpatient service, ads in local newspapers and an Internet website) | Patients with PD, with or without AG; patients with MDD and PAs | None; patients with any other current/lifetime Axis I diagnosis were excluded | Unstructured psychiatric clinical interview conducted by experienced psychiatrists, according to DSM-IV criteria |
| Suppes et al., 2014 [28] | Three-site, double-blind, placebo-controlled, flexible doses, randomized | 8 weeks | Outpatients | Patients with lifetime BD (I/II/NOS) and with lifetime PD, with or without AG or GAD | Not reported | Structured Clinical Interview for DSM-IV-TR (SCID). Qualification of interviewer: not reported |
| Sheehan et al., 2009 [29] | Three-site, double-blind, placebo-controlled, flexible doses, randomized | 8 weeks | Outpatients recruited from three University sites by ads | Patients with lifetime BD (I/II/NOS) and with lifetime PD, with or without AG or GAD | Not reported | Mini-International Neuropsychiatric Interview (MINI Plus) for DSM-IV. Qualification of interviewer: not reported |
| Sheehan et al., 2013 [30] | Three-site, double-blind, placebo-controlled, flexible doses, randomized | 8 weeks | Outpatients | Patients with lifetime BD (I/II/not otherwise specified) and with lifetime PD, with or without AG or GAD | Not reported | Mini-International Neuropsychiatric Interview (MINI Plus) for DSM-IV. Qualification of interviewer: not reported |
| Authors, Year [Reference] | Number of Randomized Patients | Treatments (n = Number of Participants) | Daily Dose Range (mg) | Mean Daily Dose (mg, (SD) or Range) | Completer Population (n = Number of Participants) | |
| Goddard et al., 2015 [26] | 27 | Adjunctive treatment to SSRI/SNRI stable dose (i.e., baseline SSRI/SNRI doses were held constant throughout the 8-week trial): quetiapine XR (n = 14), PLB (n = 13). No other psychotropic medications were allowed during the study (urine toxicology: yes) | Quetiapine XR 50–400 | 150 (106) | n = 21 (78% of the randomized group) | |
| Prosser et al., 2009 [27] | 56 | Monotherapy: risperidone (n = 33) or paroxetine (standard-of-care) (n = 23). The observed randomization distribution did not significantly deviate from the expected distribution on a 1: 1 basis (i.e., 28 subjects per treatment group). No other psychotropic medications were allowed during the study (urine toxicology: no). Period of withdrawal from previous medication: not reported | Risperidone: 0.125–16, paroxetine: 30–60 | Risperidone: 0.53 (range 0.125–1.0), paroxetine: all participants received 30 mg, except one who received 40 mg | n = 29 (51.8% of the randomized group). Risperidone: n = 20 (60.6%); paroxetine: n = 9 (39.1%) (no significant difference in the retention rate between the two treatment groups) | |
| Suppes et al., 2014 [28] | 49 | Monotherapy: ziprasidone (n = 25, % female = 76, mean age (SD) = 36.7 (17.7) years), PLB (n = 24, % female = 70.8, mean age (SD) = 34.6 (12.2) years). Participants discontinued any psychotropic medication for at least 1 week before baseline (if fluoxetine or depot antipsychotics: for at least 4 weeks). Adjunctive lorazepam (up to 2 mg die) allowed during the first 2 weeks of the study. Zolpidem/zaleplon for insomnia and benztropine for EPs allowed throughout the study | Ziprasidone 40–160 | 146.7 (20.7) | n = 23 (46.9% of the randomized group). Ziprasidone: n = 6 (24%); PLB: n = 17 (70.8%) (p = 0.001) | |
| Sheehan et al., 2009 [29] | 111 | Monotherapy: risperidone (n = 54, % female = 66.7, mean age (SD) = 35.1 (12.4) years, participants with lifetime PD: n = 45, 83.3%), PLB (n = 57, % female = 61.4; mean age (SD) = 38.4 (12.8) years, participants with lifetime PD: n = 35, 61.4%). Participants discontinued any psychotropic medication for at least 1 week before baseline (if fluoxetine or depot antipsychotics: for at least 4 weeks). Adjunctive lorazepam allowed during the first 1 week (up to 2 mg die) and the second week (up to 1 mg die) of the study. Zolpidem/zaleplon for insomnia allowed throughout the study | Risperidone 0.5–4 | 2.5 (1.1) | n = 63 (56.7% of the randomized group). Risperidone: n = 27 (50%); PLB: n = 36 (63%) | |
| Sheehan et al., 2013 [30] | 149 | Monotherapy: quetiapine XR (n = 49, % female = 57.1, mean age (SD) = 41.4 (12.1) years, participants with current PD: n = 37), divalproex XR (n = 49, % female = 55.1%, mean age (SD) = 37.5 (12.0) years, participants with current PD: n = 37), PLB (n = 51, % female = 64.7; mean age (SD)= 37.6 (11.6) years, participants with current PD: n = 39). Participants discontinued any psychotropic medication for at least 1 week before baseline (if fluoxetine or depot antipsychotics: for at least 4 weeks). Adjunctive lorazepam allowed during the first 1 week (up to 2 mg die) and the second week (up to 1 mg die) of the study. Zolpidem/zaleplon for insomnia allowed throughout the study | Quetiapine XR 50–300, divalproex XR 500–3000 | Quetiapine XR: 186.4 (100.3), divalproex XR: 1991 (866) | n = 108 (72.5% of the randomized group). Quetiapine XR: n = 38 (77.5%); divalproex XR: n = 35 (71.4%); PLB: n = 35 (68.6%) | |
| Authors, Year [Reference] | ITT Population (n = Number of Participants) | Significant Baseline Differences in Socio-Demographic/Clinical Characteristic between Treatment Groups | Main Outcome Measures and Results | |||
| Goddard et al., 2015 [26] | n = 26 (quetiapine XR, n = 13, % female = 77; mean age (SD) = 35.5 (9.6) years; PLB, n = 13, % female = 62, mean age (SD) = 35.5 (16.8) years). LOCF imputation was used for participants who withdrew prematurely | None | PDSS total scores; PDSS Item 1 (panic attack frequency) score; rates of responders (i.e., ≥50% improvement from baseline PDSS total score); rate of remitters (i.e., PDSS total score ≤4) at endpoint. Both in the ITT and completer populations: significant improvement in panic symptoms over the trial (main effect of time, p < 0.0001), but no significant drug/PLB differences. | |||
| Prosser et al., 2009 [27] | n = 56 (risperidone, n = 33, % female = 76; mean age (SD) = 38.8 (9.7) years, PD diagnosis n = 24 (73%); paroxetine, n = 23, % female = 65, mean age (SD) = 42.6 (14.3) years, PD diagnosis n = 19 (83%). LOCF imputation was used for participants who withdrew prematurely | Significantly higher HAM-D scores in the paroxetine group than risperidone group (p = 0.049) | CGI, HAMA, HAMD scores, PDSS total scores, PDSS Items 1 (panic attack frequency) and 2 (panic attack severity) scores, SPAS-P score. In the ITT population: significant improvement in all of the outcome measures over the trial, except for SPAS-P score. No significant differences between risperidone and paroxetine. Baseline HAMD scores significantly correlated with both baseline and midpoint/endpoint outcome HAMA scores and with midpoint outcome PDSS total scores. No analyses in completer population. No analyses in the subgroup with PD. | |||
| Suppes et al., 2014 [28] | n = 46 (ziprasidone, n = 23; PLB, n = 23). LOCF imputation was used for participants who withdrew prematurely | Significantly higher SSTS scores in the ziprasidone group than PLB group (p = 0.037) | CGI-21 Anxiety, SDS scores. In the ITT population: significant improvement in both measures over the trial, but no significant differences between ziprasidone and PLB. No analyses in completer population. No analyses in the subgroup with PD. | |||
| Sheehan et al., 2009 [29] | n = 102 (risperidone, n = 49; PLB, n = 53). LOCF imputation was used for participants who withdrew prematurely | Significantly higher rate of participants with a mixed mood state (59% vs. 40%, p < 0.05) and lifetime PD (83.3% vs. 61.4%, p < 0.01) in the risperidone group than the PLB group | CGI-21 Anxiety score. No difference in improvement between risperidone and PLB. Over the trial, the improvement was similar for patients with and without PD. Within the subgroup with PD, the PLB group showed a trend towards greater improvement (p < 0.07). No analyses in the completer population. | |||
| Sheehan et al., 2013 [30] | n = 144 (quetiapine XR n = 47, divalproex XR n = 46; PLB, n = 51). LOCF imputation was used for participants who withdrew prematurely | None | CGI-21 Anxiety score. The quetiapine XR group had a numerically higher improvement compared to divalproex XR and PLB, but it did not reach statistical significance (p < 0.07). No analyses in the completer population were available. | |||
| Authors, Year [Reference] | Secondary Outcome Measures and Other Results | Side Effects/Tolerability | Funding | |||
| Goddard et al., 2015 [26] | CGI-S, CGI-I, HAMA, HAMD, PSQI scores: significant improvement over the trial (p < 0.0001; PSQI (sleep quality item) p < 0.05), but no significant drug/PLB differences. | Three patients in the quetiapine XR group discontinued early due to medication-related SEs (sedation, n = 1; derealization, n = 2). No significant difference in SEs emerged between quetiapine XR and PLB, including sedation/somnolence, extrapyramidal SEs and akathisia (BARS, SAS), weight gain and blood glucose levels. | AstraZeneca | |||
| Prosser et al., 2009 [27] | Preliminary evidence of significantly faster decrease of HAMA and HAMD in the risperidone group than the paroxetine group (t-tests, without corrections for multiple comparisons). | No significant difference between the two groups in the number of participants who discontinued early due to intolerable (unspecified) SEs (n = 2 in the risperidone group; n = 1 in the paroxetine group). The reason for attrition was not collected for 7 participants who discontinued early. No other information available on SEs. | Partial support by a grant from New York State Empire Clinical Research Investigation Award | |||
| Suppes et al., 2014 [28] | PGI-21, HAMA, SPS, CGI-BP, YMRS, MADRS, SSTS, SIS scores. No significant differences between ziprasidone and PLB. | Compared to PLB, a significantly higher number of participants in the ziprasidone group withdrew from the study due to AEs/SEs (n = 9 and 2, respectively, p = 0.02). The ziprasidone group reported significantly more sleep disturbance (p = 0.040), sedation/somnolence (p = 0.049), weight gain (p = 0.035) and higher increase of the AIMS scores (p = 0.003) than PLB. No difference in akathisia (BARS) was found between the two groups. | Pfizer | |||
| Sheehan et al., 2009 [29] | SPS, HAMA, PG-21 Anxiety, YMRS, IDS, CGIBP, SDS scores. No difference between risperidone and PLB in improvement on all outcome measures. Over the trial, improvement of the HAMA score was similar for patients with and without PD. Within the subgroup with PD, the PLB group had a significantly lower mean endpoint HAMA score than the risperidone group (p < 0.007), but no analyses on the specific panic symptom scale (SPS) were available. | One participant in the risperidone group and one in the PLB group discontinued early due to treatment-related adverse events (risperidone: one episode of heightened anxiety and anger; PLB: multiple symptoms). Drowsiness was the only side effect that was two times more frequent in the risperidone group than the PLB group. Extrapyramidal symptoms (BARS, SAS, AIMS) did not significantly differ between the two groups. Weight gain was numerically higher in the risperidone group, but without statistical significance. | Janssen Pharmaceutica | |||
| Sheehan et al., 2013 [30] | HAMA, SPS, PGI-21 Anxiety, YMRS, MADRS, CGIBP, SIS, RISC, SSTS, SDS scores. The quetiapine XR group had a significantly greater improvement on HAMA and SPS compared to both divalproex XR and PLB groups (p < 0.05). No significant differences between groups were found on PGI-21 Anxiety, YMRS, SSTS, SIS and RISC scores. The quetiapine XR group had a significantly greater improvement on the MADRS score compared to both divalproex and PLB groups (p < 0.05) and on the SDS and CGIBP depression scores compared to divalproex XR (p < 0.05 and p < 0.04, respectively). In the subgroup with current PD (n = 113), the quetiapine XR group had a significantly greater improvement on HAMA and SPS compared to the divalproex XR group (p < 0.05). | One participant in the quetiapine XR group, 3 in the divalproex XR group and one in the PLB group discontinued early due to treatment-related adverse events. The most common SEs in the three groups were: drowsiness, dry mouth, nausea, tingling, increased appetite, sedation, headache. Participants in quetiapine XR reported significantly higher rates of dry mouth compared to both divalproex XR and PLB (p < 0.006). Participants in quetiapine XR and divalproex XR reported greater weight gain compared to PLB (p < 0.001, p < 0.03, respectively). Extrapyramidal symptoms (BARS, SAS, AIMS) did not significantly differ by treatment group. | AstraZeneca | |||
| Authors, Year | Selection Bias | Performance Bias | Detection Bias | Attrition Bias | Reporting Bias | ||
| Random Sequence Generation | Allocation Concealment | Blinding of Participants | Blinding of Personnel | Blinding of Outcome Assessors | Incomplete Outcome Data | Selective Reporting | |
| Goddard et al., 2015 [26] | U Participants were randomized sequentially. | L Pharmacy-controlled randomization; identical-appearing PLB/quetiapine tablets; coordinator (not involved in patients ratings) managed the medication bottles. | L | L | L | L | L |
| Prosser et al., 2009 [27] | L Computer number random generator (SPSS 12.0.1). | U | H Participants not blinded. | U | U | H High attrition rate. | L |
| Suppes et al., 2014 [28] | U Randomized block design. | U | U Blinded guess of treatment performed. Participants were accurate 71.4% of the time; those in the PLB group and the ziprasidone group were accurate 100% and 66.7% of the time, respectively. | U Blinded guess of treatment performed. Treating clinicians were accurate 47.6% of the time; within the ziprasidone group: 66.7% of the time. | U Blinded guess of treatment performed. Raters were accurate 42.3% of the time. | H High attrition rate; significant difference in the retention rate between the two treatment groups. | L |
| Sheehan et al., 2009 [29] | U Not reported. | U | U | U | U | H High attrition rate. | L |
| Sheehan et al., 2013 [30] | U Not reported. | L Study medications were encapsulated using a double-dummy design. | L | U | U | L | L |
| Authors, Year | Sampling Bias | Other Bias | |||||
| Recruitment Strategies | Inclusion/Exclusion Criteria | Power Calculation | Adjunctive Medication | Adjunctive Bias | |||
| Goddard et al., 2015 [26] | H Mixed recruitment strategies | H Mixed criteria to define SSRI/SNRI resistance (inclusion criterion). | Power calculation performed. Study powered to detect large effect sizes. H for small-to-moderate effects. | L Not permitted; urine toxicology performed to detect surreptitious use of benzodiazepines. | |||
| Prosser et al., 2009 [27] | H Mixed recruitment strategies | H Sample with mixed diagnoses (no separate analyses in the PD subgroup). Lack of specific panic symptom measures as inclusion criteria. | U No power calculation reported. | U Not permitted; urine toxicology not declared. | U for previous medications: period of withdrawal not reported. H for medication initiation: no titration in one of the two treatment groups (paroxetine); H for baseline imbalance: higher baseline depression severity in one of the two groups (paroxetine), related to outcome measures. | ||
| Suppes et al., 2014 [28] | U | H Sample with mixed diagnoses (no separate analyses in the PD subgroup). Lack of specific panic symptom measures as inclusion criteria. | Power calculation performed. Study powered to detect large effect sizes. H for small-to-moderate effects. | L Some adjunctive medications permitted. No differences in adjunctive medication distribution between the two treatment groups. | |||
| Sheehan et al., 2009 [29] | L | H Sample with mixed diagnoses (no separate analyses on panic symptoms in the PD subgroup). Lack of specific panic symptom measures as inclusion criteria. | U No power calculation reported. | L Some adjunctive medications permitted. No significant influence on the results was found. | H for baseline imbalance: higher rate of participants with mixed mood state and with PD in one of the two groups (risperidone). | ||
| Sheehan et al., 2013 [30] | U | H Lack of specific panic symptom measures as inclusion criteria. | U No power calculation reported. | L Some adjunctive medications permitted. No differences in adjunctive medication distribution between groups. | |||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perna, G.; Alessandra, A.; Raffaele, B.; Elisa, M.; Giuseppina, D.; Paolo, C.; Maria, N.; Daniela, C. Is There Room for Second-Generation Antipsychotics in the Pharmacotherapy of Panic Disorder? A Systematic Review Based on PRISMA Guidelines. Int. J. Mol. Sci. 2016, 17, 551. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040551
Perna G, Alessandra A, Raffaele B, Elisa M, Giuseppina D, Paolo C, Maria N, Daniela C. Is There Room for Second-Generation Antipsychotics in the Pharmacotherapy of Panic Disorder? A Systematic Review Based on PRISMA Guidelines. International Journal of Molecular Sciences. 2016; 17(4):551. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040551
Chicago/Turabian StylePerna, Giampaolo, Alciati Alessandra, Balletta Raffaele, Mingotto Elisa, Diaferia Giuseppina, Cavedini Paolo, Nobile Maria, and Caldirola Daniela. 2016. "Is There Room for Second-Generation Antipsychotics in the Pharmacotherapy of Panic Disorder? A Systematic Review Based on PRISMA Guidelines" International Journal of Molecular Sciences 17, no. 4: 551. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040551