
Smad3 Sensitizes Hepatocelluar Carcinoma Cells to Cisplatin by Repressing Phosphorylation of AKT

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
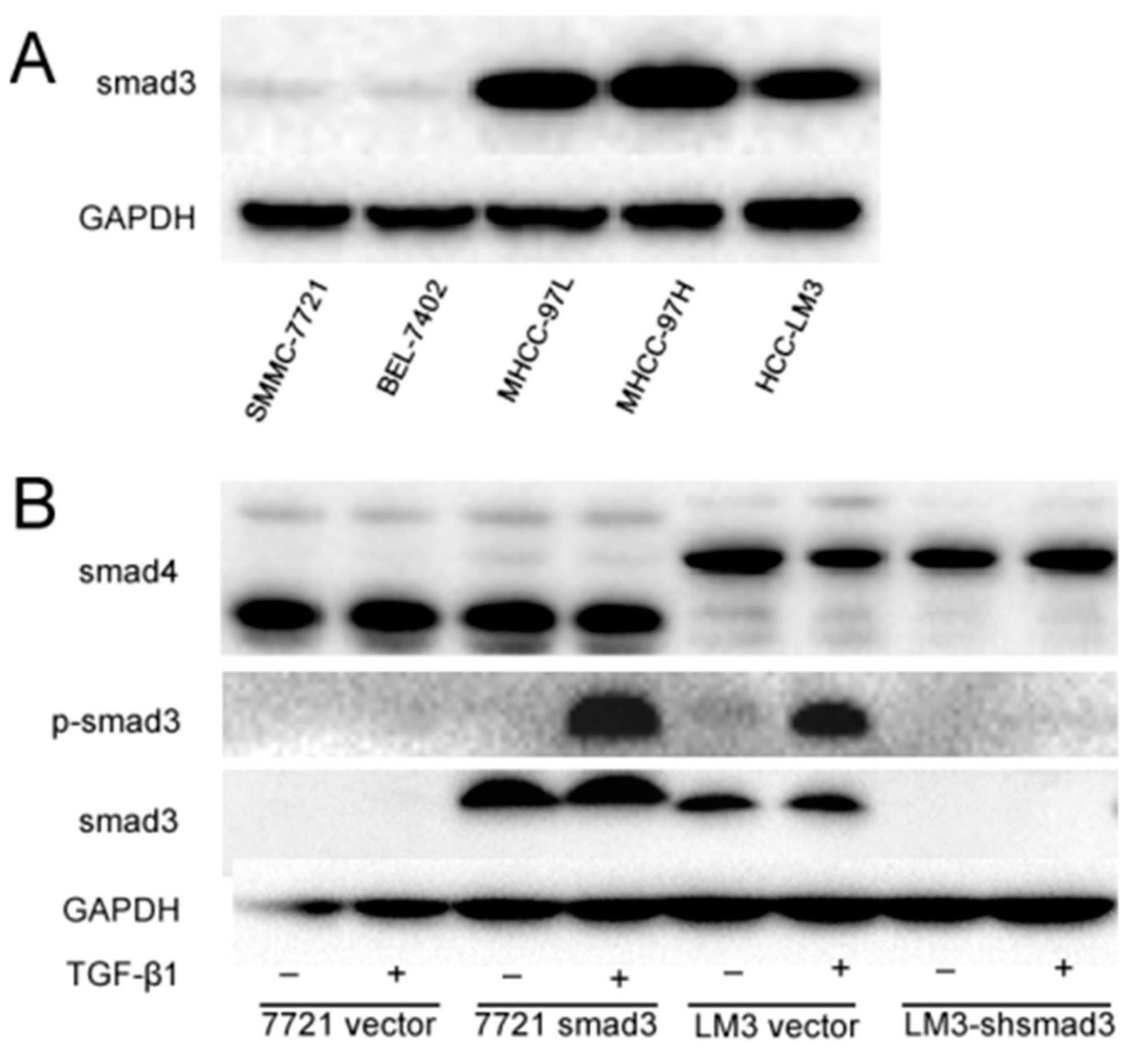
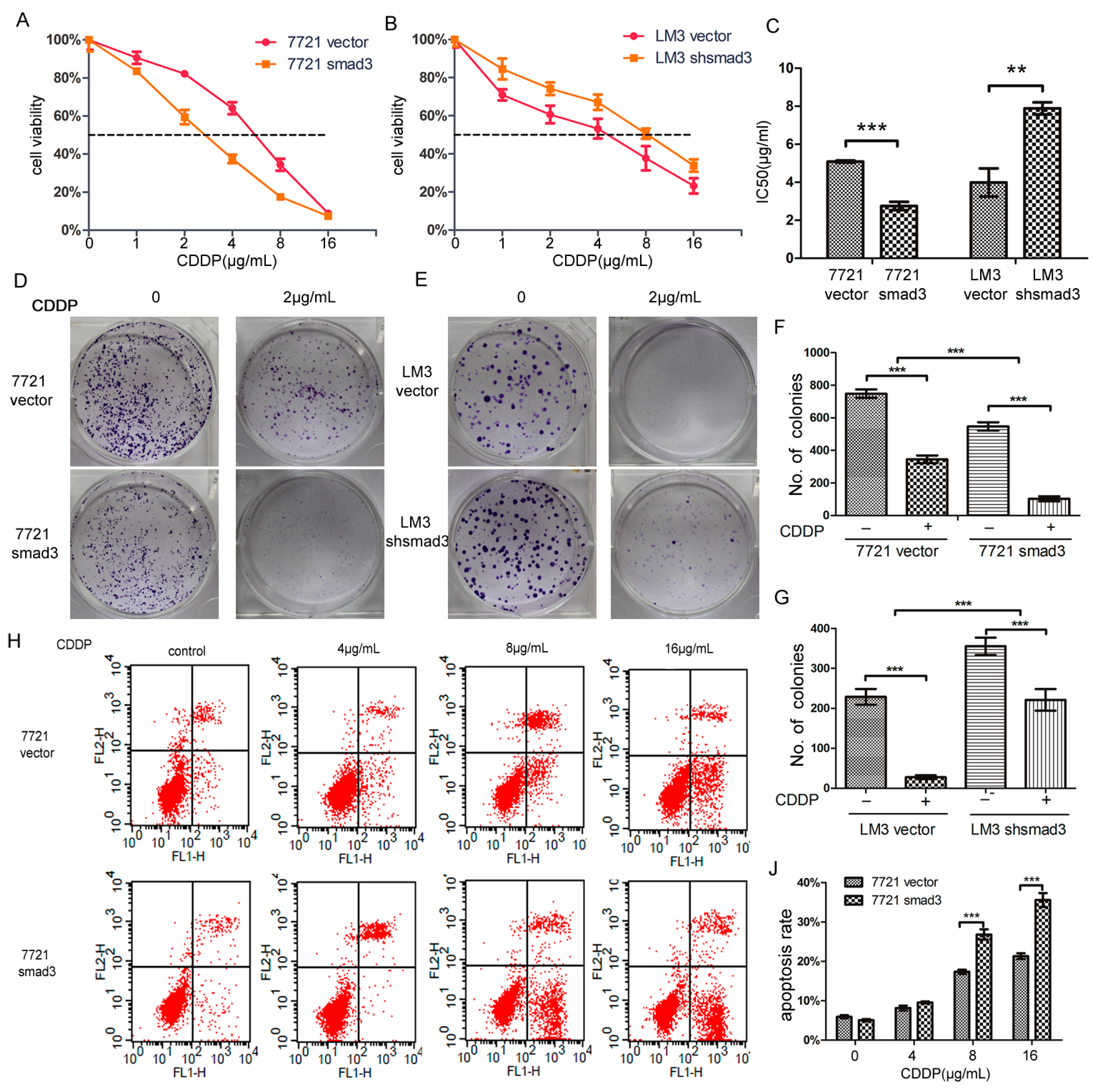
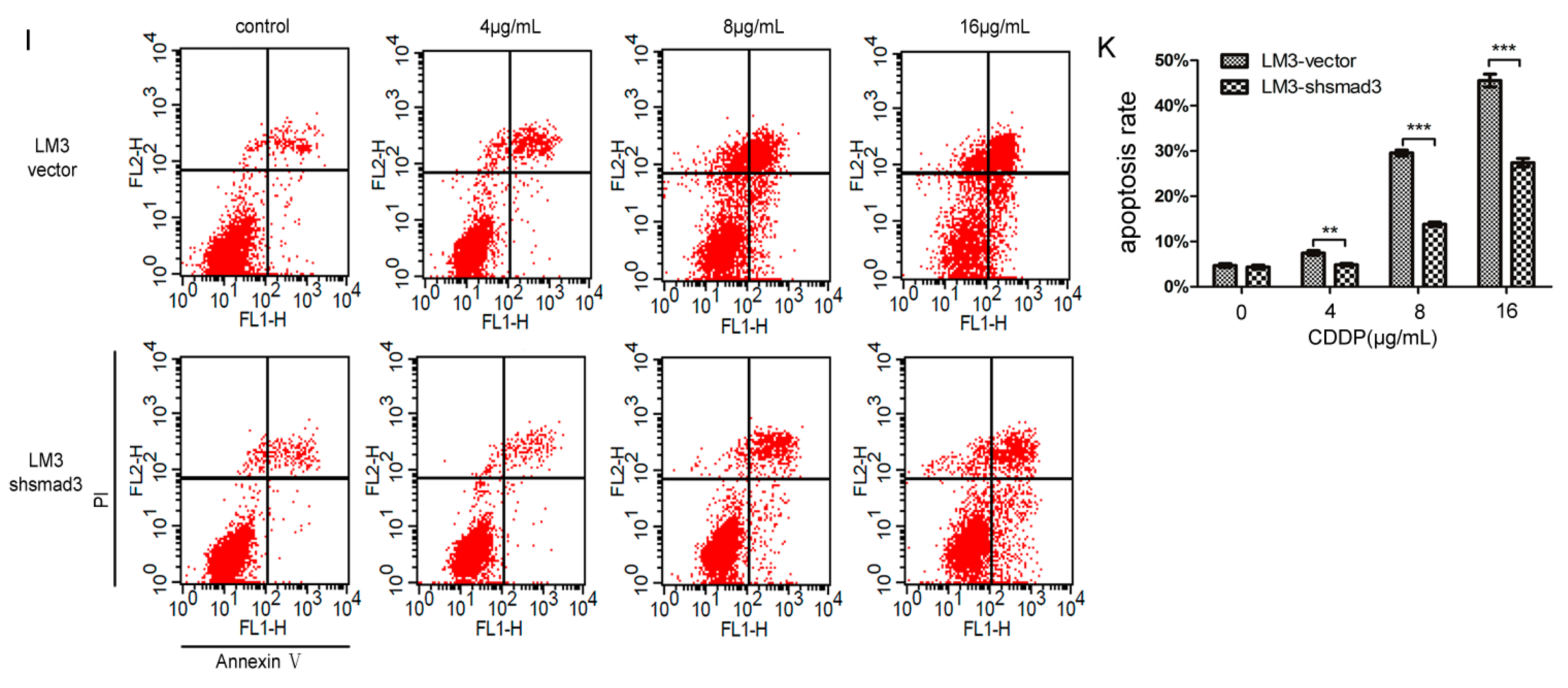
2.1. Smad3 Increases the Sensitivity of Heptocelluar Carcinoma (HCC) to Cisplatin in Vitro
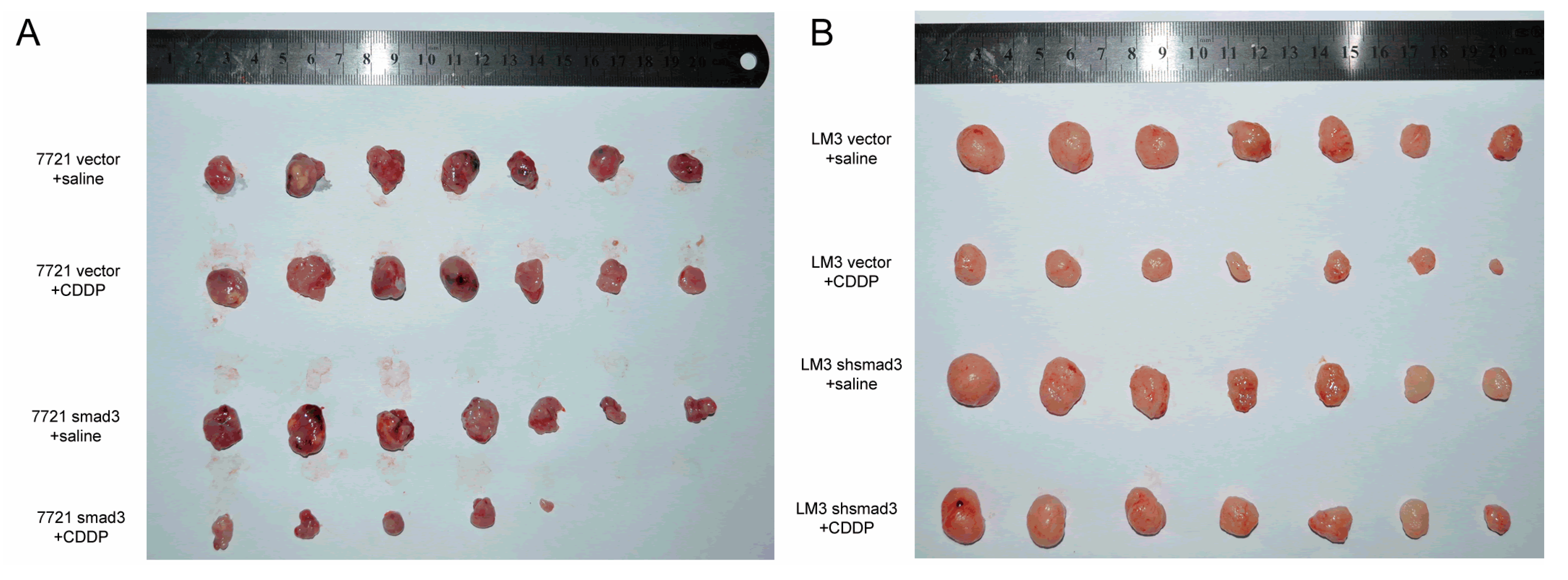
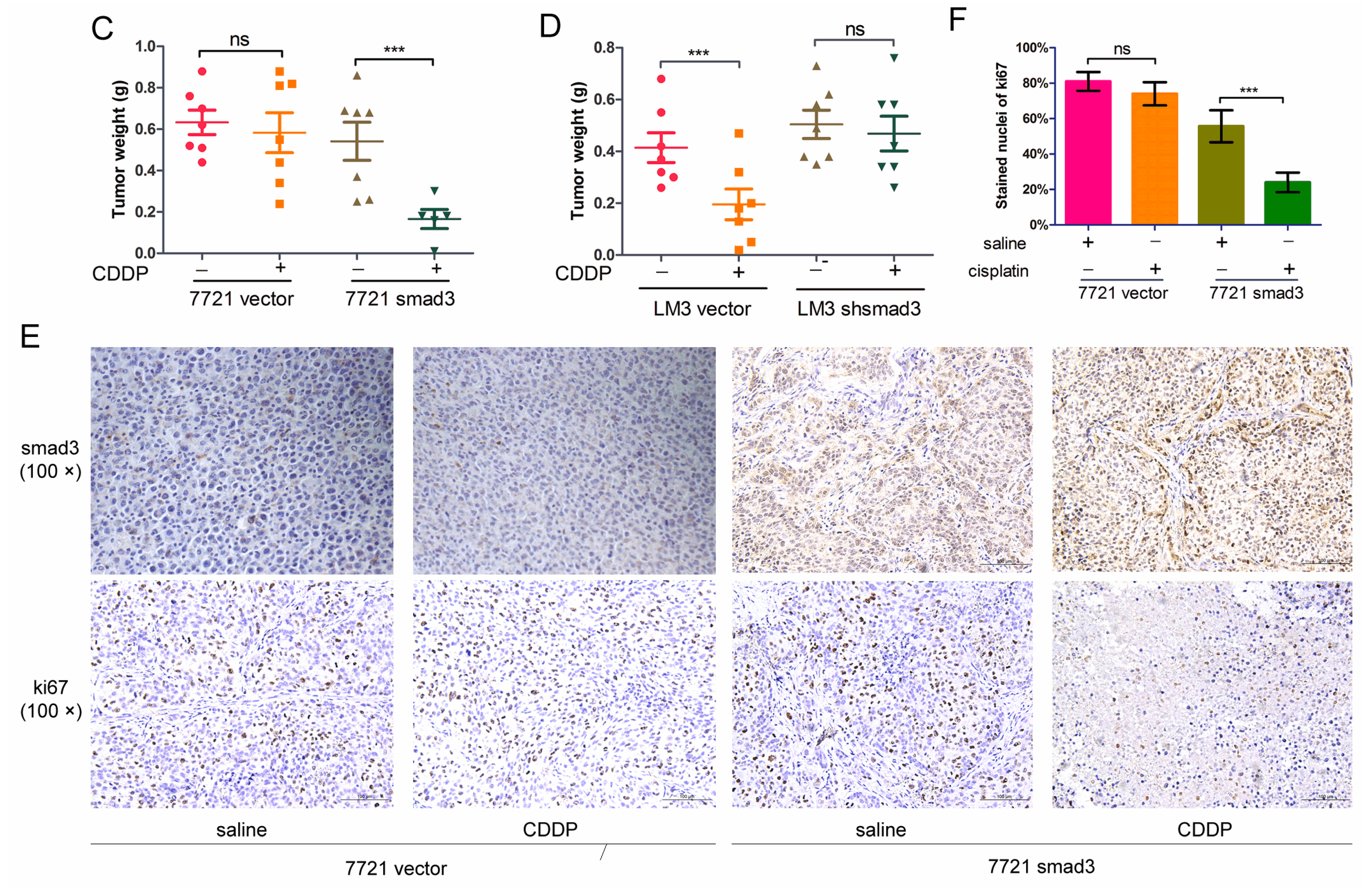
2.2. Smad3 Increases the Sensitivity of HCC to Cisplatin in Vivo
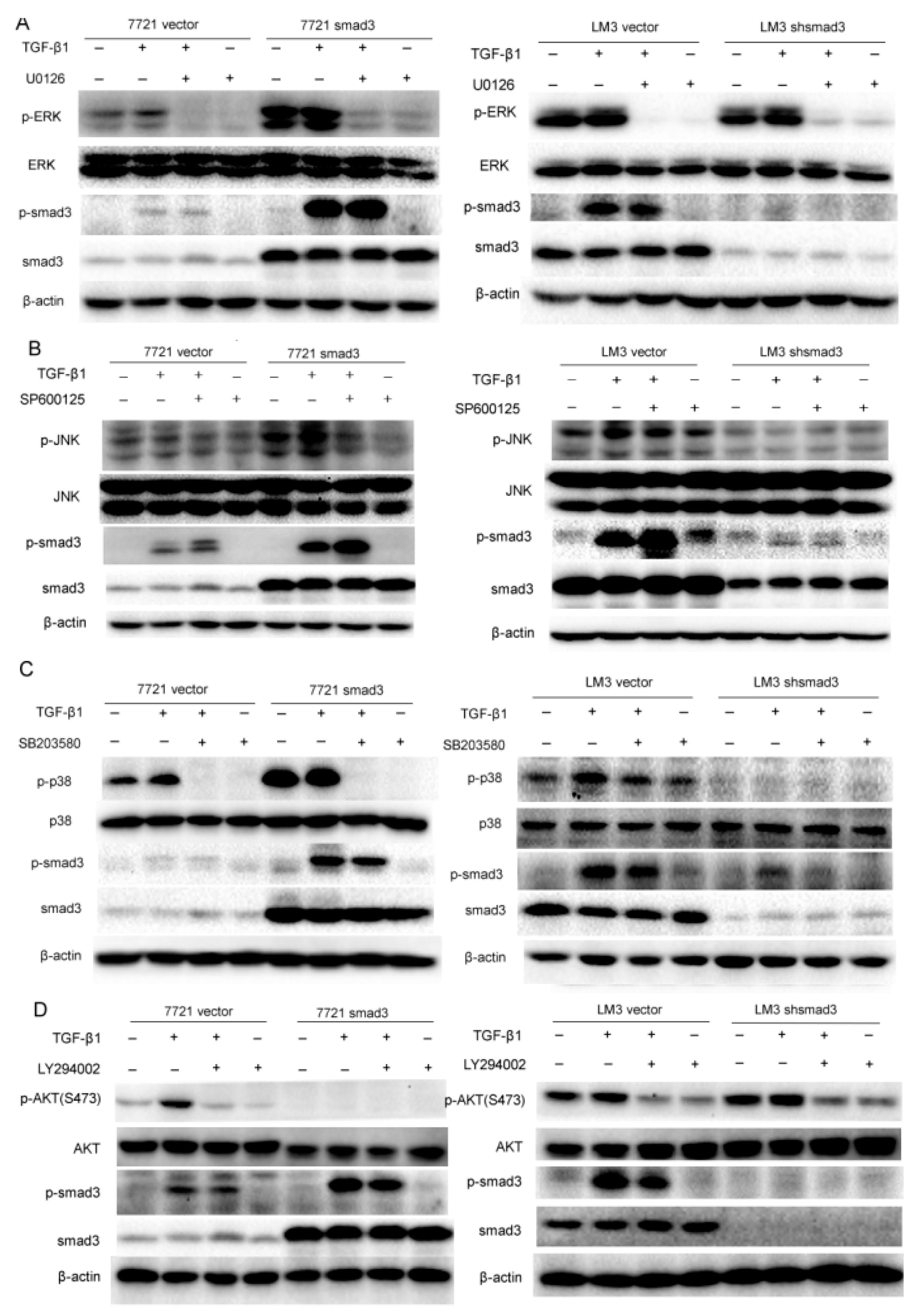
2.3. Smad3 Activates MAPK but Represses AKT Signaling
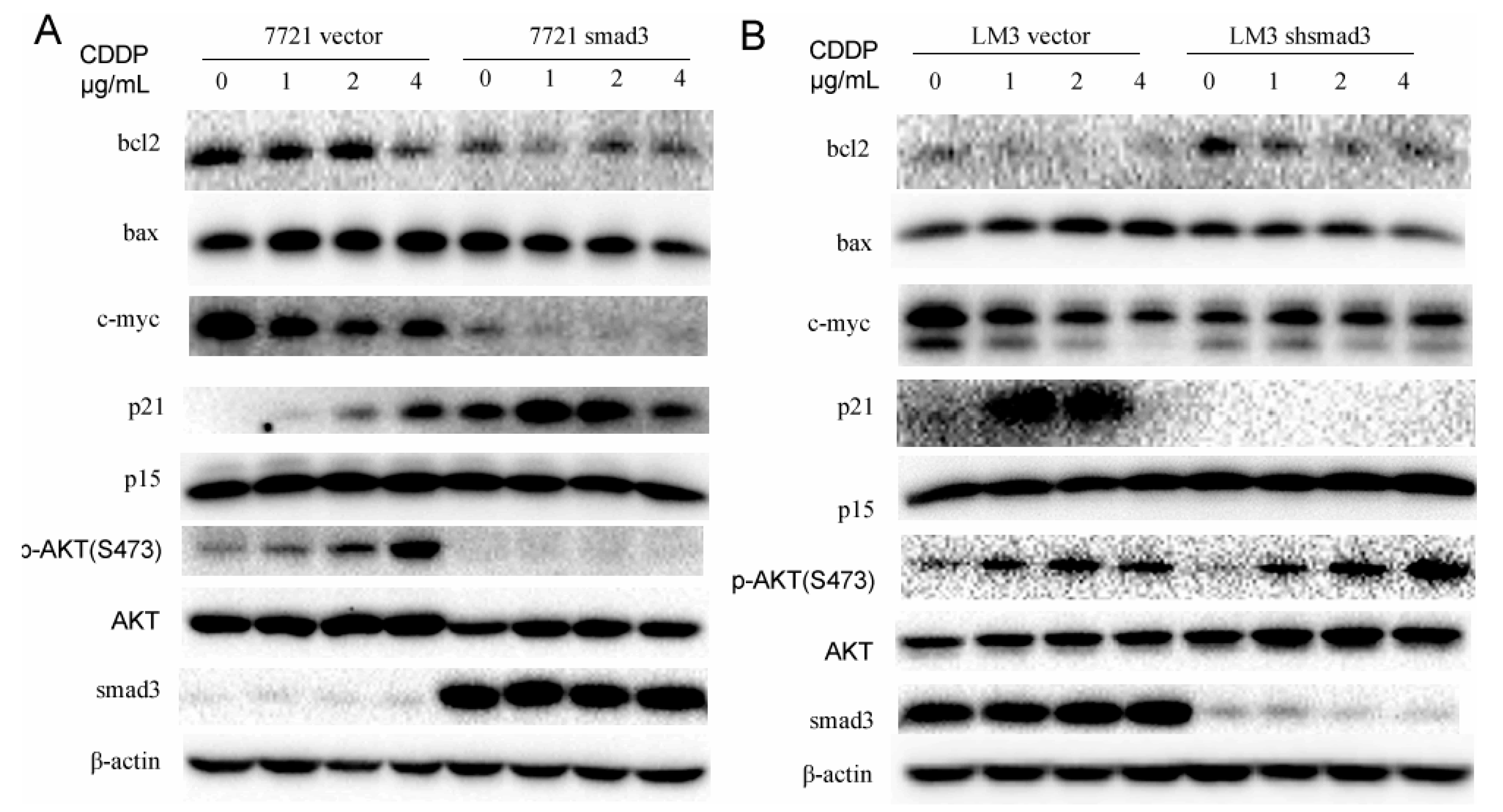
2.4. Smad3 Represses AKT Phosphorylation and Regulates Apoptosis-Related Proteins in the Presence of Cisplatin
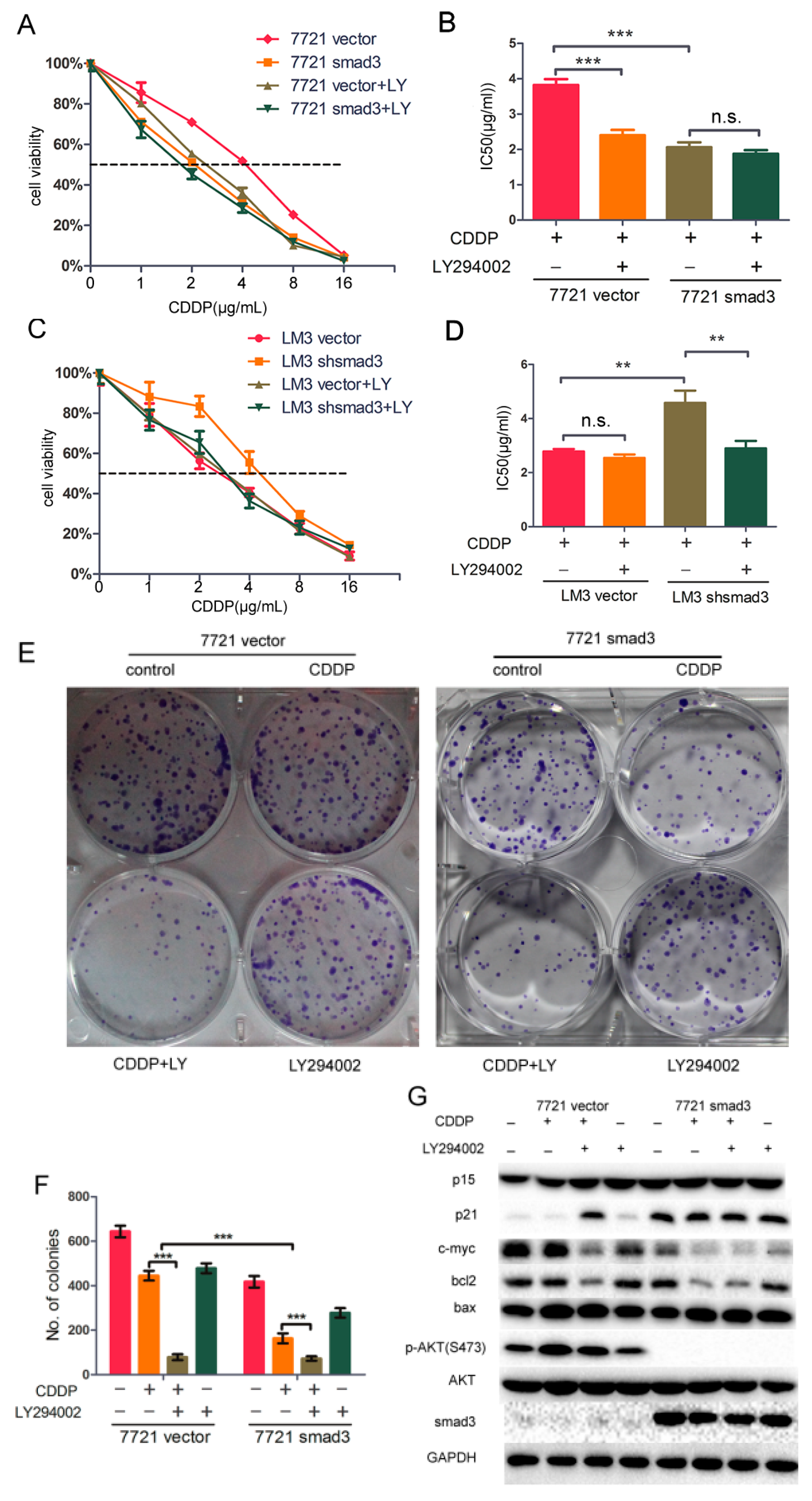
2.5. LY294002 Restores Chemosensitivity of HCC in Smad3-Defeciency Cells
3. Discussion
4. Methods and Materials
4.1. Cell Lines and Culture
4.2. Materials
4.3. Retrovirus Production, Virus Infection and Establishment of Stable Cell Clones
4.4. Cell Viability Assay
4.5. Colony Formation Assay
4.6. Flow-Cytometric Analysis
4.7. Tumorigenicity Assay
4.8. Western Blotting Assay
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Bruix, J.; Sherman, M. Practice guidelines committee, american association for the study of liver diseases. Management of hepatocellular carcinoma. Hepatology 2005, 42, 1208–1236. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Bruix, J.; Gores, G.J.; Mazzaferro, V. Hepatocellular carcinoma: Clinical frontiers and perspectives. Gut 2014, 63, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Kishi, Y.; Hasegawa, K.; Sugawara, Y.; Kokudo, N. Hepatocellular carcinoma: Current management and future development-improved outcomes with surgical resection. Int. J. Hepatol. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Poon, R.T.; Fan, S.T.; Lo, C.M.; Liu, C.L.; Wong, J. Long-term survival and pattern of recurrence after resection of small hepatocellular carcinoma in patients with preserved liver function: Implications for a strategy of salvage transplantation. Ann. Surg. 2002, 235, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.; Meyer, T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J. Hepatol. 2012, 56, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGF-β signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, A.; Curley, S.A.; Wu, X.; Brown, P.; Hwang, J.P.; Shetty, K.; Yao, Z.X.; He, A.R.; Li, S.; Katz, L.; et al. Hepatic stem cells and transforming growth factor β in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Smad phospho-isoforms direct context-dependent TGF-β signaling. Cytokine Growth Factor Rev. 2013, 24, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Stathopoulos, G.P. Cisplatin: Process and future. J. BUON 2013, 18, 564–569. [Google Scholar] [PubMed]
- Brozovic, A.; Osmak, M. Activation of mitogen-activated protein kinases by cisplatin and their role in cisplatin-resistance. Cancer Lett. 2007, 251, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chen, A.Y.; Ye, X.; Li, B.; Rojanasakul, Y.; Rankin, G.O.; Chen, Y.C. Myricetin inhibits proliferation of cisplatin-resistant cancer cells through a p53-dependent apoptotic pathway. Int. J. Oncol. 2015, 47, 1494–1502. [Google Scholar] [CrossRef] [PubMed]
- Muscella, A.; Urso, L.; Calabriso, N.; Vetrugno, C.; Rochira, A.; Storelli, C.; Marsigliante, S. Anti-apoptotic effects of protein kinase c-δ and c-fos in cisplatin-treated thyroid cells. Br. J. Pharmacol. 2009, 156, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Echevarria-Vargas, I.M.; Valiyeva, F.; Vivas-Mejia, P.E. Upregulation of miR-21 in cisplatin resistant ovarian cancer via JNK-1/c-Jun pathway. PLoS ONE 2014, 9, e97094. [Google Scholar]
- Yang, Y.; Zhang, P.; Zhao, Y.; Yang, J.; Jiang, G.; Fan, J. Decreased MicroRNA-26a expression causes cisplatin resistance in human non-small cell lung cancer. Cancer Biol. Ther. 2015. [Google Scholar] [CrossRef] [PubMed]
- Sussman, R.T.; Ricci, M.S.; Hart, L.S.; Sun, S.Y.; El-Deiry, W.S. Chemotherapy-resistant side-population of colon cancer cells has a higher sensitivity to TRAIL than the non-SP, a higher expression of c-myc and TRAIL-receptor DR4. Cancer Biol. Ther. 2007, 6, 1490–1495. [Google Scholar] [CrossRef] [PubMed]
- Gordian, E.; Li, J.; Pevzner, Y.; Mediavilla-Varela, M.; Luddy, K.; Ohaegbulam, K.; Daniel, K.G.; Haura, E.B.; Munoz-Antonia, T. Transforming growth factor β signaling overcomes dasatinib resistance in lung cancer. PLoS ONE 2014, 9, e114131. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.Y.; Jin, G.N.; Wang, W.; Chen, W.X.; Wu, Y.H.; Ai, X.; Chen, L.; Zhang, W.G.; Liang, H.F.; Laurence, A.; et al. Reduced expression of transcriptional intermediary factor 1 γ promotes metastasis and indicates poor prognosis of hepatocellular carcinoma. Hepatology 2014, 60, 1620–1636. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Sherman, M. American association for the study of liver diseases. Management of hepatocellular carcinoma: An update. Hepatology 2011, 53, 1020–1022. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Millet, C.; Zhang, Y.E. Roles of smad3 in TGF-β signaling during carcinogenesis. Crit. Rev. Eukaryot. Gene Expr. 2007, 17, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Frederick, J.P.; Liberati, N.T.; Waddell, D.S.; Shi, Y.; Wang, X.F. Transforming growth factor β-mediated transcriptional repression of c-myc is dependent on direct binding of smad3 to a novel repressive smad binding element. Mol. Cell. Biol. 2004, 24, 2546–2559. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.H.; Lin, X.; Derynck, R. Smad2, smad3 and smad4 cooperate with sp1 to induce p15(Ink4b) transcription in response to TGF-β. EMBO J. 2000, 19, 5178–5193. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.D.; Guh, J.Y.; Chiou, S.J.; Chen, H.C.; Hung, W.C.; Chuang, L.Y. Sp1 and smad3 are required for high glucose-induced p21(WAF1) gene transcription in LLC-PK1 cells. J. Cell. Biochem. 2007, 102, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.A.; Zhang, G.M.; Feigenbaum, L.; Zhang, Y.E. Smad3 reduces susceptibility to hepatocarcinoma by sensitizing hepatocytes to apoptosis through downregulation of bcl2. Cancer Cell 2006, 9, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.; Neve, E.P.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A snail1-smad3/4 transcriptional repressor complex promotes TGF-β mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Lin, X.; Chiu, W.T.; Chen, Y.H.; Yu, G.; Liu, M.; Feng, X.H.; Sawaya, R.; Medema, R.H.; Hung, M.C.; et al. Sustained activation of smad3/smad4 by FOXM1 promotes TGF-β-dependent cancer metastasis. J. Clin. Investig. 2014, 124, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, R.K.; Nordheim, A.; Dittmer, J. Interfering with TGF-β-induced smad3 nuclear accumulation differentially affects TGF-β-dependent gene expression. Mol. Cancer 2003, 2. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.N.; Ding, W.Q.; Guo, X.J.; Yuan, X.W.; Wang, D.M.; Song, J.G. Epigenetic regulation of smad2 and smad3 by profilin-2 promotes lung cancer growth and metastasis. Nat. Commun. 2015, 6, 8230. [Google Scholar] [CrossRef] [PubMed]
- Tufegdzic Vidakovic, A.; Rueda, O.M.; Vervoort, S.J.; Sati Batra, A.; Goldgraben, M.A.; Uribe-Lewis, S.; Greenwood, W.; Coffer, P.J.; Bruna, A.; Caldas, C. Context-specific effects of TGF-β/smad3 in cancer are modulated by the epigenome. Cell Rep. 2015, 13, 2480–2490. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.X.; Shi, J.; Zhang, Y.; Zhao, J.S.; Long, L.Y.; Chen, T.W.; Zhang, E.B.; Feng, Y.Y.; Bao, W.D.; Deng, Y.Z.; et al. Sorafenib enriches epithelial cell adhesion molecule-positive tumor initiating cells and exacerbates a subtype of hepatocellular carcinoma through TSC2-AKT cascade. Hepatology 2015, 62, 1791–1803. [Google Scholar] [CrossRef] [PubMed]
- Zhai, B.; Jiang, X.; He, C.; Zhao, D.; Ma, L.; Xu, L.; Jiang, H.; Sun, X. Arsenic trioxide potentiates the anti-cancer activities of sorafenib against hepatocellular carcinoma by inhibiting AKT activation. Tumour Biol. 2015, 36, 2323–2334. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.J.; Wohlschlaeger, J.; Lang, H.; Sotiropoulos, G.C.; Malago, M.; Steveling, K.; Reis, H.; Cicinnati, V.R.; Schmid, K.W.; Baba, H.A. Activation of the ERK and AKT signalling pathway predicts poor prognosis in hepatocellular carcinoma and ERK activation in cancer tissue is associated with hepatitis C virus infection. J. Hepatol. 2008, 48, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Qi, Z.; Liu, B.; Ren, Y.; Li, H.; Yang, G.; Zhang, Q. RY-2f, an isoflavone analog, overcomes cisplatin resistance to inhibit ovarian tumorigenesis via targeting the PI3K/AKT/mTOR signaling pathway. Oncotarget 2015, 6, 25281–25294. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Yan, L.; Patnaik, A.; Fearen, I.; Olmos, D.; Papadopoulos, K.; Baird, R.D.; Delgado, L.; Taylor, A.; Lupinacci, L.; et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J. Clin. Oncol. 2011, 29, 4688–4695. [Google Scholar] [CrossRef] [PubMed]
- Molife, L.R.; Yan, L.; Vitfell-Rasmussen, J.; Zernhelt, A.M.; Sullivan, D.M.; Cassier, P.A.; Chen, E.; Biondo, A.; Tetteh, E.; Siu, L.L.; et al. Phase 1 trial of the oral AKT inhibitor MK-2206 plus carboplatin/paclitaxel, docetaxel, or erlotinib in patients with advanced solid tumors. J. Hematol. Oncol. 2014, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Okusaka, T.; Kasugai, H.; Shioyama, Y.; Tanaka, K.; Kudo, M.; Saisho, H.; Osaki, Y.; Sata, M.; Fujiyama, S.; Kumada, T.; et al. Transarterial chemotherapy alone versus transarterial chemoembolization for hepatocellular carcinoma: A randomized phase III trial. J. Hepatol. 2009, 51, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Otsuji, K.; Takai, K.; Nishigaki, Y.; Shimizu, S.; Hayashi, H.; Imai, K.; Suzuki, Y.; Hanai, T.; Ideta, T.; Miyazaki, T.; et al. Efficacy and safety of cisplatin versus miriplatin in transcatheter arterial chemoembolization and transarterial infusion chemotherapy for hepatocellular carcinoma: A randomized controlled trial. Hepatol. Res. 2015, 45, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Choy, L.; Skillington, J.; Derynck, R. Roles of autocrine TGF-β receptor and smad signaling in adipocyte differentiation. J. Cell Biol. 2000, 149, 667–682. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Dorn, D.C.; Erdjument-Bromage, H.; Tempst, P.; Moore, M.A.; Massague, J. Hematopoiesis controlled by distinct TIF1γ and smad4 branches of the TGF-β pathway. Cell 2006, 125, 929–941. [Google Scholar] [CrossRef] [PubMed]








© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, H.-H.; Chen, L.; Liang, H.-F.; Li, G.-Z.; Zhang, B.-X.; Chen, X.-P. Smad3 Sensitizes Hepatocelluar Carcinoma Cells to Cisplatin by Repressing Phosphorylation of AKT. Int. J. Mol. Sci. 2016, 17, 610. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040610
Zhou H-H, Chen L, Liang H-F, Li G-Z, Zhang B-X, Chen X-P. Smad3 Sensitizes Hepatocelluar Carcinoma Cells to Cisplatin by Repressing Phosphorylation of AKT. International Journal of Molecular Sciences. 2016; 17(4):610. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040610
Chicago/Turabian StyleZhou, Hong-Hao, Lin Chen, Hui-Fang Liang, Guang-Zhen Li, Bi-Xiang Zhang, and Xiao-Ping Chen. 2016. "Smad3 Sensitizes Hepatocelluar Carcinoma Cells to Cisplatin by Repressing Phosphorylation of AKT" International Journal of Molecular Sciences 17, no. 4: 610. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040610