
Targeted Gene Knockin in Porcine Somatic Cells Using CRISPR/Cas Ribonucleoproteins

Abstract
:1. Introduction
2. Results
2.1. Targeted Knockin of Pseudo-attP Sites Downstream of Porcine COL1A Gene in Somatic Cells Using CRISPR/Cas System
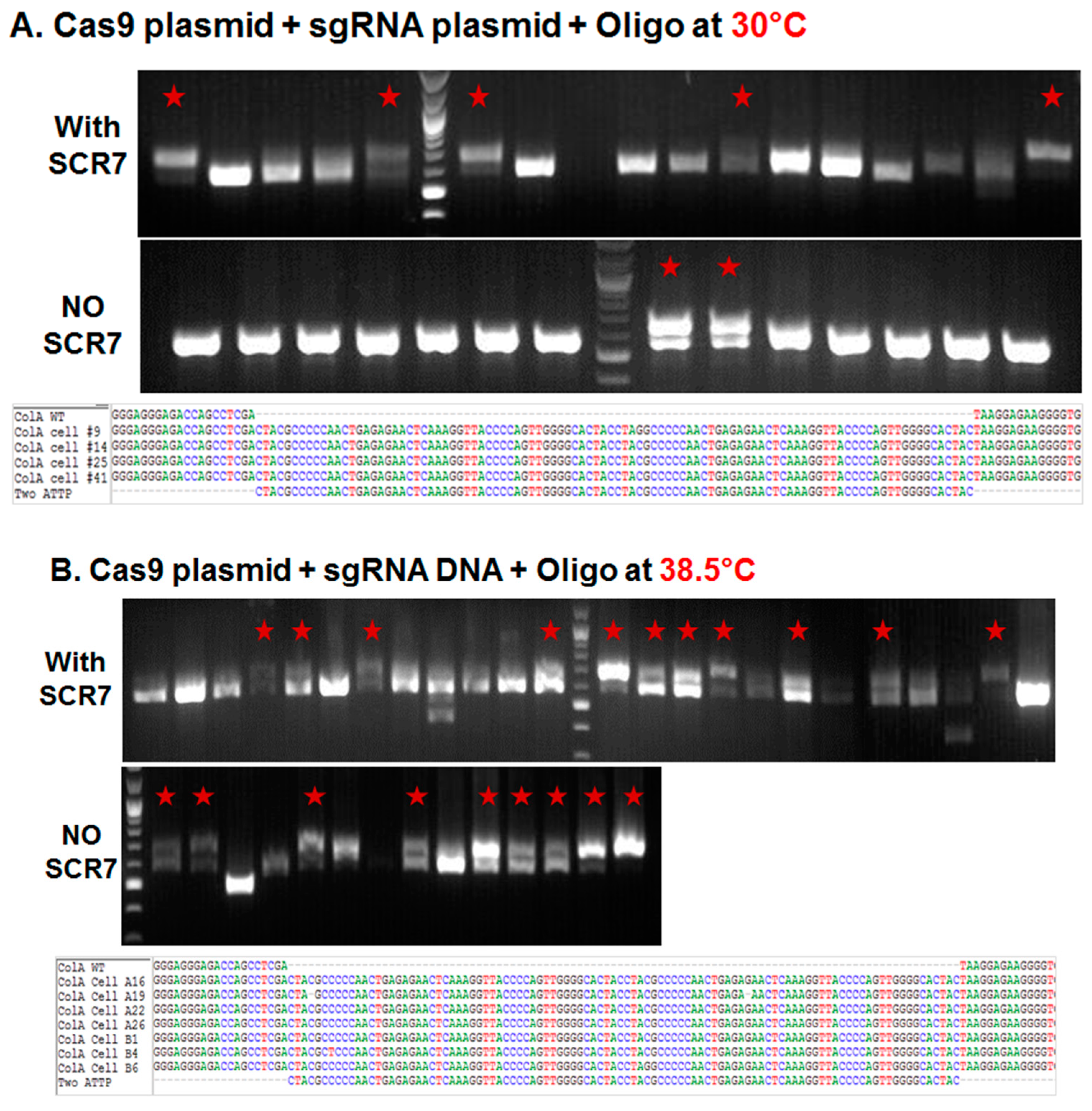
2.1.1. Influence of Temperature on Colony Formation and HDR Outcome
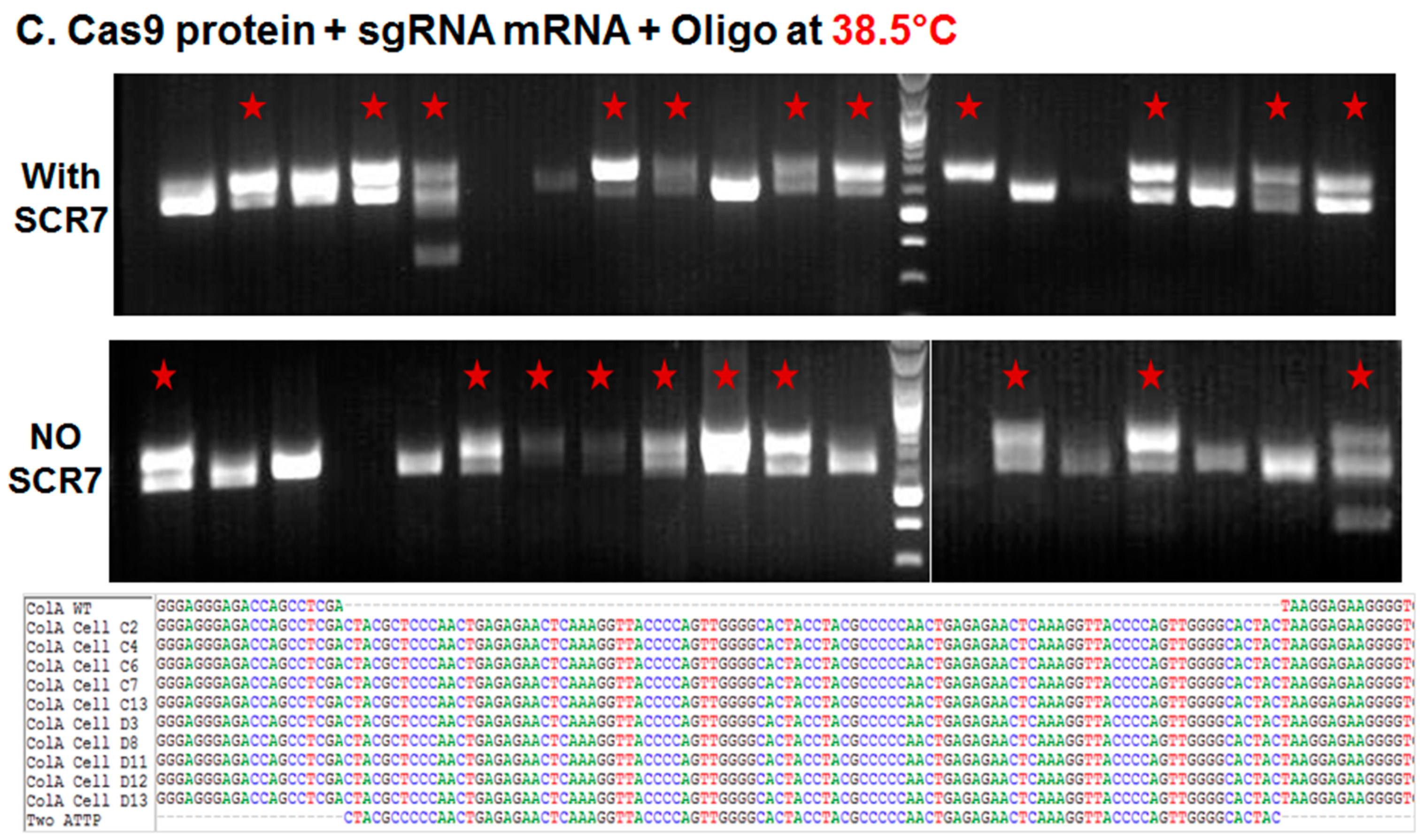
2.1.2. Comparison of Cas9:GFP/sgRNA Plasmid versus Ribonucleoprotein Delivery
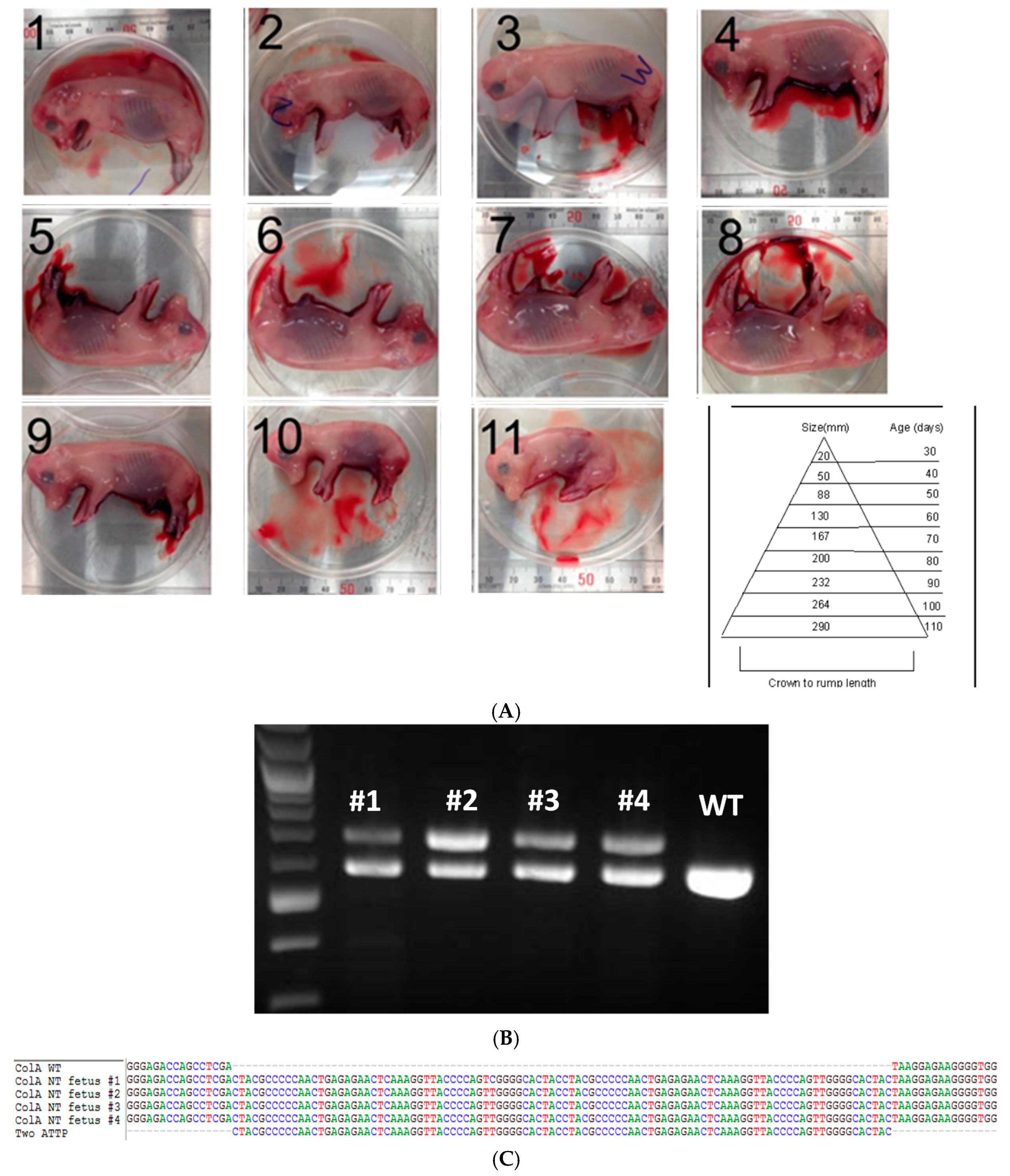
2.2. Somatic Cell Nuclear Transfer to Generate Clonal COL1A Targeted Fetuses and Fibroblast Lines
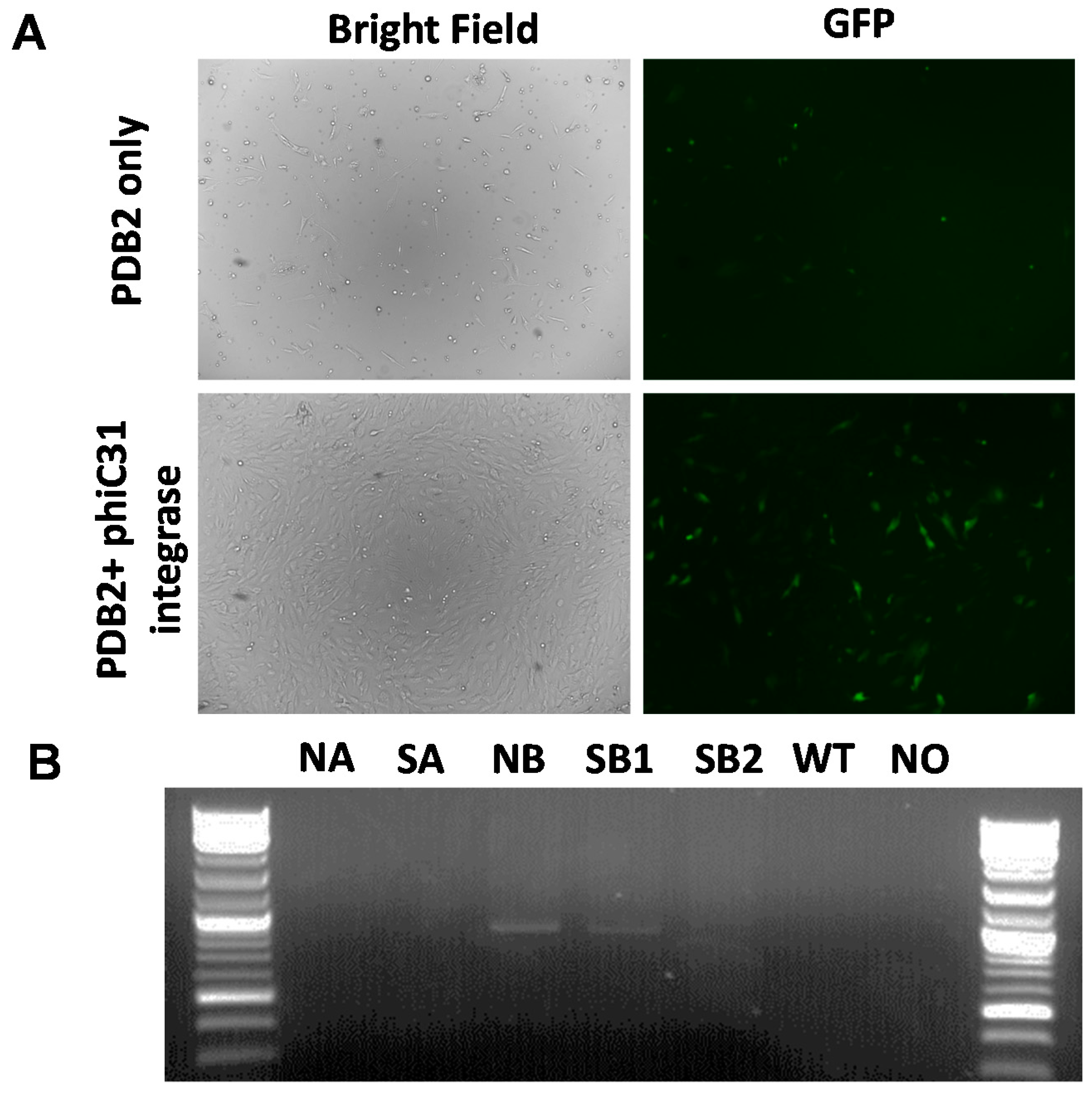
2.3. Phic31 Integrase Mediated Integration of Functional GFP Transgene into the attP Modified COL1A Locus
3. Discussion
4. Materials and Methods
4.1. Plasmid Construction and Production of sgRNA
Nucleofection and Knockin Experiments
4.2. Single Cell Sorting and Culture For Colony Screening
4.3. Somatic Cell Nuclear Transfer (SCNT)
4.4. Genotyping of Fibroblast Cell Colonies, and Edited Nuclear Transfer Fetuses
4.5. Targeted phiC31 Integrase Mediated Integration of GFP Transgene into Pseudo attP Sites in COL1A Locus
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CRISPR/Cas | Clustered regulated interspaced short palindromic repeat and CRISPR-associated (Cas) nuclease system |
| SCNT | Somatic cell nuclear transfer |
| COL1A | Collagen 1A locus |
| DSB | Double strand break |
References
- Whitelaw, C.B.; Sheets, T.P.; Lillico, S.G.; Telugu, B.P. Engineering large animal models of human disease. J. Pathol. 2016, 238, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Grubb, B.R.; Boucher, R.C. Pathophysiology of gene-targeted mouse models for cystic fibrosis. Physiol. Rev. 1999, 79, S193–S214. [Google Scholar] [PubMed]
- Montier, T.; Delepine, P.; Pichon, C.; Ferec, C.; Porteous, D.J.; Midoux, P. Non-viral vectors in cystic fibrosis gene therapy: Progress and challenges. Trends Biotechnol. 2004, 22, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Carvalho-Oliveira, I.; Scholte, B.J.; Penque, D. What have we learned from mouse models for cystic fibrosis? Expert Rev. Mol. Diagn. 2007, 7, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Wilke, M.; Buijs-Offerman, R.M.; Aarbiou, J.; Colledge, W.H.; Sheppard, D.N.; Touqui, L.; Bot, A.; Jorna, H.; de Jonge, H.R.; Scholte, B.J. Mouse models of cystic fibrosis: Phenotypic analysis and research applications. J. Cyst. Fibros. 2011, 10 (Suppl. 2), S152–S171. [Google Scholar] [CrossRef]
- Park, K.E.; Telugu, B.P. Role of stem cells in large animal genetic engineering in the TALENs-CRISPR era. Reprod. Fertil. Dev. 2013, 26, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Moehle, E.A.; Rock, J.M.; Lee, Y.L.; Jouvenot, Y.; DeKelver, R.C.; Gregory, P.D.; Urnov, F.D.; Holmes, M.C. Targeted gene addition into a specified location in the human genome using designed zinc finger nucleases. Proc. Natl. Acad. Sci. USA 2007, 104, 3055–3060. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xin, J.; Fan, N.; Zou, Q.; Huang, J.; Ouyang, Z.; Zhao, Y.; Zhao, B.; Liu, Z.; Lai, S.; et al. Generation of CRISPR/Cas9-mediated gene-targeted pigs via somatic cell nuclear transfer. Cell. Mol. Life Sci. 2015, 72, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, L.; Du, Y.; Xie, F.; Li, L.; Liu, Y.; Liu, C.; Wang, S.; Zhang, S.; Huang, X.; et al. Efficient generation of gene-modified pigs harboring precise orthologous human mutation via CRISPR/Cas9-induced homology-directed repair in zygotes. Hum. Mutat. 2016, 37, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, K.M.; Lee, K.; Benne, J.A.; Beaton, B.P.; Spate, L.D.; Murphy, S.L.; Samuel, M.S.; Mao, J.; O’Gorman, C.; Walters, E.M.; et al. Use of the CRISPR/Cas9 system to produce genetically engineered pigs from in vitro-derived oocytes and embryos. Biol. Reprod. 2014, 91. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, J.; Cao, C.; Huang, J.; Hai, T.; Wang, Y.; Zheng, Q.; Zhang, H.; Qin, G.; Miao, X.; et al. Efficient CRISPR/Cas9-mediated biallelic gene disruption and site-specific knockin after rapid selection of highly active sgRNAs in pigs. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cao, C.; Huang, J.; Yao, J.; Hai, T.; Zheng, Q.; Wang, X.; Zhang, H.; Qin, G.; Cheng, J.; et al. One-step generation of triple gene-targeted pigs using CRISPR/Cas9 system. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wang, Y.; Yuan, Y.; Zhang, W.; Ren, Z.; Jin, Y.; Liu, X.; Xiong, Q.; Chen, Q.; Zhang, M.; et al. Generation of B cell-deficient pigs by highly efficient CRISPR/Cas9-mediated gene targeting. J. Genet. Genomics 2015, 42, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.; Wei, S.; Zhao, B.; Ouyang, Z.; Zhang, Q.; Fan, N.; Liu, Z.; Zhao, Y.; Yan, Q.; Zhou, X.; et al. Generation of knock-in pigs carrying oct4-tdtomato reporter through CRISPR/Cas9-mediated genome engineering. PLoS ONE 2016, 11, e0146562. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kuhn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; Choi, V.M.; Xia, D.F.; Vo, T.D.; Gregory, P.D.; Holmes, M.C. Transient cold shock enhances zinc-finger nuclease-mediated gene disruption. Nat. Methods 2010, 7, 459–460. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A tale nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.R.; Lee, N.C.; Xu, Z.; Smith, M.C. Serine recombinases as tools for genome engineering. Methods 2011, 53, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Brown, W.R.; McEwan, A.R.; Rowley, P.A. Site-specific recombination by phiC31 integrase and other large serine recombinases. Biochem. Soc. Trans. 2010, 38, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Keravala, A.; Portlock, J.L.; Nash, J.A.; Vitrant, D.G.; Robbins, P.D.; Calos, M.P. PhiC31 integrase mediates integration in cultured synovial cells and enhances gene expression in rabbit joints. J. Gene Med. 2006, 8, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Groth, A.C.; Olivares, E.C.; Thyagarajan, B.; Calos, M.P. A phage integrase directs efficient site-specific integration in human cells. Proc. Natl. Acad. Sci. USA 2000, 97, 5995–6000. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. eLife 2013, 2, e00471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, Y.H.; Park, C.H.; Jang, G.H.; Jeong, Y.I.; Hwang, I.S.; Jeong, Y.W.; Kim, Y.K.; Shin, T.; Kim, N.H.; Hyun, S.H.; et al. Production of multiple transgenic yucatan miniature pigs expressing human complement regulatory factors, human CD55, CD59, and H-transferase genes. PLoS ONE 2013, 8, e63241. [Google Scholar]
- Luo, Y.; Wang, Y.; Liu, J.; Lan, H.; Shao, M.; Yu, Y.; Quan, F.; Zhang, Y. Production of transgenic cattle highly expressing human serum albumin in milk by phiC31 integrase-mediated gene delivery. Transgenic Res. 2015, 24, 875–883. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cas9 & sgRNA Combination | Treatment with SCR7 | Initial Culture Temperature | Number of Single Cell Sorted Wells | Number of Colonies Formed | % of Colonies Formed/Sorted Wells | Number of Targeted Colonies | Percentage of Targeted Colonies/Colonies Formed |
|---|---|---|---|---|---|---|---|
| Plasmid | None | 30 °C | 960 | 16 | 1.70% | 2 | 13% |
| Plasmid | SCR7 | 30 °C | 960 | 41 | 4.30% | 18 | 44% |
| Plasmid | None | 38.5 °C | 376 | 14 | 3.70% | 8 | 57% |
| Plasmid | SCR7 | 38.5 °C | 376 | 39 | 10.40% | 17 | 44% |
| Ribonucleoprotein | None | 38.5 °C | 376 | 58 | 15.40% | 18 | 31% |
| Ribonucleoprotein | SCR7 | 38.5 °C | 376 | 61 | 16.20% | 19 | 31% |
| Target | Primer | Sequence |
|---|---|---|
| Gene Knockin Primers | COL1A Forward | AGCCAGGCTGCCTTGTTTG |
| COL1A Reverse | GCCAACCTCCCCTTTGCACT | |
| pDB2 Integration Primers | EGFPC | CATGGTCCTGCTGGAGTTCGTG |
| COL1A Reverse | AGCCAGGCTGCCTTGTTTG |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, K.-E.; Park, C.-H.; Powell, A.; Martin, J.; Donovan, D.M.; Telugu, B.P. Targeted Gene Knockin in Porcine Somatic Cells Using CRISPR/Cas Ribonucleoproteins. Int. J. Mol. Sci. 2016, 17, 810. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17060810
Park K-E, Park C-H, Powell A, Martin J, Donovan DM, Telugu BP. Targeted Gene Knockin in Porcine Somatic Cells Using CRISPR/Cas Ribonucleoproteins. International Journal of Molecular Sciences. 2016; 17(6):810. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17060810
Chicago/Turabian StylePark, Ki-Eun, Chi-Hun Park, Anne Powell, Jessica Martin, David M. Donovan, and Bhanu P. Telugu. 2016. "Targeted Gene Knockin in Porcine Somatic Cells Using CRISPR/Cas Ribonucleoproteins" International Journal of Molecular Sciences 17, no. 6: 810. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17060810