
Circulating Tumor Cells (CTC) and Cell-Free DNA (cfDNA) Workshop 2016: Scientific Opportunities and Logistics for Cancer Clinical Trial Incorporation

 , , ,
, , ,
Abstract
:1. Introduction
- (1)
- To highlight Canadian work in the areas of CTCs and cfDNA;
- (2)
- To provide an update on current technologies including advantages and limitations, potential clinical utility for analysis of CTCs and cfDNA, and recent translational breakthroughs;
- (3)
- To consider optimal clinical trial designs for CTC and cfDNA incorporation, including discussion of the best standard operating procedures (SOPs) for collection and analysis; and
- (4)
- To provide networking opportunities for clinical and translational researchers, and facilitate new collaborations in the area of CTCs/cfDNA and cancer clinical trials.
2. Theme 1: Technological and Methodological Advances in Circulating Tumor Cells (CTCs) and Cell-Free DNA (cfDNA) Detection and Analysis
3. Theme 2: CTCs versus cfDNA: Comparable or Complimentary Biomarkers?
4. Theme 3: Importance of Heterogeneity
5. Theme 4: Considerations for Incorporation of CTCs and cfDNA into Clinical Trials
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Joosse, S.A.; Gorges, T.M.; Pantel, K. Biology, detection, and clinical implications of circulating tumor cells. EMBO Mol. Med. 2015, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sethi, N.; Kang, Y. Unravelling the complexity of metastasis—Molecular understanding and targeted therapies. Nat. Rev. Cancer 2011, 11, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Canadian Cancer Society. Canadian Cancer Statistics, 2015; Canadian Cancer Society: Toronto, ON, Canada, 2015; Available online: http: www.cancer.ca (accessed on 6 September 2016).
- Joosse, S.A.; Pantel, K. Genetic traits for hematogeneous tumor cell dissemination in cancer patients. Cancer Metastasis Rev. 2016, 35, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Pantel, K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Alix-Panabières, C. Real-time liquid biopsy in cancer patients: fact or fiction? Cancer Res. 2013, 73, 6384–6388. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Schwarzenbach, H.; Pantel, K. Circulating tumor cells and circulating tumor DNA. Annu. Rev. Med. 2012, 63, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Kidess, E.; Jeffrey, S.S. Circulating tumor cells versus tumor-derived cell-free DNA: Rivals or partners in cancer care in the era of single-cell analysis? Genome Med. 2013, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Brown, P. The Cobas® EGFR Mutation Test v2 assay. Future Oncol. 2016, 12, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Pantel, K. Challenges in circulating tumour cell research. Nat. Rev. Cancer 2014, 14, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; Scher, H.I.; Montgomery, R.B.; Parker, C.; Miller, M.C.; Tissing, H.; Doyle, G.V.; Terstappen, L.W.; Pienta, K.J.; Raghavan, D. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2008, 14, 6302–6309. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.J.; Punt, C.J.A.; Iannotti, N.; Saidman, B.H.; Sabbath, K.D.; Gabrail, N.Y.; Picus, J.; Morse, M.; Mitchell, E.; Miller, M.C.; et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 3213–3221. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, D. Nucleic acid-based methods for the detection of cancer. Science 1997, 278, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Nawroz, H.; Koch, W.; Anker, P.; Stroun, M.; Sidransky, D. Microsatellite alterations in serum DNA of head and neck cancer patients. Nat. Med. 1996, 2, 1035–1037. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Stroun, M.; Magnenat, J.L.; Nicod, L.P.; Kurt, A.M.; Lyautey, J.; Lederrey, C.; Anker, P. Microsatellite alterations in plasma DNA of small cell lung cancer patients. Nat. Med. 1996, 2, 1033–1035. [Google Scholar] [CrossRef] [PubMed]
- Norton, S.E.; Lechner, J.M.; Williams, T.; Fernando, M.R. A stabilizing reagent prevents cell-free DNA contamination by cellular DNA in plasma during blood sample storage and shipping as determined by digital PCR. Clin. Biochem. 2013, 46, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, C.; Nicolazzo, C.; Gradilone, A. Circulating tumor cells isolation: The “post-EpCAM era”. Chin. J. Cancer Res. 2015, 27, 461–470. [Google Scholar] [PubMed]
- Beltran, H.; Jendrisak, A.; Landers, M.; Mosquera, J.M.; Kossai, M.; Louw, J.; Krupa, R.; Graf, R.P.; Schreiber, N.A.; Nanus, D.M.; et al. The initial detection and partial characterization of circulating tumor cells in neuroendocrine prostate cancer. Clin. Cancer Res. 2016, 22, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Mohamadi, R.M.; Besant, J.D.; Mepham, A.; Green, B.; Mahmoudian, L.; Gibbs, T.; Ivanov, I.; Malvea, A.; Stojcic, J.; Allan, A.L.; et al. Nanoparticle-mediated binning and profiling of heterogeneous circulating tumor cell subpopulations. Angew. Chem. Int. Ed. Engl. 2015, 54, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Muhanna, N.; Mepham, A.; Mohamadi, R.M.; Chan, H.; Khan, T.; Akens, M.; Besant, J.D.; Irish, J.; Kelley, S.O. Nanoparticle-based sorting of circulating tumor cells by epithelial antigen expression during disease progression in an animal model. Nanomedicine 2015, 11, 1613–1620. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, N.; Hu, P.; Bedard, P.; Clemons, M.; McCready, D.; Done, S.J. Identification of genomic signatures in circulating tumor cells from breast cancer. Int. J. Cancer 2015, 137, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Adebayo Awe, J.; Xu, M.C.; Wechsler, J.; Benali-Furet, N.; Cayre, Y.E.; Saranchuk, J.; Drachenberg, D.; Mai, S. Three-dimensional telomeric analysis of isolated circulating tumor cells (CTCs) defines CTC subpopulations. Transl. Oncol. 2013, 6, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.A.; Volik, S.V.; Wyatt, A.W.; Haegert, A.; le Bihan, S.; Bell, R.H.; Anderson, S.A.; McConeghy, B.; Shukin, R.; Bazov, J.; et al. Androgen receptor gene aberrations in circulating cell-free dna: biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin. Cancer Res. 2015, 21, 2315–2324. [Google Scholar] [CrossRef] [PubMed]
- Lallous, N.; Volik, S.V.; Awrey, S.; Leblanc, E.; Tse, R.; Murillo, J.; Singh, K.; Azad, A.A.; Wyatt, A.W.; LeBihan, S.; et al. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biol. 2016, 17, 10. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, A.W.; Azad, A.A.; Volik, S.V.; Annala, M.; Beja, K.; McConeghy, B.; Haegert, A.; Warner, E.W.; Mo, F.; Brahmbhatt, S.; et al. Genomic alterations in cell-free DNA and enzalutamide resistance in castration-resistant prostate cancer. JAMA Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Chedgy, E.C.P.; Annala, M.; Beja, K.; Warner, E.W.; Gleave, M.E.; Chi, K.N.; Wyatt, A.W. Moving toward personalized care: Liquid biopsy predicts response to cisplatin in an unusual case of BRCA2-Null neuroendocrine prostate cancer. Clin. Genitourin. Cancer 2016, 14, e233–e236. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, M.; Yu, S.; Bushell, K.; Fornika, D.; Nielsen, J.S.; Nelson, B.H.; Mann, K.K.; Assouline, S.; Johnson, N.A.; Morin, R.D. Multiplex droplet digital PCR quantification of recurrent somatic mutations in diffuse large B-cell and follicular lymphoma. Clin. Chem. 2016, 62, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Assouline, S.E.; Nielsen, T.H.; Yu, S.; Alcaide, M.; Chong, L.; MacDonald, D.; Tosikyan, A.; Kukreti, V.; Kezouh, A.; Petrogiannis-Haliotis, T.; et al. Phase 2 study of panobinostat+/− rituximab in relapsed diffuse large B cell lymphoma and biomarkers predictive of response. Blood 2016. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Bratman, S.V.; Newman, A.M.; Alizadeh, A.A.; Diehn, M. Potential clinical utility of ultrasensitive circulating tumor DNA detection with CAPP-Seq. Expert Rev. Mol. Diagn. 2015, 15, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Spiliotaki, M.; Mavroudis, D.; Kapranou, K.; Markomanolaki, H.; Kallergi, G.; Koinis, F.; Kalbakis, K.; Georgoulias, V.; Agelaki, S. Evaluation of proliferation and apoptosis markers in circulating tumor cells of women with early breast cancer who are candidates for tumor dormancy. Breast Cancer Res. 2014, 16, 485. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, A.J.; Wentzel, J.F.; Aucamp, J.; van Dyk, E.; du Plessis, L.; Pretorius, P.J. Characterization of the cell-free DNA released by cultured cancer cells. Biochim. Biophys. Acta 2016, 1863, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Mohamadi, R.M.; Ivanov, I.; Stojcic, J.; Nam, R.K.; Sargent, E.H.; Kelley, S.O. Sample-to-answer isolation and mRNA Profiling of circulating tumor cells. Anal. Chem. 2015, 87, 6258–6264. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Lu, D.; Schreiber, N.A.; Louw, J.; Graf, R.P.; Vargas, H.A.; Johnson, A.; Jendrisak, A.; Bambury, R.; Danila, D.; et al. Association of AR-V7 on circulating tumor cells as a treatment-specific biomarker with outcomes and survival in castration-resistant prostate cancer. JAMA Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Alix-Panabières, C. Functional studies on viable circulating tumor cells. Clin. Chem. 2016, 62, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Thress, K.S.; Brant, R.; Carr, T.H.; Dearden, S.; Jenkins, S.; Brown, H.; Hammett, T.; Cantarini, M.; Barrett, J.C. EGFR mutation detection in ctDNA from NSCLC patient plasma: A cross-platform comparison of leading technologies to support the clinical development of AZD9291. Lung Cancer 2015, 90, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Lowes, L.E.; Lock, M.; Rodrigues, G.; D’Souza, D.; Bauman, G.; Ahmad, B.; Venkatesan, V.; Allan, A.L.; Sexton, T. The significance of circulating tumor cells in prostate cancer patients undergoing adjuvant or salvage radiation therapy. Prostate Cancer Prostatic Dis. 2015, 18, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef] [PubMed]
- Roschewski, M.; Dunleavy, K.; Pittaluga, S.; Moorhead, M.; Pepin, F.; Kong, K.; Shovlin, M.; Jaffe, E.S.; Staudt, L.M.; Lai, C.; et al. Circulating tumour DNA and CT monitoring in patients with untreated diffuse large B-cell lymphoma: A correlative biomarker study. Lancet Oncol. 2015, 16, 541–549. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.M.; Chan, J.Y.K.; Zhang, Z.; Wang, H.; Khan, Z.; Bishop, J.A.; Westra, W.; Koch, W.M.; Califano, J.A. Saliva and plasma quantitative polymerase chain reaction-based detection and surveillance of human papillomavirus-related head and neck cancer. JAMA Otolaryngol. Head Neck Surg. 2014, 140, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Wang, W.Y.; Chen, K.Y.; Wei, Y.H.; Liang, W.M.; Jan, J.S.; Jiang, R.S. Quantification of plasma Epstein-Barr virus DNA in patients with advanced nasopharyngeal carcinoma. N. Engl. J. Med. 2004, 350, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Clinical Trials.gov Registry and Database. U.S. National Institutes of Health. Available online: https://clinicaltrials.gov/ (accessed on 6 August 2016).
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, J.; Nordgren, H.; Sjöström, J.; Wester, K.; Villman, K.; Bengtsson, N.O.; Ostenstad, B.; Lundqvist, H.; Blomqvist, C. HER2 expression in breast cancer primary tumours and corresponding metastases. Original data and literature review. Br. J. Cancer 2004, 90, 2344–2348. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.Y.; Roudier, M.P.; True, L.D. Heterogeneity in primary and metastatic prostate cancer as defined by cell surface CD profile. Am. J. Pathol. 2004, 165, 1543–1556. [Google Scholar] [CrossRef]
- Gancberg, D.; di Leo, A.; Cardoso, F.; Rouas, G.; Pedrocchi, M.; Paesmans, M.; Verhest, A.; Bernard-Marty, C.; Piccart, M.J.; Larsimont, D. Comparison of HER-2 status between primary breast cancer and corresponding distant metastatic sites. Ann. Oncol. 2002, 13, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.A.; Jonas, S.K.; Le-Marer, N.; Patel, H.; Wharton, R.Q.; Tarragona, A.; Ivison, A.; Allen-Mersh, T.G. P53 mutations in primary and metastatic tumors and circulating tumor cells from colorectal carcinoma patients. Clin. Cancer Res. 2000, 6, 3499–3504. [Google Scholar] [PubMed]
- Riethdorf, S.; Müller, V.; Zhang, L.; Rau, T.; Loibl, S.; Komor, M.; Roller, M.; Huober, J.; Fehm, T.; Schrader, I.; et al. Detection and HER2 expression of circulating tumor cells: Prospective monitoring in breast cancer patients treated in the neoadjuvant GeparQuattro trial. Clin. Cancer Res. 2010, 16, 2634–2645. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A mathematical theory of communication. Bell Tech. J. 1948, 27, 379–423, 623–656. [Google Scholar] [CrossRef]
- Scher, H.I.; Graf, R.P.; Schreiber, N.A.; McLaughlin, B.; Lu, D.; Louw, J.; Jendrisak, A.; Greene, S.; Rodriguez, A.; Dugan, L.; et al. AR-V7 and CTC heterogeneity biomarkers additively to predict patient (pt) outcomes with taxanes relative to approved AR targeted therapy. J. Clin. Oncol. 2016, 34, 5013. [Google Scholar]
- Scher, H.I.; Jendrisak, A.; Graf, R.P.; Schreiber, N.A.; McLaughlin, B.; Greene, S.; Rodriguez, A.; Louw, J.; Dugan, L.; Leitz, L.; et al. CTC phenotype classifier to identify mCRPC patients (pts) with high genomic instability CTCs and to predict failure of androgen ecreptor signaling (AR Tx) and taxane (T) systemic therapies. J. Clin. Oncol. 2016, 34, 5044. [Google Scholar]
- Bidard, F.C.; Fehm, T.; Ignatiadis, M.; Smerage, J.B.; Alix-Panabières, C.; Janni, W.; Messina, C.; Paoletti, C.; Müller, V.; Hayes, D.F.; et al. Clinical application of circulating tumor cells in breast cancer: Overview of the current interventional trials. Cancer Metastasis Rev. 2013, 32, 179–188. [Google Scholar] [CrossRef] [PubMed]


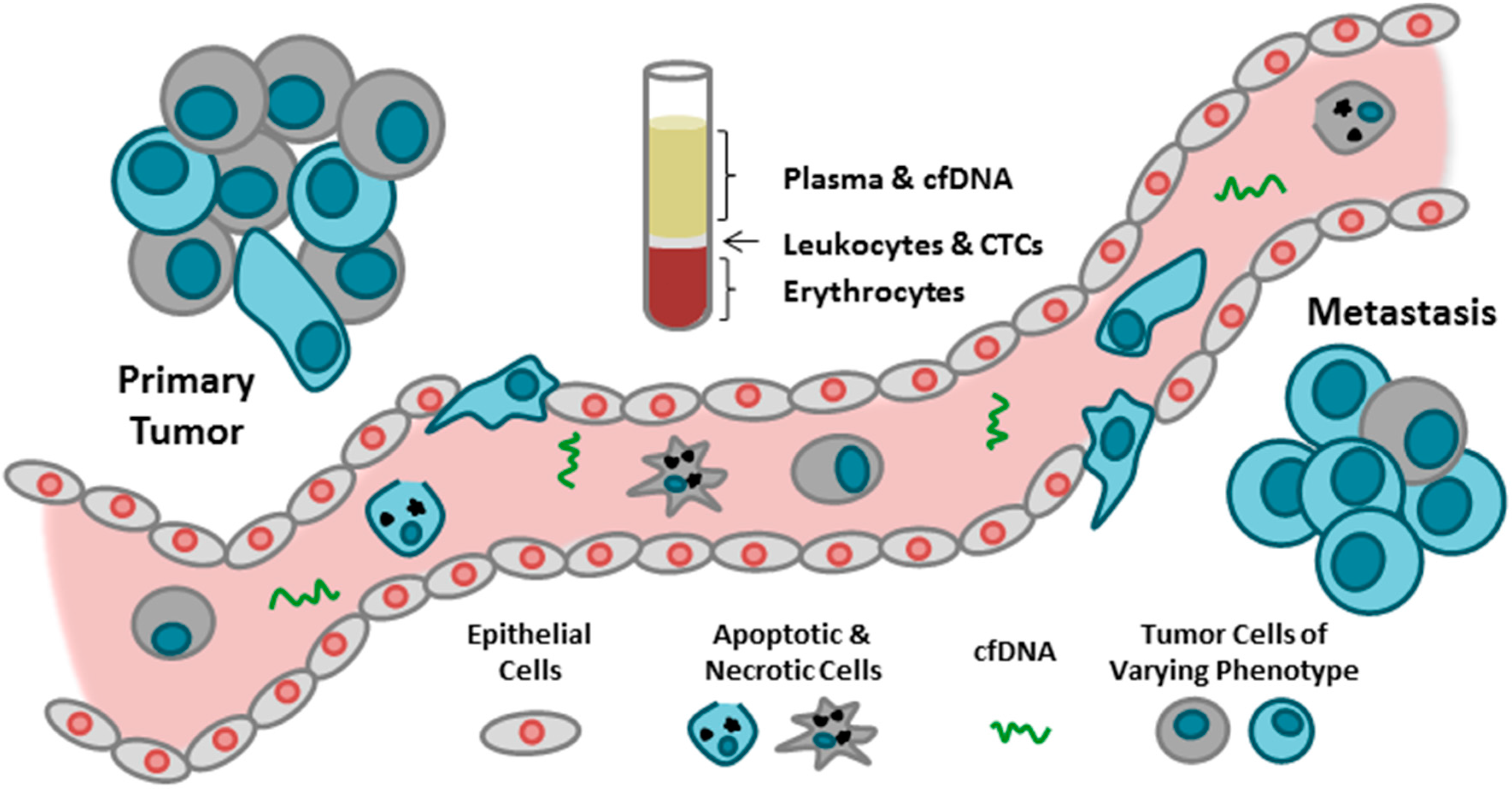{kind=link}
| Comparison | CTCs | cfDNA |
|---|---|---|
| Origin | Intact cells (not necessarily viable) [34] | Necrotic/apoptotic cells [33] and/or actively secreted from intact cells [35] |
| Definition | Tumor cells derived from primary/metastatic sites [6] | Fragmented DNA in circulation [6] |
| Capture & Analysis Techniques | Enrichment: size/density-, immunomagnetic-, or microfluidic-based [10] Detection: protein- or nucleic acid-based [10] | Enrichment: plasma collection [33] Detection: PCR-, or sequencing-based [33] |
| Advantages | Extensive downstream analysis (DNA, RNA, protein, functional assays) [22,36,37,38] Assessment of single cells [39] Clinically-validated technology available (CellSearch® system; metastatic breast, prostate, & colorectal cancers) [11,12,13] Captured viable cells can be used for in vitro culture or in vivo animal studies [38] | Easy to isolate/enrich from whole blood [33] Amenable to long-term storage for subsequent analysis [33] High-sensitivity read-out [28] Clinically validated test for EGFR mutations in non-small cell lung cancer [9,40] |
| Disadvantages | Low cell numbers in non-metastatic setting [41] Challenging to store long-term and subsequently analyze (Lowes, L.E., unpublished) Both detection and enrichment steps require highly sensitive and often expensive technology [6] | Limited (pre-)analytical/analytical SOPs, assay validation, & appropriate prognostic/predictive read-out (may be disease/mutation specific) [33] Limited downstream analysis (DNA only) Currently only feasible in high tumor burden setting [6] Need known target mutations to confirm cfDNA originated from tumor cells [6] |
| Recent Technological & Methodological Advances |
|
| Considerations for Incorporation of CTCs and cfDNA into Clinical Trials: |
|
| Moving Forward: General Considerations for the Future Use of CTCs and cfDNA |
|
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lowes, L.E.; Bratman, S.V.; Dittamore, R.; Done, S.; Kelley, S.O.; Mai, S.; Morin, R.D.; Wyatt, A.W.; Allan, A.L. Circulating Tumor Cells (CTC) and Cell-Free DNA (cfDNA) Workshop 2016: Scientific Opportunities and Logistics for Cancer Clinical Trial Incorporation. Int. J. Mol. Sci. 2016, 17, 1505. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091505
Lowes LE, Bratman SV, Dittamore R, Done S, Kelley SO, Mai S, Morin RD, Wyatt AW, Allan AL. Circulating Tumor Cells (CTC) and Cell-Free DNA (cfDNA) Workshop 2016: Scientific Opportunities and Logistics for Cancer Clinical Trial Incorporation. International Journal of Molecular Sciences. 2016; 17(9):1505. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091505
Chicago/Turabian StyleLowes, Lori E., Scott V. Bratman, Ryan Dittamore, Susan Done, Shana O. Kelley, Sabine Mai, Ryan D. Morin, Alexander W. Wyatt, and Alison L. Allan. 2016. "Circulating Tumor Cells (CTC) and Cell-Free DNA (cfDNA) Workshop 2016: Scientific Opportunities and Logistics for Cancer Clinical Trial Incorporation" International Journal of Molecular Sciences 17, no. 9: 1505. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091505