
Reactivities of Quinone Methides versus o-Quinones in Catecholamine Metabolism and Eumelanin Biosynthesis

Abstract
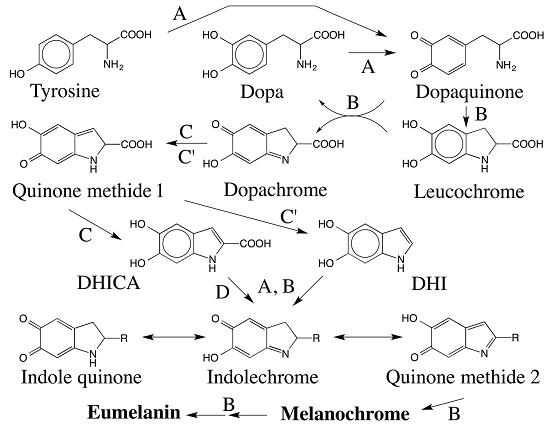
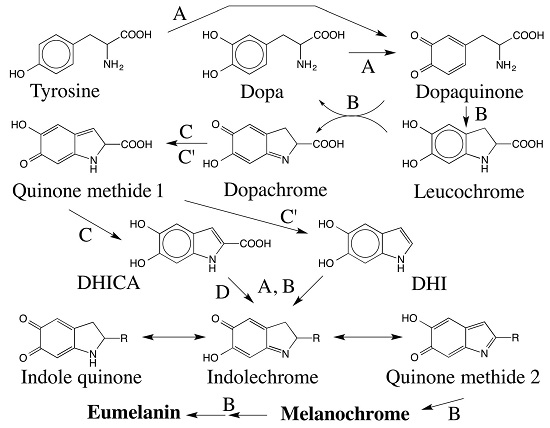
:
{kind=link}
{kind=link}
{kind=link}
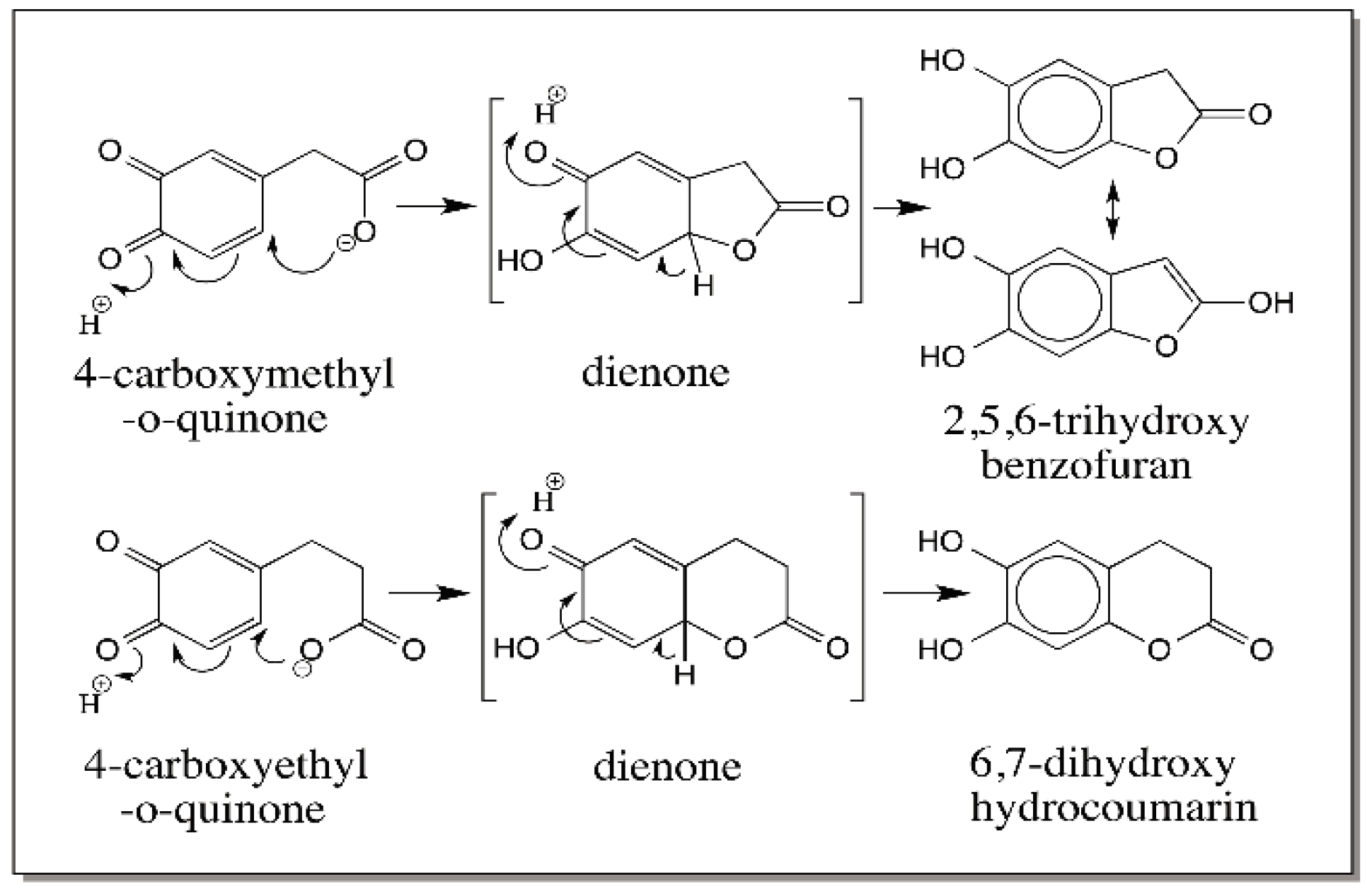{kind=link}
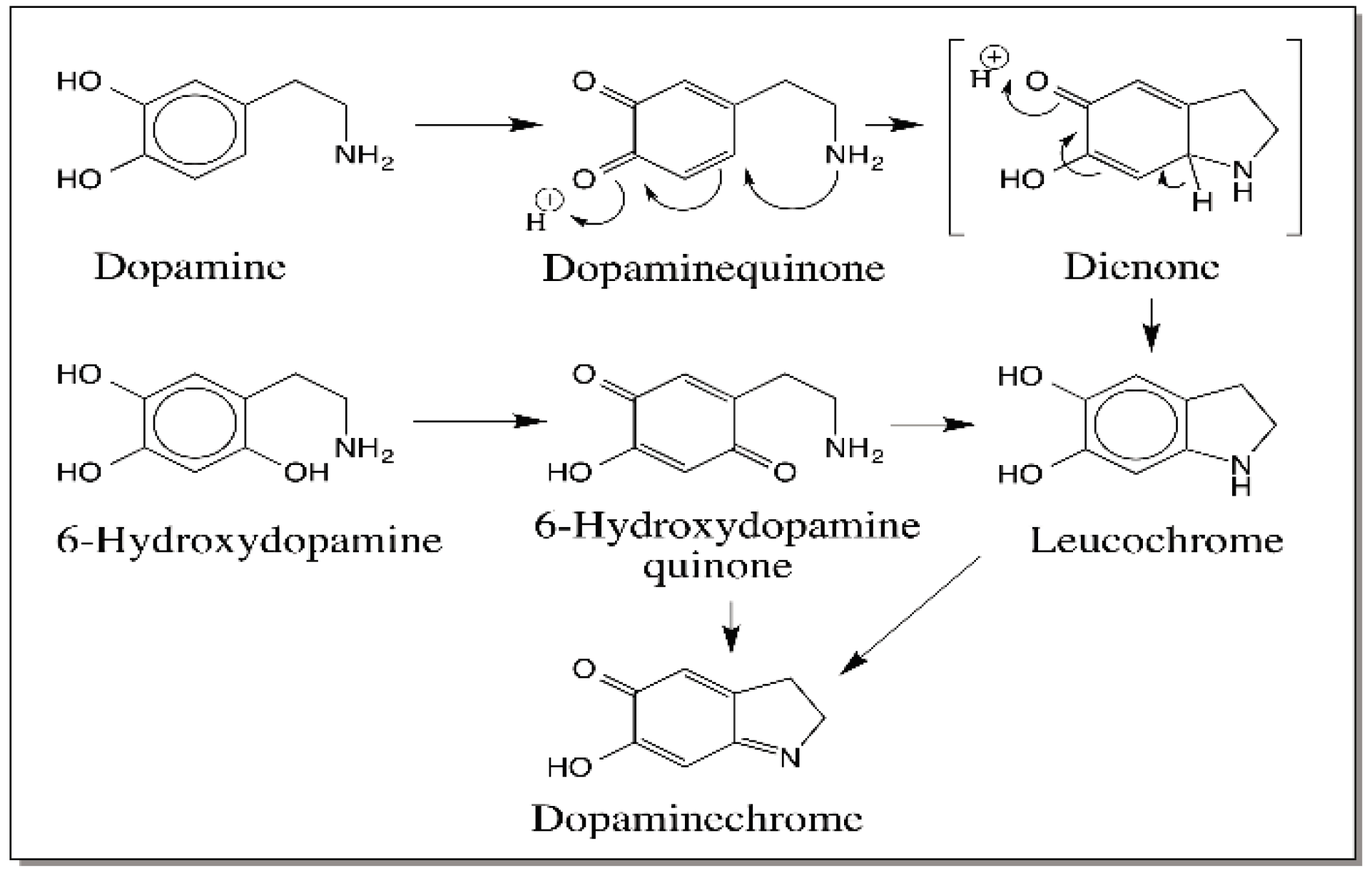{kind=link}
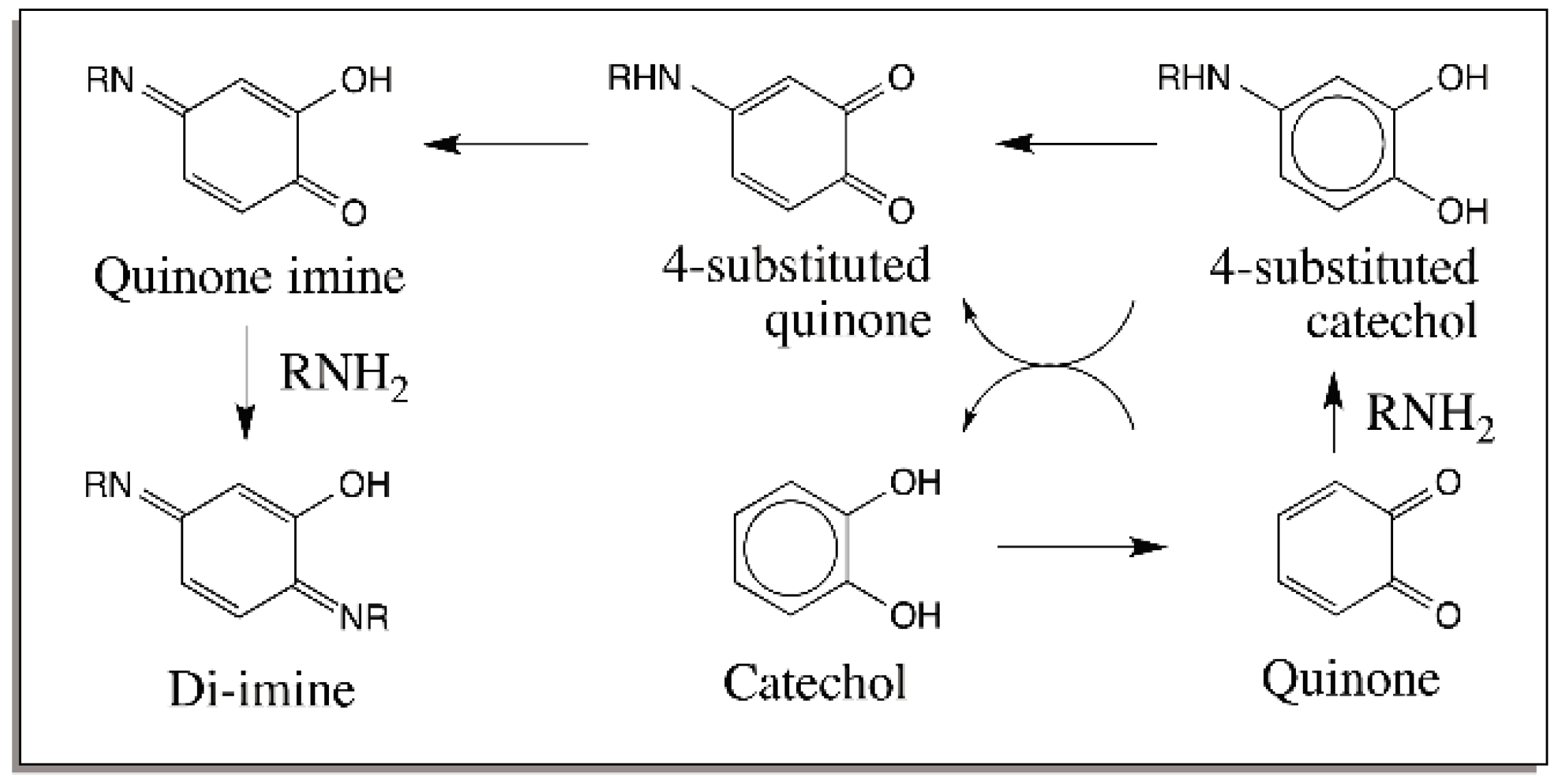{kind=link}
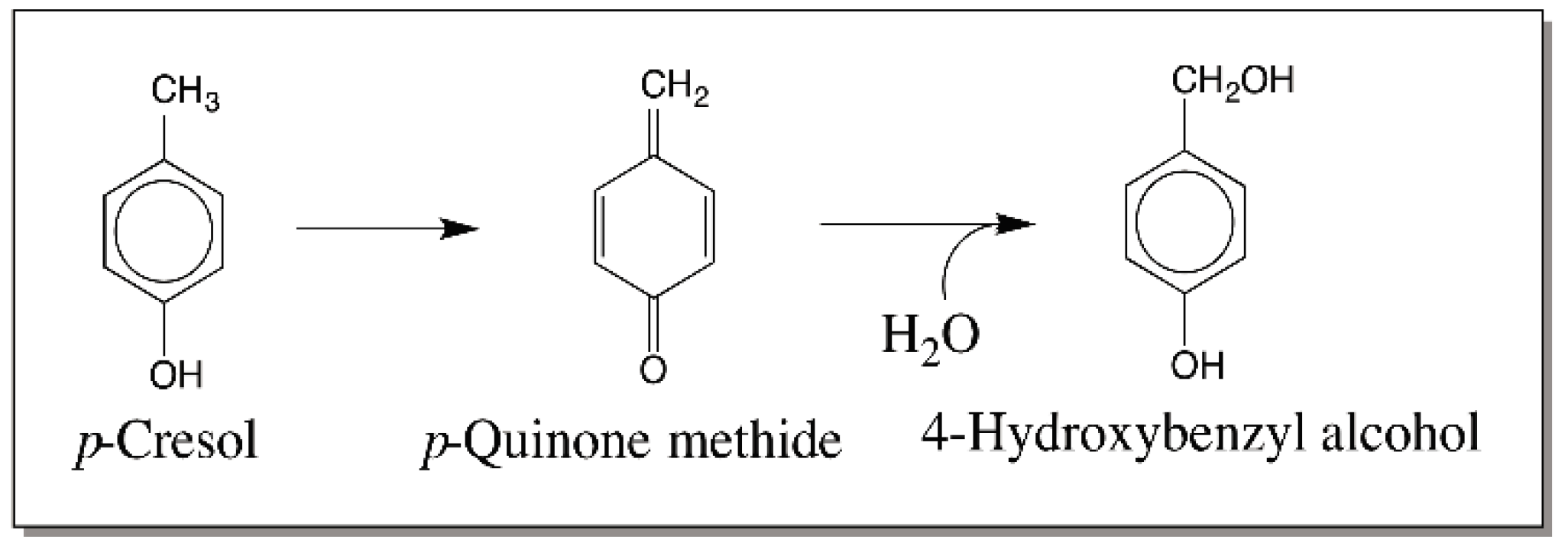{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
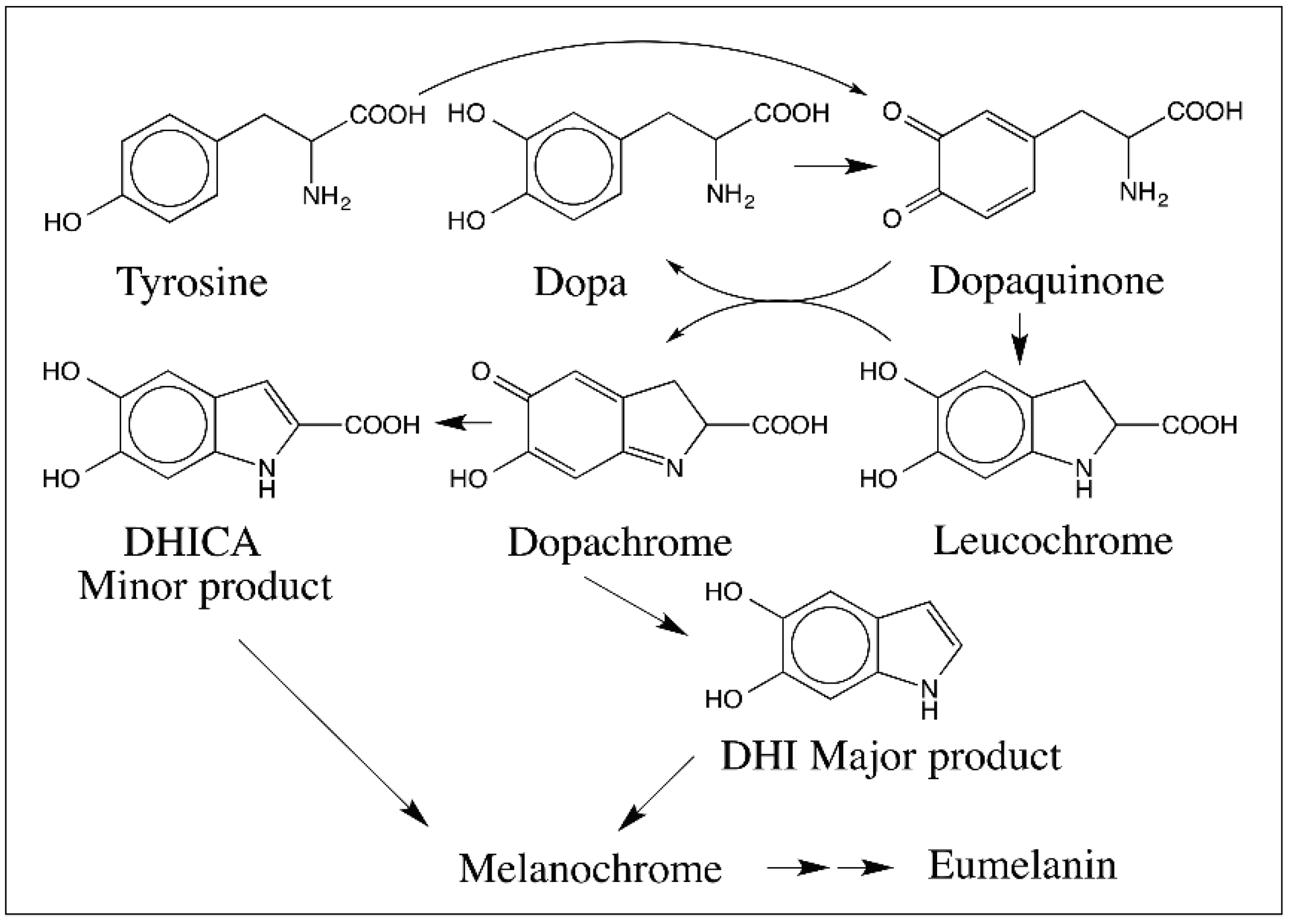
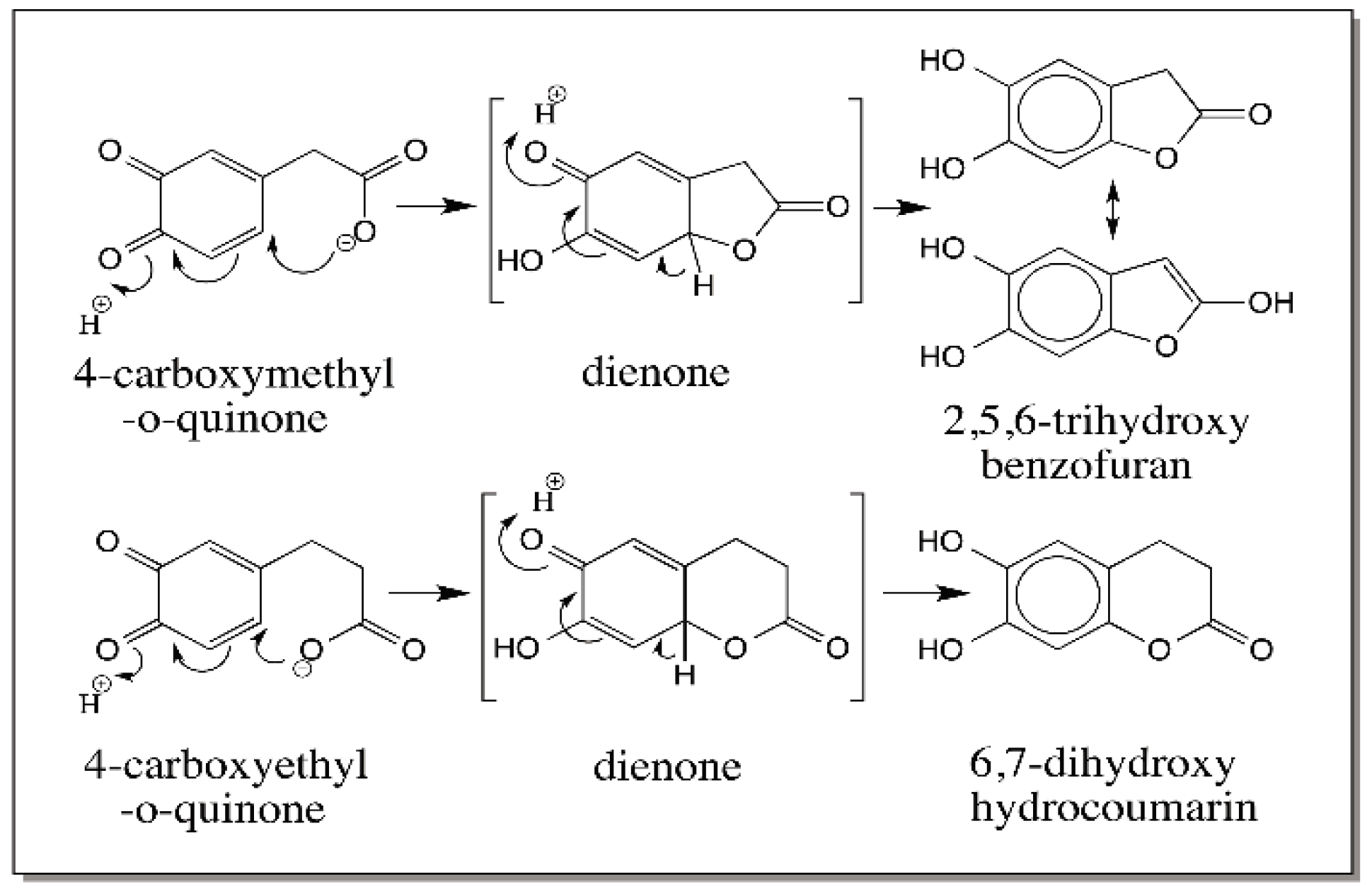
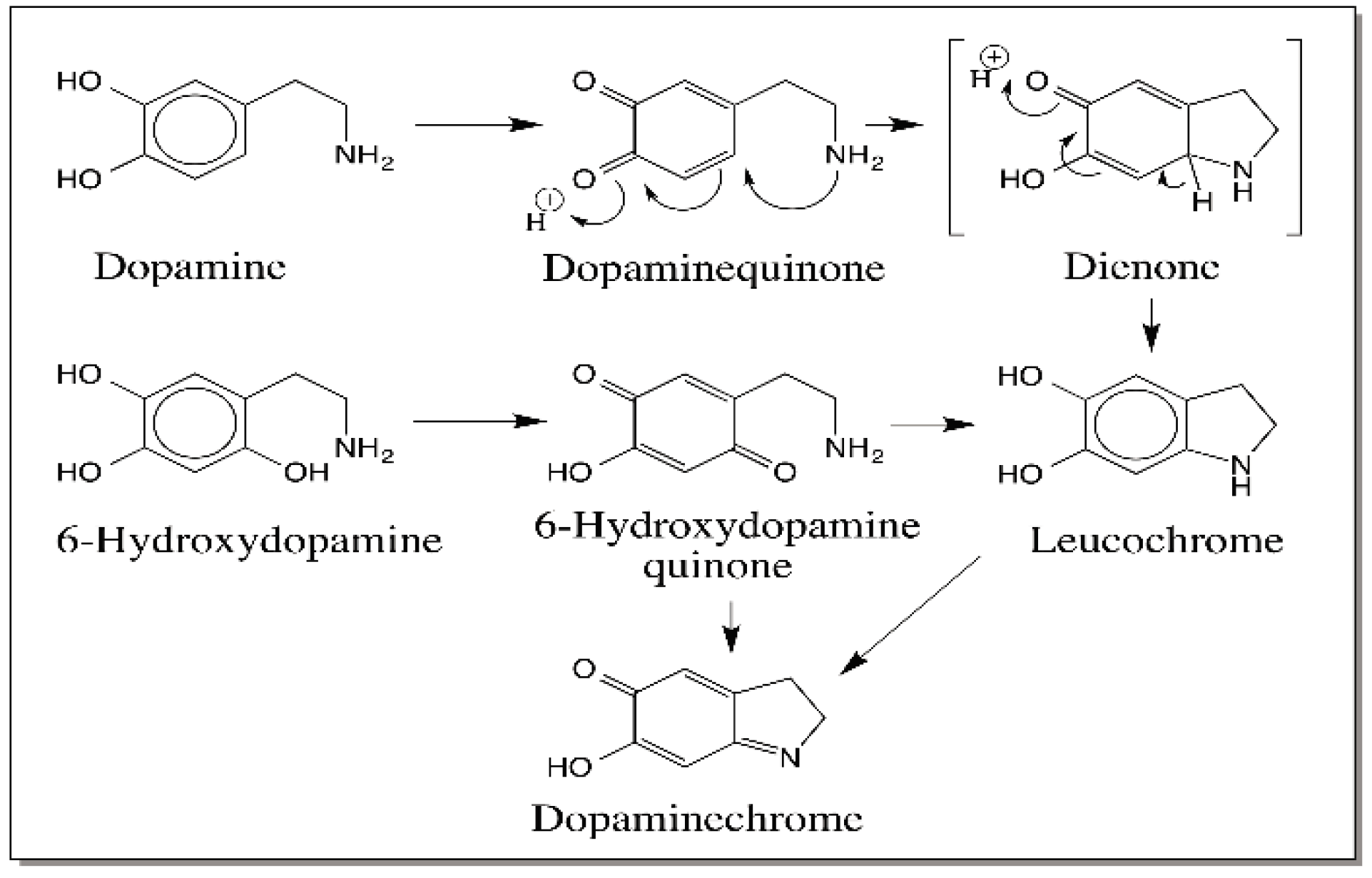
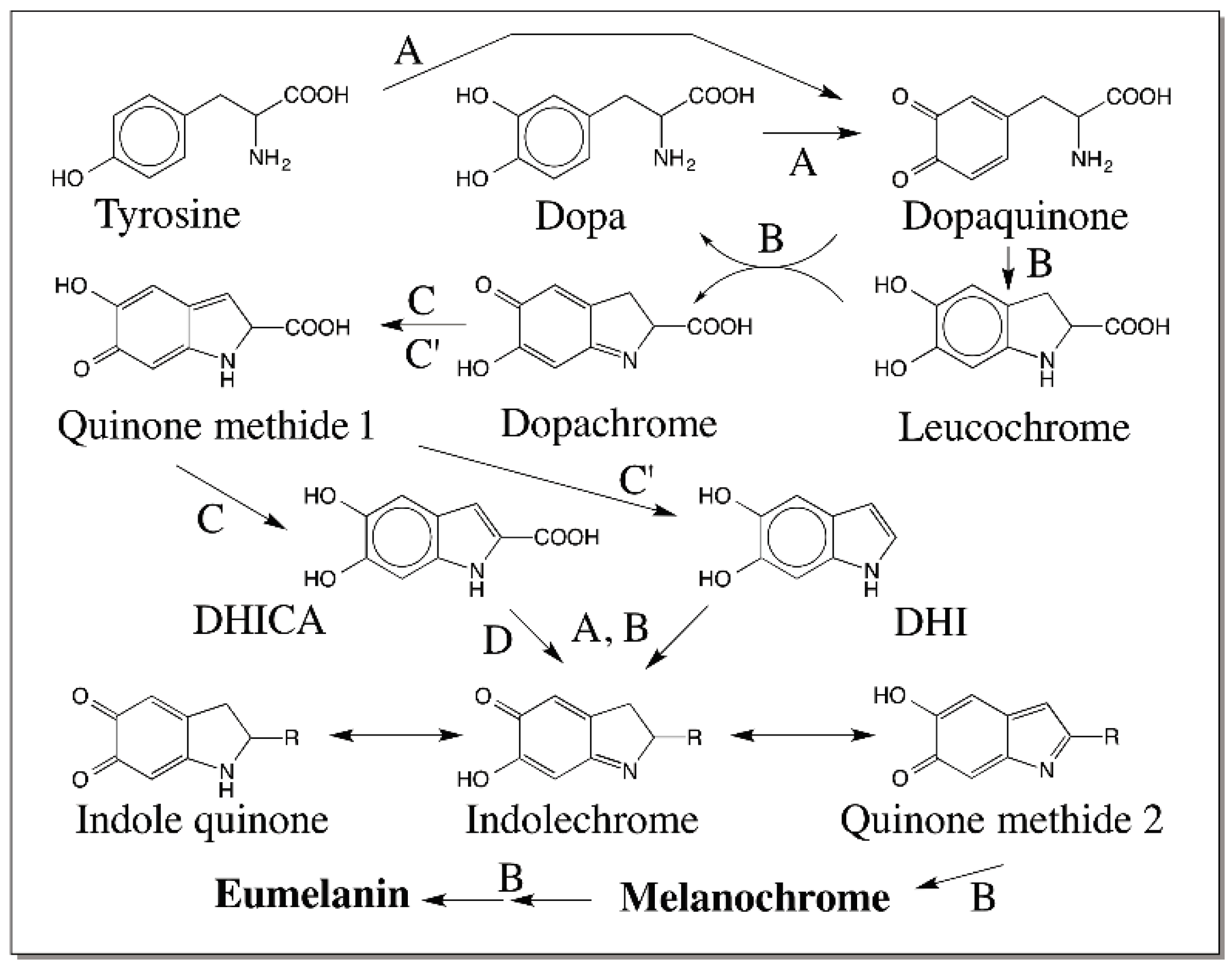{kind=link}
1. Introduction
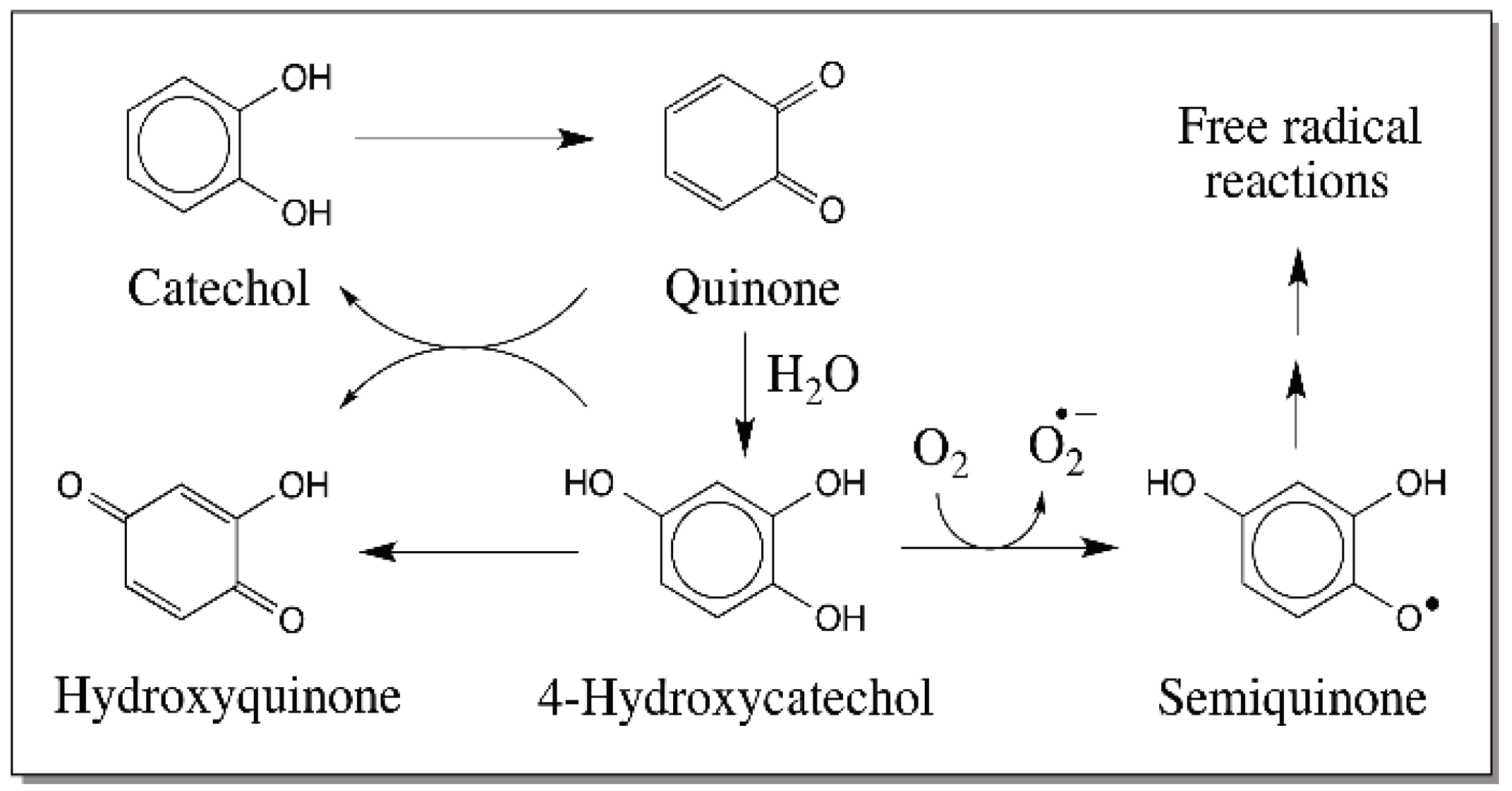
2. Reactions of Quinones
3. Quinone Methides—General Consideration
4. Predominance of Quinone to Quinone Methide Tautomerism in Simple Catechols
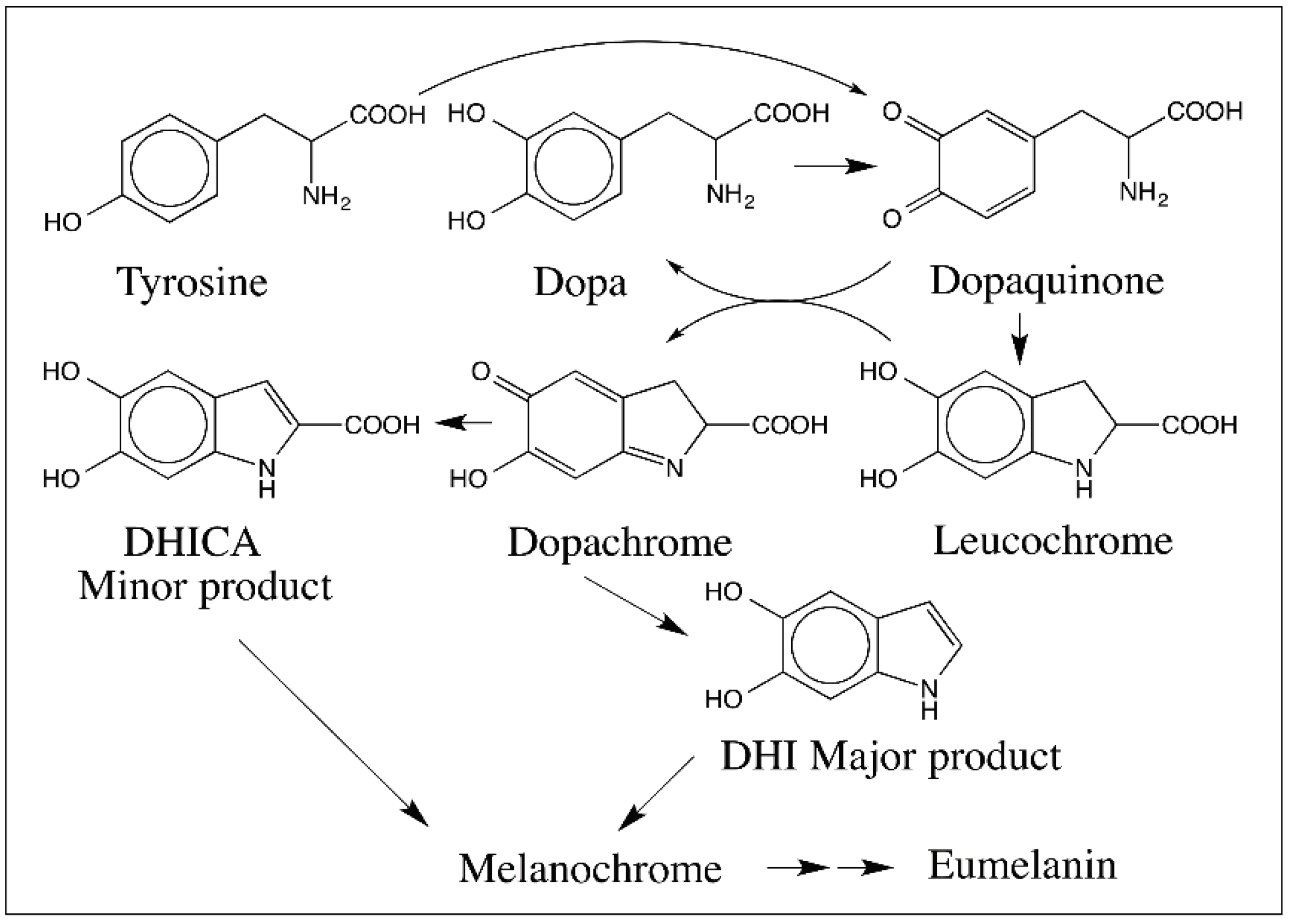
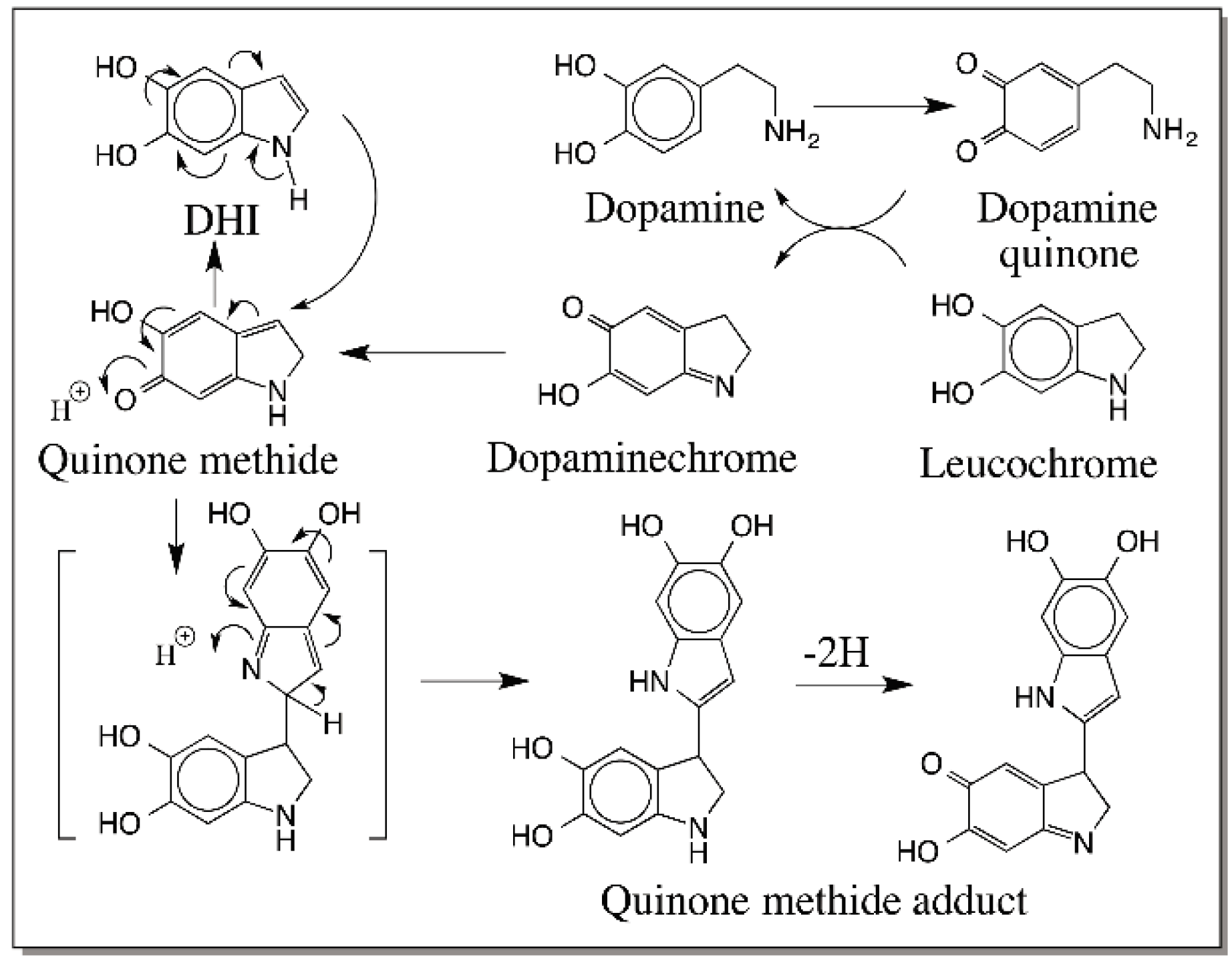
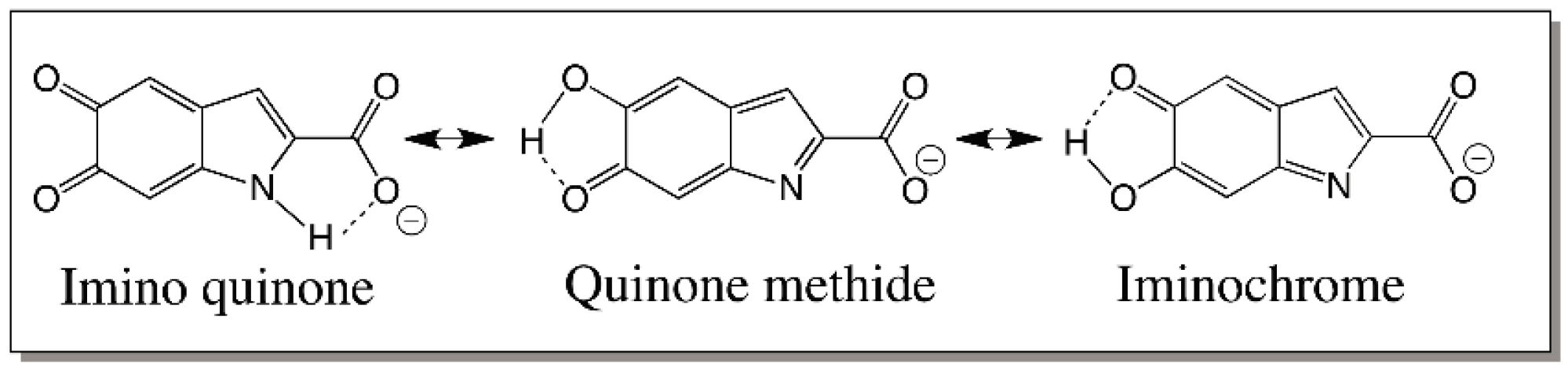
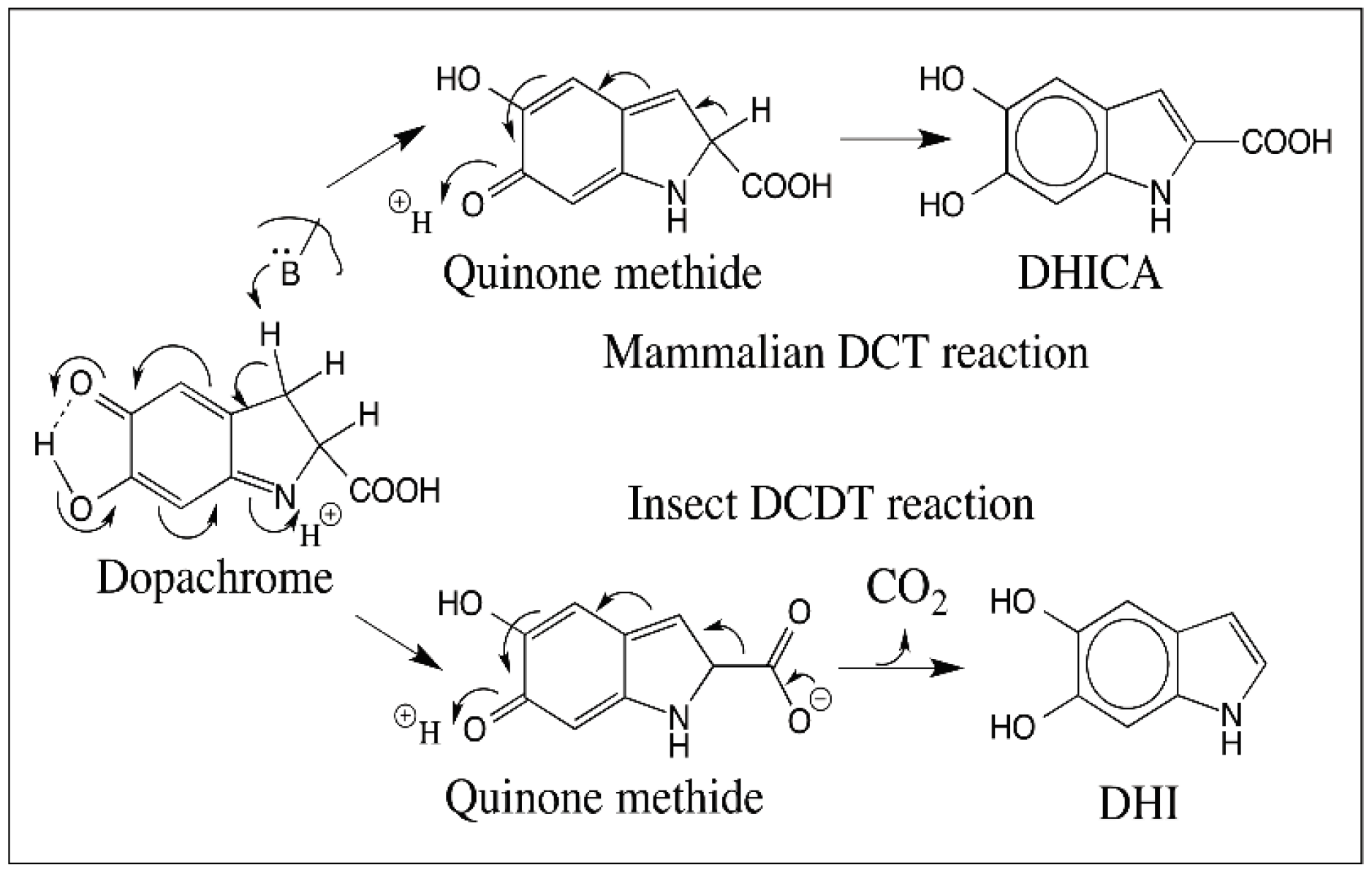
5. Dopachrome Conversion and Quinone Methide Formation in Melanin Biochemistry
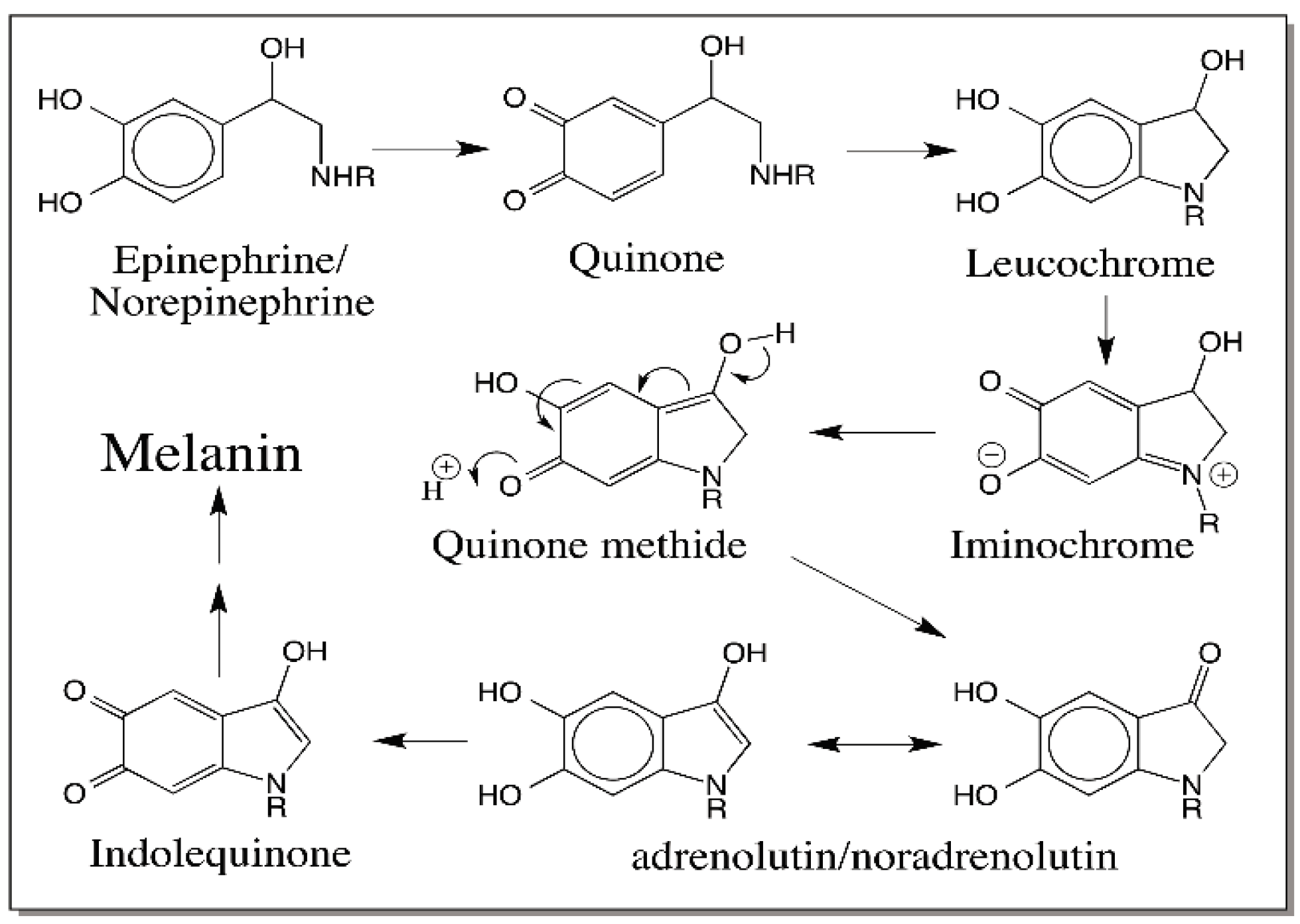
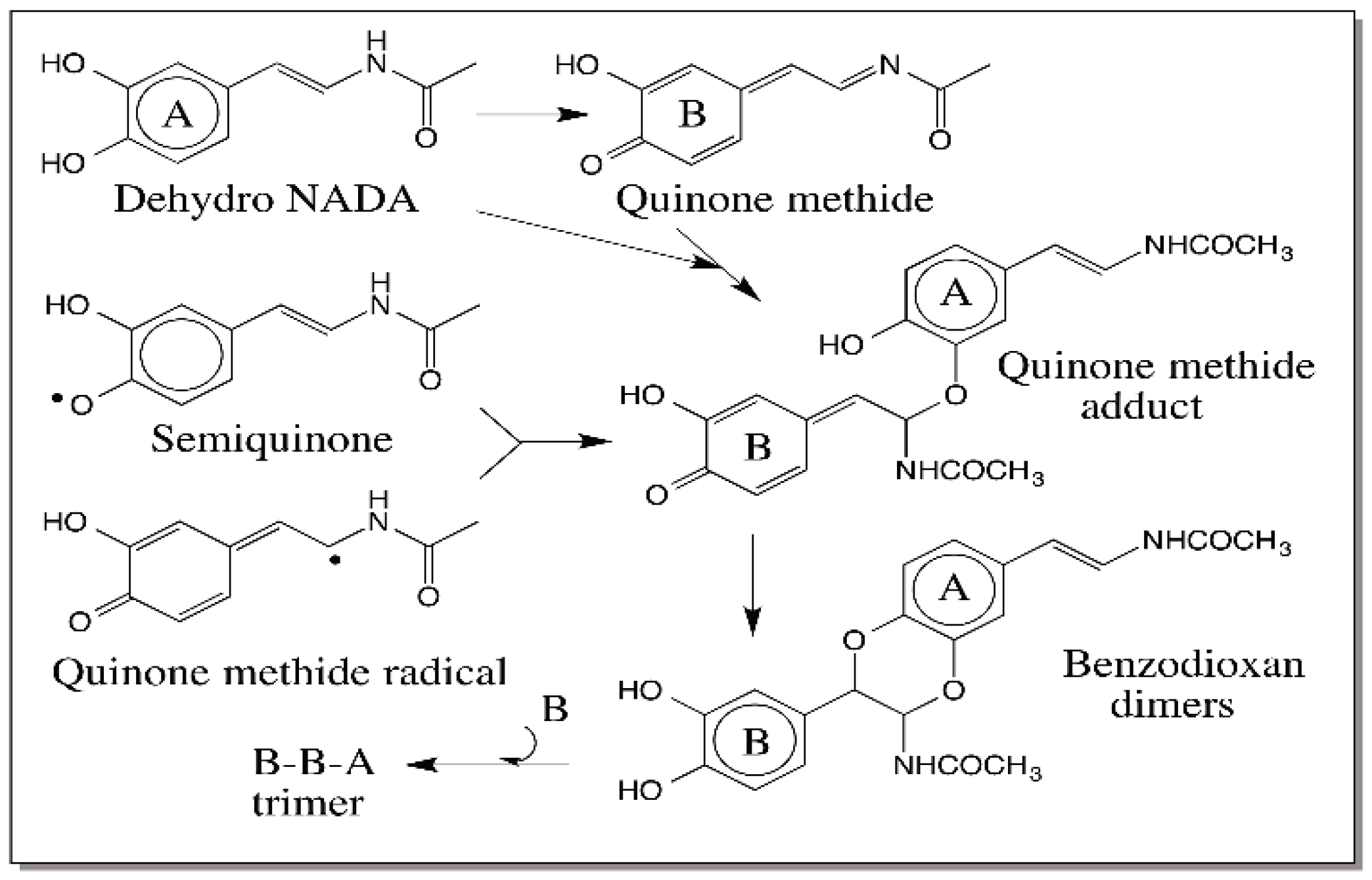
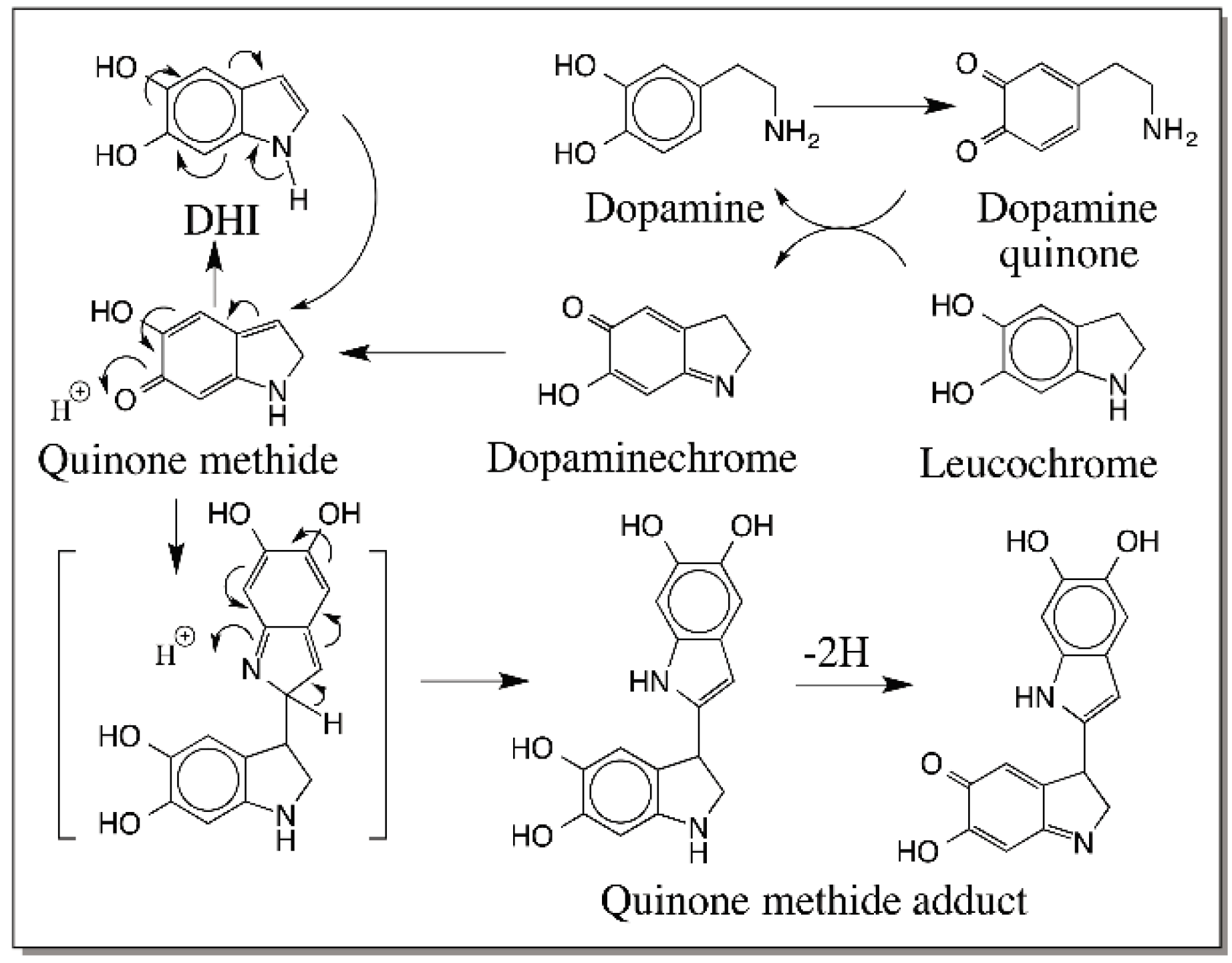
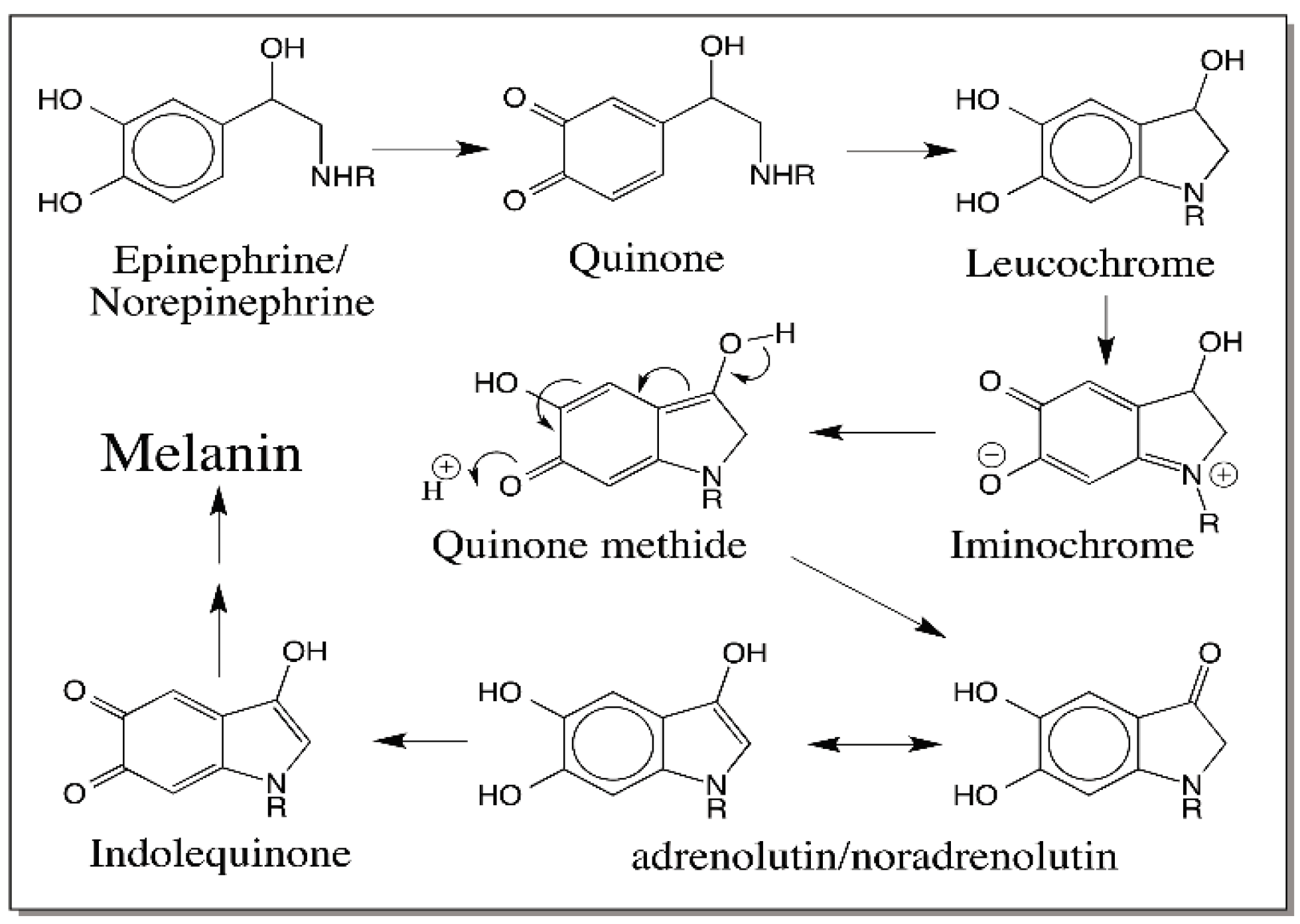
6. Quinone Methide Intermediate in Other Catecholamine Reactions
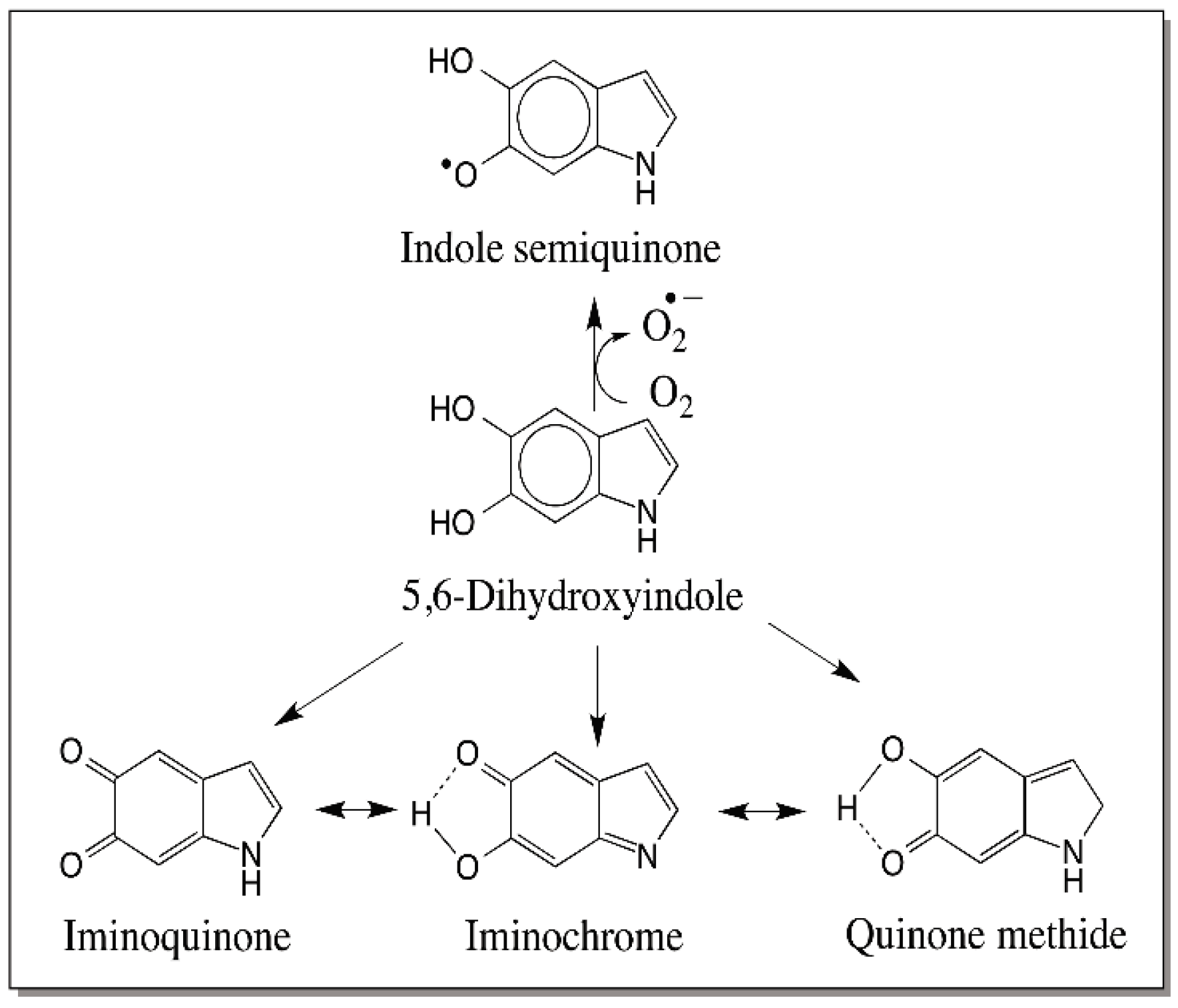
7. Reactivity of Dihydroxyindoles
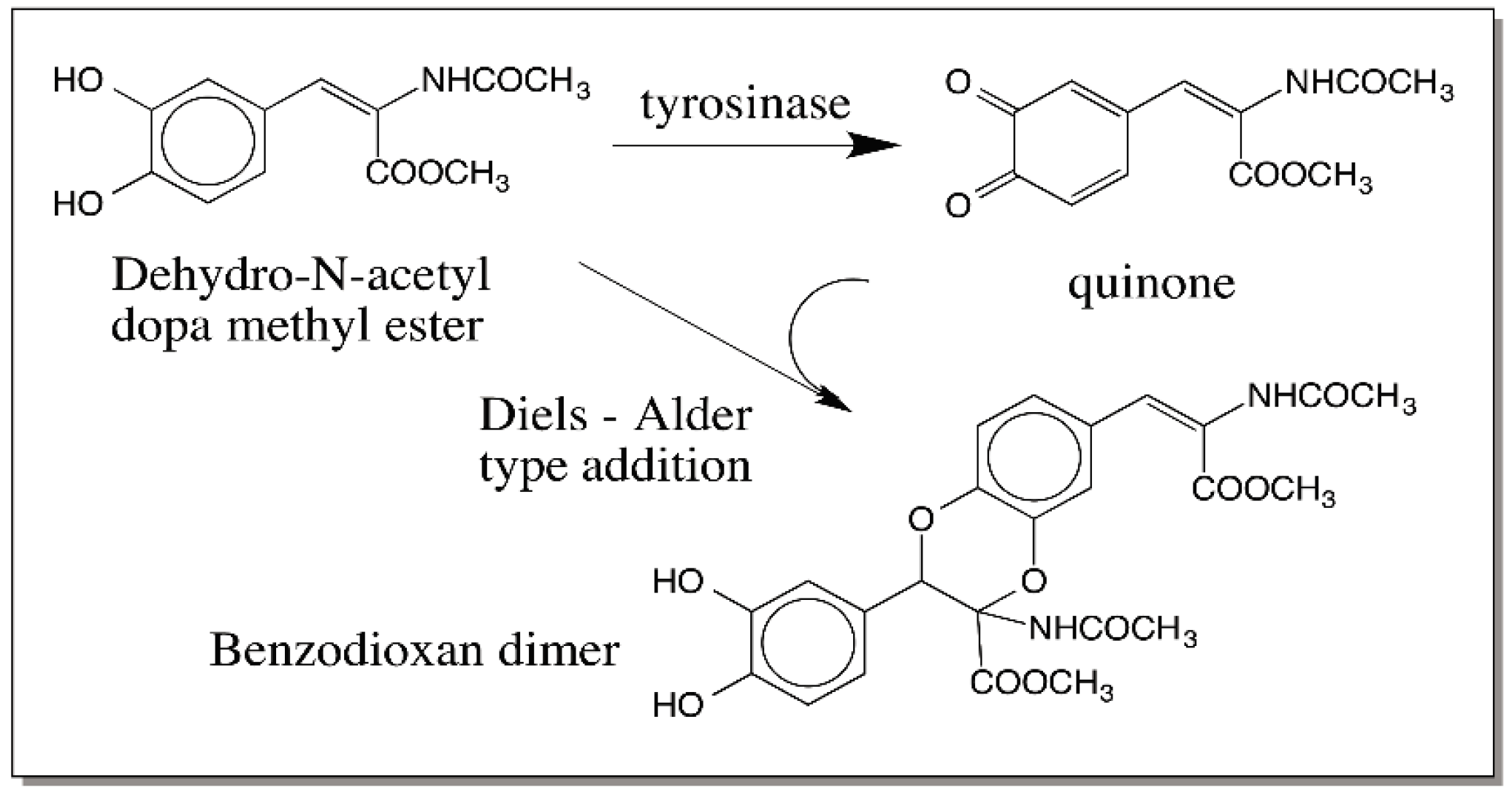
8. Comparative Oxidation Chemistry of 1,2-Dehydro-N-acyldopamines and Dihydroxyindoles
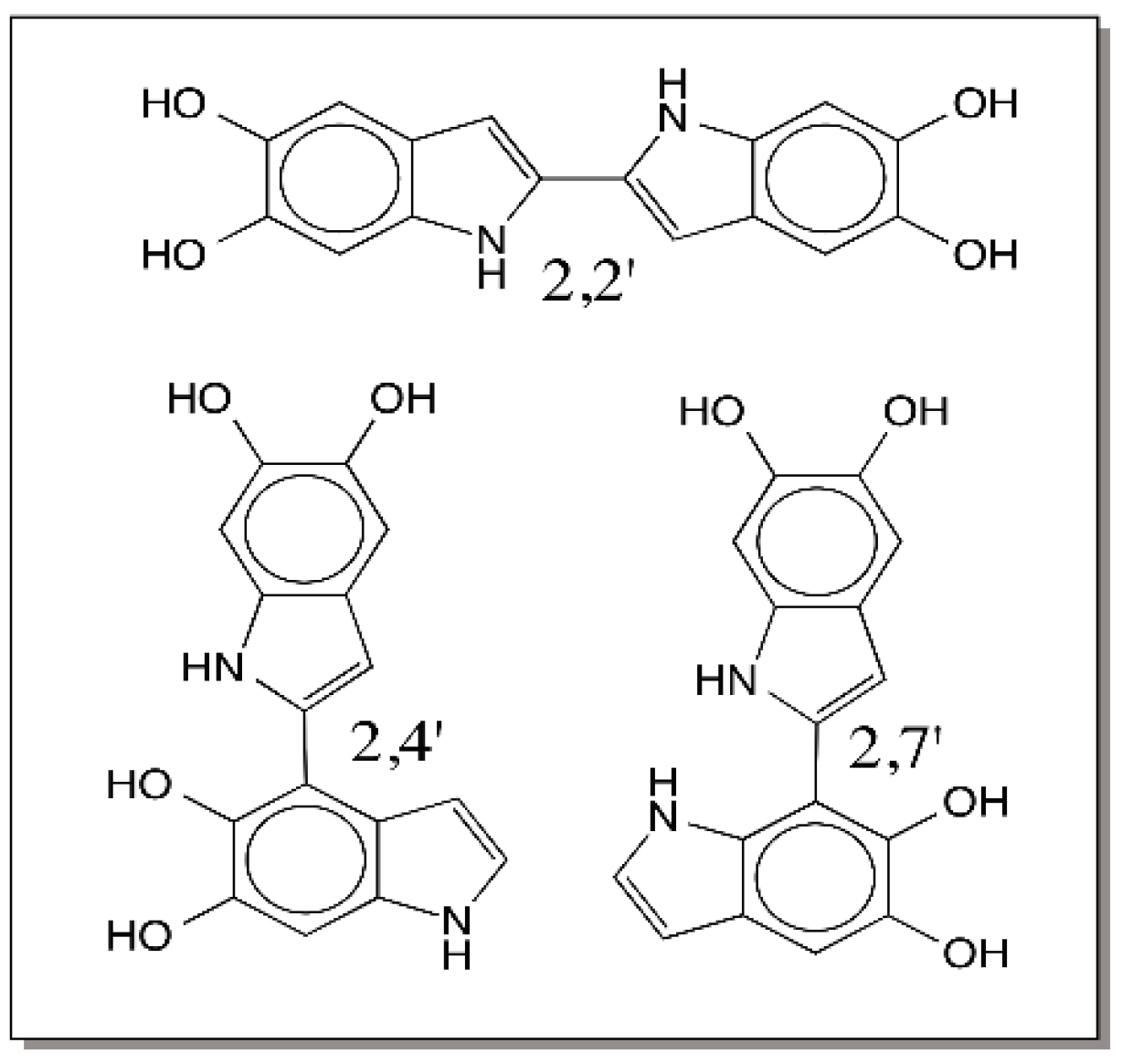
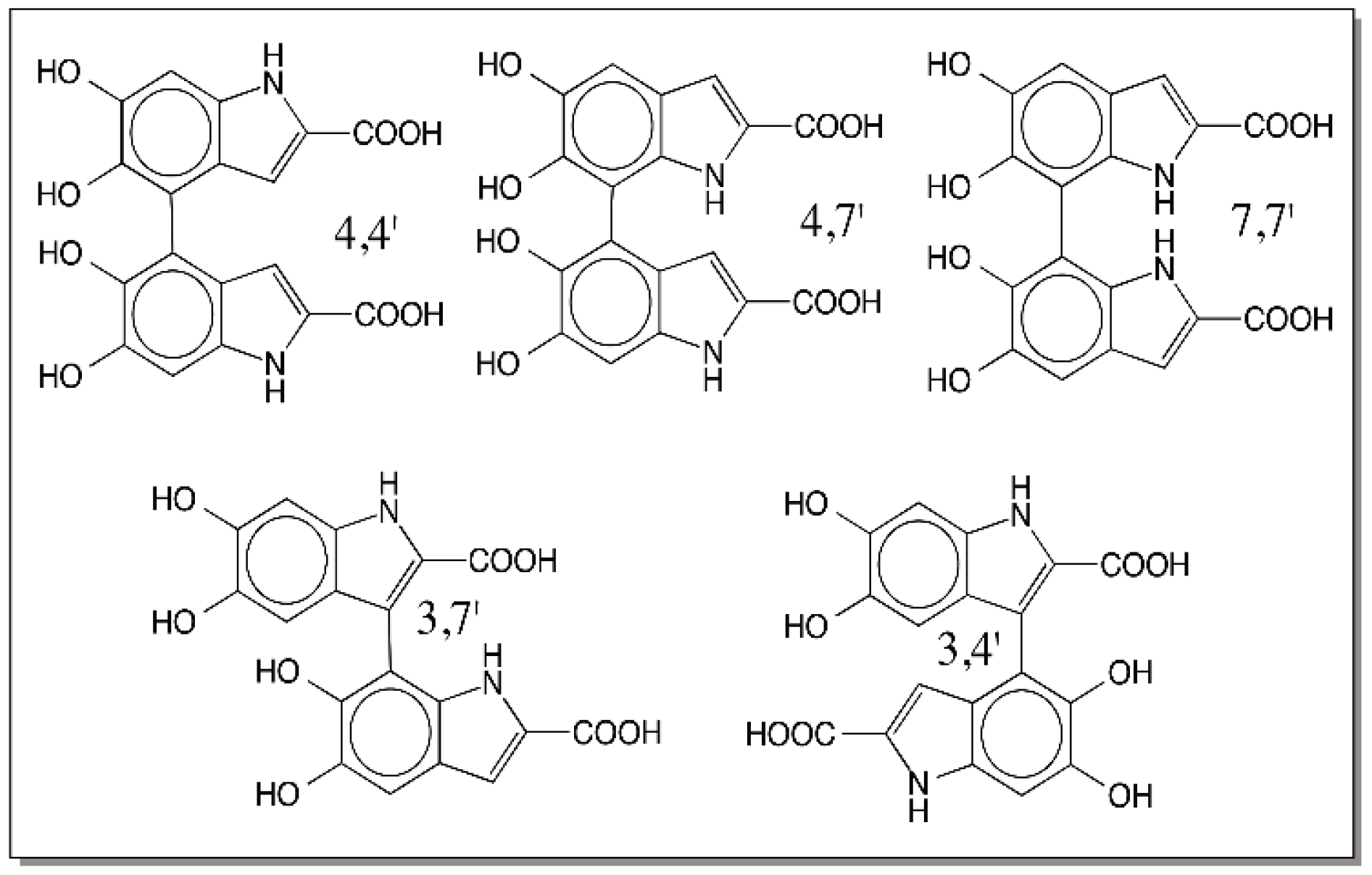
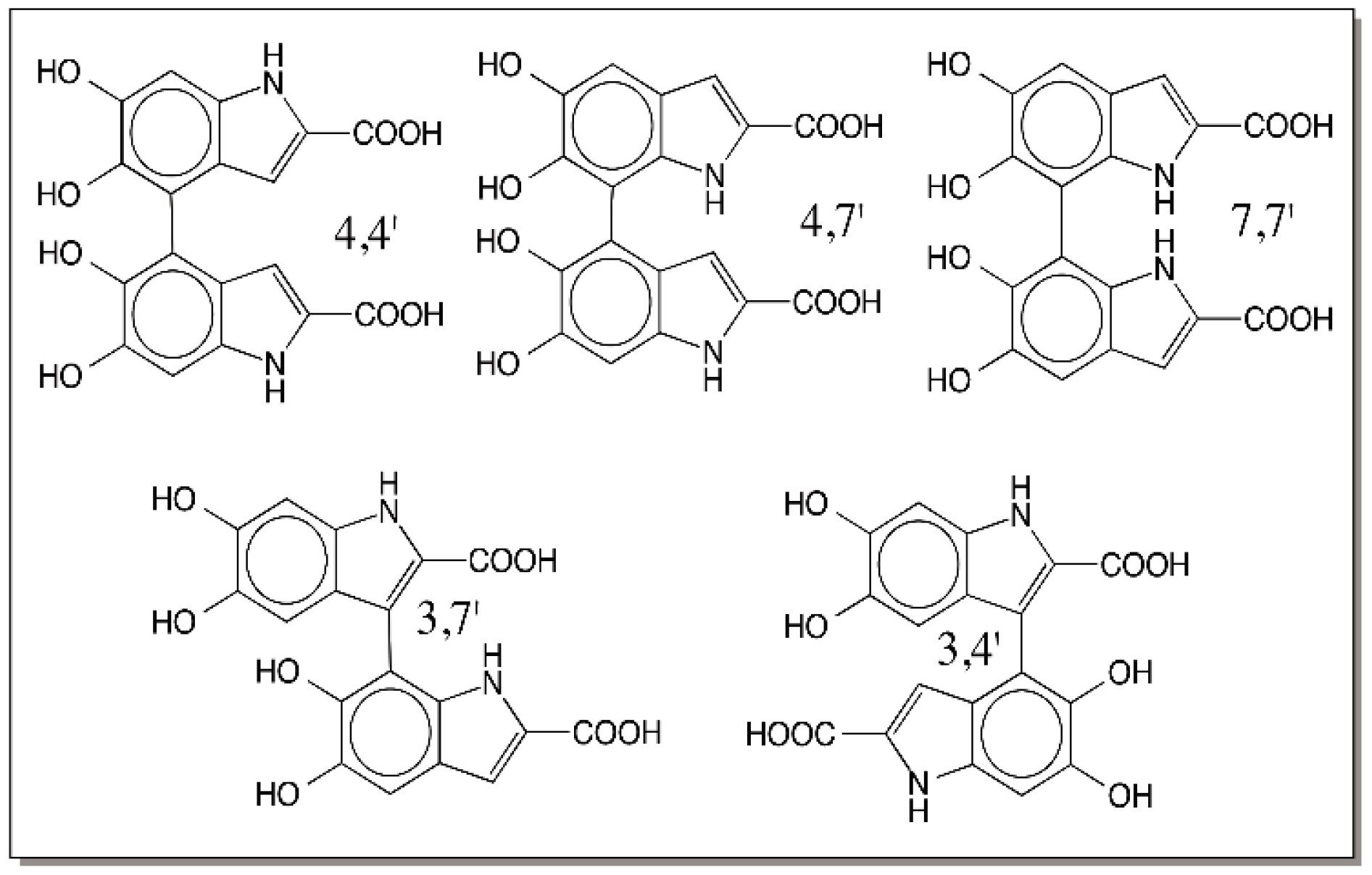
9. Oligomerization of DHI and DHICA and the Final Eumelanogenic Pathway
10. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| DHI | 5,6-dihydroxyindole |
| DHICA | 5,6-dihydroxyindole-2-carboxylic acid |
| Dehydro NADA | 1,2-dehydro-N-acetyldopamine |
| DCT | Dopachrome tautomerase |
| DCDT | Dopachrome decarboxylase/tautomerase |
References
- Prota, G. Melanins and Melanogenesis; Academic Press: New York, NY, USA, 1992; pp. 1–290. [Google Scholar]
- Hill, H.Z. The function of melanin or six blind people examine an elephant. BioEssays 1992, 14, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M. Comparative biochemistry of eumelanogenesis and the protective roles of phenoloxidase and melanin in insects. Pigment Cell Res. 2002, 15, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Ito, S. A chemist’s view of melanogenesis. Pigment Cell Res. 2003, 16, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Hearing, V.J.; Tsukamoto, K. Enzymatic control of pigmentation in mammals. FASEB J. 1991, 2, 2902–2909. [Google Scholar]
- Raper, H.S. The tyrosinase-tyrosine reaction. Production of l-3,4-dihydroxyphenylalanine from tyrosine. Biochem. J. 1926, 20, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Raper, H.S. The aerobic oxidases. Physiol. Rev. 1928, 8, 245–248. [Google Scholar]
- Raper, H.S. Some problems of tyrosine metabolism. J. Chem. Soc. 1938. [Google Scholar] [CrossRef]
- Mason, H.S. The chemistry of melanin. III. Mechanism of the oxidation of dihydroxyphenylalanine by tyrosinase. J. Biol. Chem. 1948, 172, 83–99. [Google Scholar] [PubMed]
- Mason, H.S. Comparative biochemistry of the phenolase complex. Adv. Enzymol. 1955, 16, 105–184. [Google Scholar]
- Korner, A.M.; Pawelek, J. Dopachrome conversion factor: A possible control point in melanin biosynthesis. J. Investig. Dermatol. 1980, 75, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Pawelek, J. Dopachrome conversion factor functions as an isomerase. Biochem. Biophys. Res. Commun. 1990, 166, 1328–1333. [Google Scholar] [CrossRef]
- Aroca, P.; Solano, F.; Garcia-Borron, J.; Lozano, J. A new spectrophotometric assay for dopachrome tautomerase. J. Biochem. Biophys. Methods 1990, 21, 35–46. [Google Scholar] [CrossRef]
- Aroca, P.; Garcia-Borron, J.; Solano, F.; Lozano, J. Regulation of mammalian melanogenesis I: Partial purification and characterization of a dopachrome converting factor: Dopachrome tautomerase. Biochim. Biophys. Acta 1990, 1035, 266–275. [Google Scholar] [CrossRef]
- Pawelek, J.M. After dopachrome? Pigment Cell Res. 1991, 4, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, K.; Jackson, I.J.; Urabe, K.; Montague, P.M.; Hearing, V. A second tyrosinase related protein, TRP-2, is a melanogenic enzyme termed DOPAchrome tautomerase. EMBO J. 1992, 11, 519–526. [Google Scholar] [PubMed]
- Aso, Y.; Kramer, K.J.; Hopkins, T.L.; Whetzel, S.Z. Properties of tyrosinase and dopa quinone imine conversion factor from pharate pupal cuticle of Manduca sexta. Insect Biochem. 1984, 14, 463–472. [Google Scholar] [CrossRef]
- Aso, Y.; Imamura, Y.; Yamasaki, N. Further studies on dopa quinone imine conversion factor from the cuticles of Manduca sexta. Insect Biochem. 1989, 19, 401–407. [Google Scholar] [CrossRef]
- Sugumaran, M.; Semensi, V. Quinone methides as new intermediates of melanin biosynthesis. J. Biol. Chem. 1991, 266, 6073–6078. [Google Scholar] [PubMed]
- Jimenez-Cervantes, C.; Solano, F.; Kobayashi, T.; Urabe, K.; Hearing, V.J.; Lozano, J.A.; Garcia-Borron, J.C. A new enzymatic function in the melanogenic pathway. The 5,6-dihydroxyindole-2-carboxylic acid oxidase activity of tyrosinase-related protein-1 (TRP1). J. Biol. Chem. 1994, 269, 17993–18001. [Google Scholar] [PubMed]
- Kobayashi, T.; Urabe, K.; Winder, A.; Jimenez-Cervantes, C.; Imokawa, G.; Brewington, T.; Solano, F.; Garcia-Borron, J.C.; Hearing, V.J. Tyrosinase-related protein-1 (TRP1) functions as DHICA oxidase in melanin biosynthesis. EMBO J. 1994, 13, 5818–5825. [Google Scholar] [PubMed]
- Jane, S.M.; Mu, D.; Wemmer, D.; Smith, A.I.; Kaur, S.; Malby, D.; Burlingame, A.I.; Klinman, J.P. A new redox cofactor in eukaryotic enzymes: 6-Hydroxydopa at the active site of bovine amine oxidase. Science 1990, 248, 981–987. [Google Scholar] [CrossRef]
- Wang, S.X.; Mure, M.; Medzihradszky, K.F.; Burlingame, A.I.; Brown, D.E.; Dooley, D.M.; Smith, A.J.; Kagan, M.; Klinman, J.P. A crosslinked cofactor in lysyl oxidase: Redox function for amino acid side chains. Science 2006, 273, 1078–1084. [Google Scholar] [CrossRef]
- Sugumaran, M.; Semensi, V.; Dali, H.; Mitchell, W. Novel oxidative transformations of 3,4-dihydroxy phenylacetic acid to 2,5,6-trihydroxybenzofuran and 3,4-dihydroxymandelic acid catalyzed by mushroom tyrosinase. Bioorg. Chem. 1989, 17, 86–95. [Google Scholar] [CrossRef]
- Sugumaran, M.; Dali, H.; Kundzicz, H.; Semensi, V. Unusual intramolecular cyclization and side chain desaturation of carboxyethyl-o-benzoquinone derivatives. Bioorg. Chem. 1989, 17, 443–453. [Google Scholar] [CrossRef]
- Abebe, A.; Kuang, Q.F.; Evans, J.; Sugumaran, M. Mass spectrometric studies shed light on unusual oxidative transformations of 1,2-dehydro-N-acetyldopa. Rapid Commun. Mass Spectrom. 2013, 27, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Dryhurst, G. Oxidation chemistry of Dopamine: Possible insights into the age-dependent loss of dopaminergic nigrostriatal neurons. Bioorg. Chem. 1993, 21, 392–410. [Google Scholar] [CrossRef]
- Blank, C.L.; Kissinger, P.T.; Adams, R.N. 5,6-Dihydroxyindole formation from oxidized 6-hydoxy dopamine. Eur. J. Pharmacol. 1972, 19, 391–394. [Google Scholar] [CrossRef]
- Huang, X.; Xu, R.; Hawley, M.D.; Kramer, K.J. Model insect cuticle sclerotization: Reactions of catecholamine quinones with the nitrogen-centered nucleophiles imidazole and N-acetyldopamine. Bioorg. Chem. 1997, 25, 179–202. [Google Scholar] [CrossRef]
- Sugumaran, M. Chemistry of cuticular sclerotization. Adv. Insect Physiol. 2010, 39, 151–209. [Google Scholar]
- Sugumaran, M. Complexities of cuticular pigmentation in insects. Pigment Cell Melanoma Res. 2009, 22, 523–525. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M. Molecular mechanisms for mammalian melanogenesis—Comparison with insect cuticular sclerotization. FEBS Lett. 1991, 293, 4–10. [Google Scholar] [CrossRef]
- Ito, S.; Prota, G. A facile one step synthesis of cysteinyldopas using mushroom tyrosinase. Experientia 1977, 33, 1118–1119. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Ito, S.; Fujita, K. Tyrosinase-catalyzed binding of 3,4-dihydroxyphenylalanine with proteins through the sulfhydryl group. Biochim. Biophys. Acta 1986, 881, 415–421. [Google Scholar] [CrossRef]
- Sugumaran, M.; Dali, H.; Semensi, V. Chemical and cuticular phenoloxidase mediated synthesis of cysteinyl catechol adducts. Arch. Insect Biochem. Physiol. 1989, 11, 127–137. [Google Scholar] [CrossRef]
- Vithayathil, P.J.; Murthy, G.S. New reaction of o-benzoquinone at the thioether group of methionine. Nature 1972, 236, 101–103. [Google Scholar] [CrossRef]
- Vithayathil, P.J.; Gupta, M.N. Reaction of methionine with some biologically important o-quinones. Ind. J. Biochem. Biophys. 1981, 18, 82–83. [Google Scholar]
- Sugumaran, M.; Nelson, E. Model sclerotization Studies: 4. Generation of N-acetylmethionyl catechol adducts during tyrosinase catalyzed oxidation of catechols in the presence of N-acetylmethionine. Arch. Insect Biochem. Physiol. 1998, 38, 44–52. [Google Scholar] [CrossRef]
- Cunane, L.M.; Chen, Z.-W.; Shamala, N.; Scott Mathews, F.; Cronin, C.N.; McIntire, W.S. Structures of the flavocytochrome p-cresol methylhydroxylase and its enzyme-substrate complex: Gated entry and proton relays support the proposed catalytic mechanism. J. Mol. Biol. 2000, 295, 357–374. [Google Scholar] [CrossRef] [PubMed]
- Senoh, S.; Witkop, B. Non-enzymatic conversions of dopamine to norepinephrine and trihydroxyphenethylamines. J. Am. Chem. Soc. 1959, 81, 6222–6231. [Google Scholar] [CrossRef]
- Senoh, S.; Creveling, C.R.; Udenfriend, S.; Witkop, B. Chemical, enzymatic and metabolic studies on the mechanism of oxidation of dopamine. J. Am. Chem. Soc. 1959, 81, 6236–6240. [Google Scholar] [CrossRef]
- Kaufman, S.; Bridgers, W.F.; Eisenberg, F.; Friedman, S. The source of oxygen in the phenylalanine hydroxylase and the dopamine-β-hydroxylase catalyzed reactions. Biochem. Biophys. Res. Commum. 1962, 9, 497–502. [Google Scholar] [CrossRef]
- Sugumaran, M.; Lipke, H. Quinone methide formation from 4-alkylcatechols. A novel reaction catalyzed by cuticular polyphenoloxidase. FEBS Lett. 1983, 155, 65–68. [Google Scholar] [CrossRef]
- Nakamura, T. On the process of enzymatic oxidation of hydroquinone. Biochem. Biophys. Res. Commun. 1960, 2, 111–113. [Google Scholar] [CrossRef]
- Sugumaran, M.; Bolton, J.L. Laccase—And not tyrosinase—Is the enzyme responsible for quinone methide production from 2,6-dimethoxy-4-allyl phenol. Arch. Biochem. Biophys. 1998, 353, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Saul, S.J.; Sugumaran, M. o-Quinone: Quinone methide isomerase—A novel enzyme which prevents the destruction of self matter by phenoloxidase generated quinones during immune response in insects. FEBS Lett. 1989, 249, 155–158. [Google Scholar] [CrossRef]
- Saul, S.J.; Sugumaran, M. 4-Alkyl-o-quinone/2-hydroxy-p-quinone methide isomerase from the larvae hemolymph of Sarcophaga bullata. I. Purification and characterization of enzyme catalyzed reaction. J. Biol. Chem. 1990, 265, 16992–16999. [Google Scholar] [PubMed]
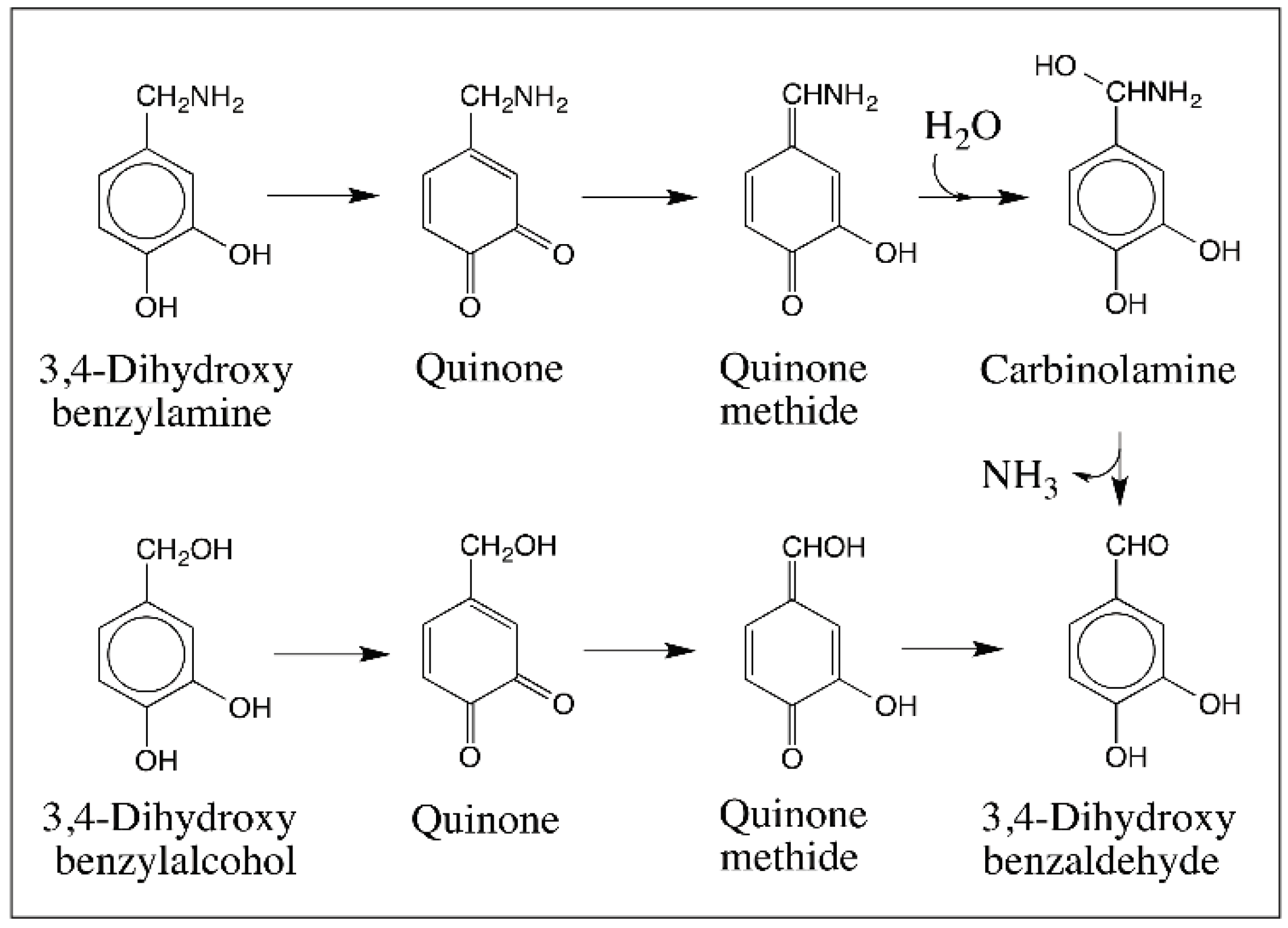
- Sugumaran, M. Oxidation of 3,4-dihydroxybenzylamine affords 3,4-dihydroxybenzaldehyde via the quinone methide intermediate. Pigment Cell Res. 1995, 8, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M.; Semensi, V.; Dali, H.; Nellaiappan, K. Oxidation of 3,4-dihydroxybenzyl alcohol: A sclerotizing precursor for cockroach ootheca. Arch. Insect Biochem. Physiol. 1991, 16, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Cooksey, C.J.; Garrett, P.J.; Land, E.J.; Ramsden, C.A.; Riley, P.A. Tyrosinase kinetics: Failure of the auto-activation mechanism of monohydric phenol oxidation by rapid formation of a quinomethane intermediate. Biochem. J. 1998, 333, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M.; Tan, S.; Sun, H.L. Tyrosinase catalyzed oxidation of 3,4-dihydroxyphenylglycine. Arch. Biochem. Biophys. 1996, 329, 175–180. [Google Scholar] [CrossRef] [PubMed]
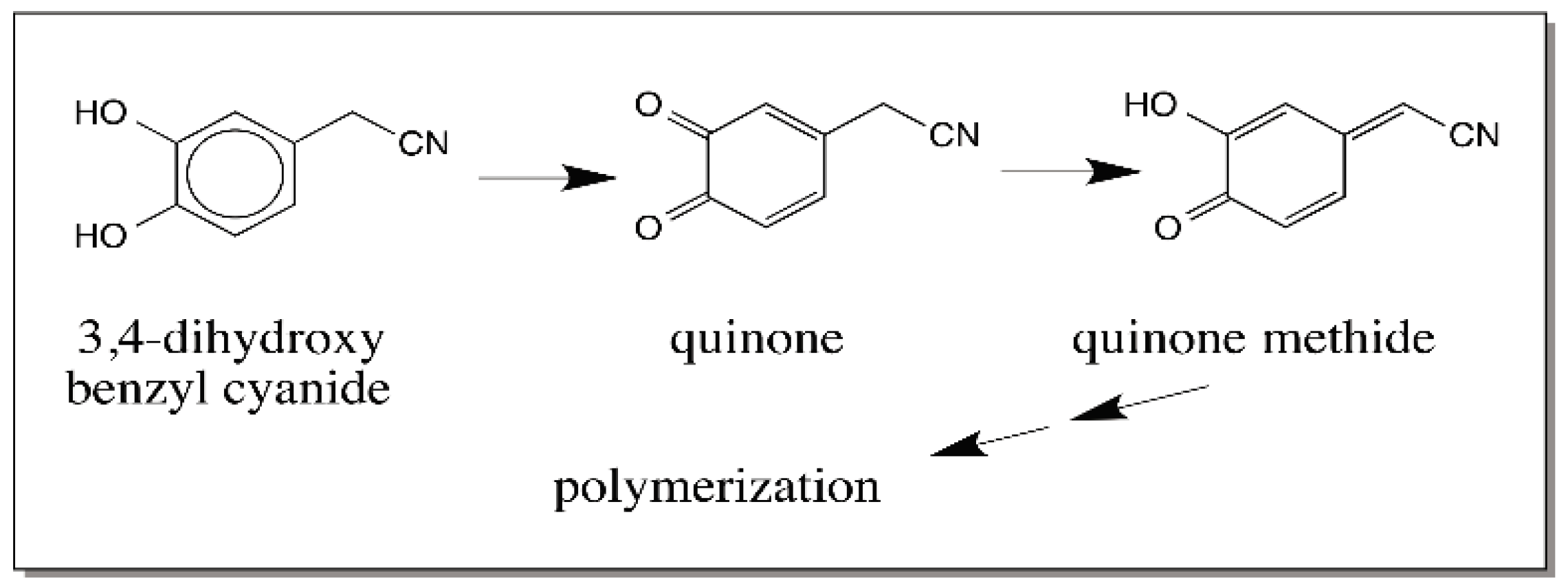
- Sugumaran, M.; Duggaraju, R.; Jayachandran, K.; Kirk, K. Formation of a new quinone methide intermediate during the oxidative transformation of 3,4-dihydroxyphenylacetic acids: Implications for eumelanin biosynthesis. Arch. Biochem. Biophys. 1999, 371, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Mefford, I.N.; Kincl, L.; Dykstra, K.H.; Simpson, J.T.; Markey, S.P.; Dietz, S.; Wightman, R.M. Facile oxidative decarboxylation of 3,4-dihydroxyphenylacetic acid catalyzed by copper and manganese ions. Biochim. Biophys. Acta 1996, 1290, 224–230. [Google Scholar] [CrossRef]
- Ito, S.; Yamanaka, Y.; Wakamatsu, K. The metabolic fate of ortho-quinones derived from catecholamine metabolites. Int. J. Mol. Sci. 2016, 17, 164. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M. Tyrosinase catalyzes an unusual oxidative decarboxylation of 3,4-dihydroxymandelate. Biochemistry 1985, 25, 4489–4492. [Google Scholar] [CrossRef]
- Cabanes, J.; Sanchez-Ferrer, A.; Bru, R.; Garcia-Carmona, F. Chemical and enzymic oxidation by tyrosinase of 3,4-dihydroxymandelate. Biochem. J. 1988, 256, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M.; Dali, H.; Semensi, V. Mechanistic studies on tyrosinase catalyzed oxidative decarboxylation of 3,4-dihydroxymandelic acid. Biochem. J. 1992, 281, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M.; Dali, H.; Semensi, V. The mechanism of tyrosinase catalyzed oxidative decarboxylation of α-(3,4-dihydroxyphenyl) lactic acid. Biochem. J. 1991, 277, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M.; Semensi, V.; Saul, S.J. On the mechanism of oxidation of 3,4-dihydroxyphenethyl alcohol and 3,4-dihydroxyphenyl glycol by cuticular phenoloxidase from Sarcophaga bullata. Arch. Insect Biochem. Physiol. 1989, 10, 13–27. [Google Scholar] [CrossRef]
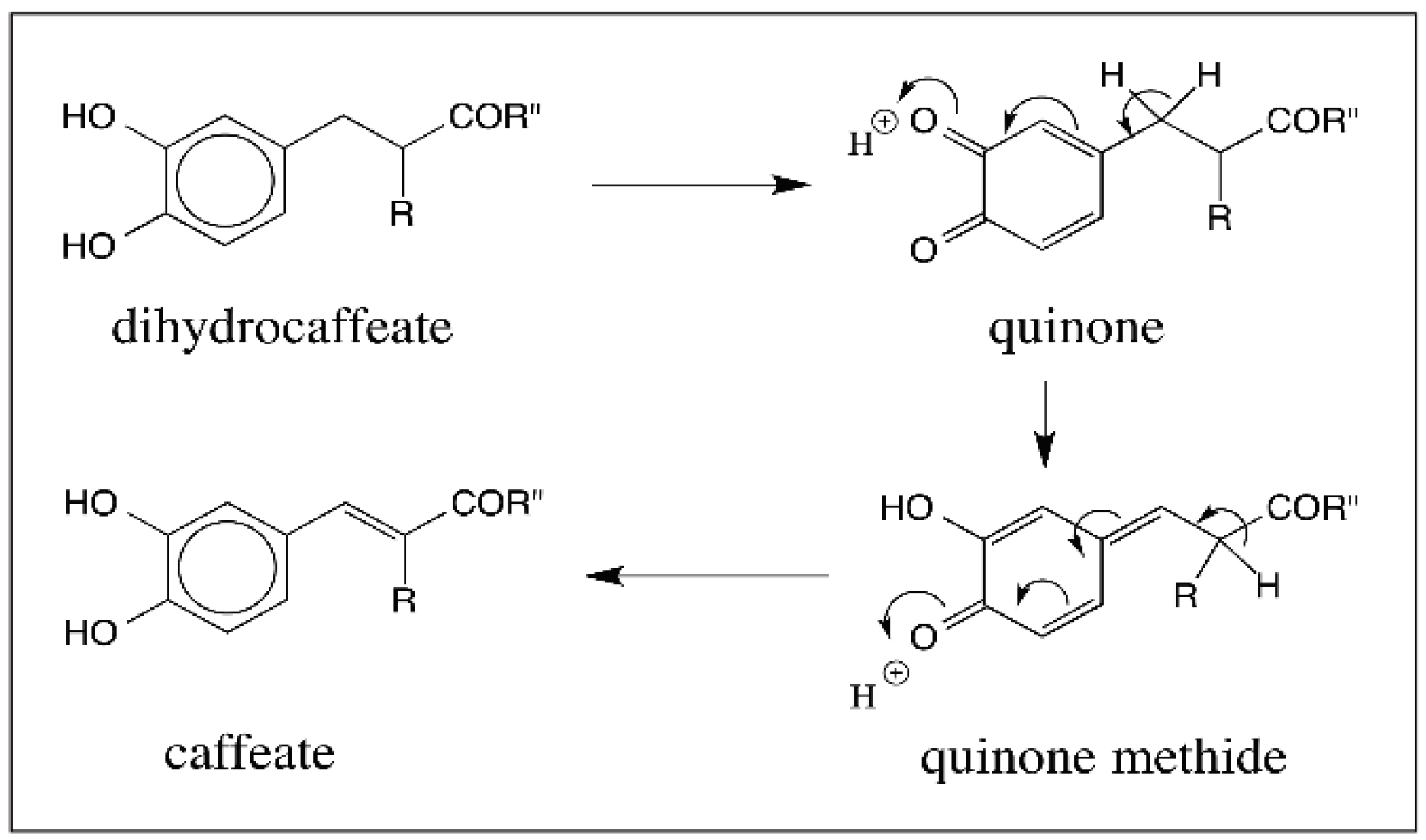
- Sugumaran, M.; Semensi, V.; Dali, H.; Saul, S.J. Nonenzymatic transformations of enzymatically generated N-acetyldopamine quinone and isomeric dihydrocaffeiyl methylamide quinone. FEBS Lett. 1989, 255, 345–349. [Google Scholar] [CrossRef]
- Taylor, S.W.; Molinski, T.F.; Rzepecki, L.M.; Waite, H.J. Oxidation of peptidyl 3,4-dihydroxy phenylalanine analogues: Implications for the biosynthesis of tunichromes and related oligopeptides. J. Nat. Prod. 1991, 54, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M.; Ricketts, D. Model sclerotization studies. 3. Cuticular enzyme catalyzed oxidation of peptidyl model tyrosine and dopa derivatives. Arch. Insect Biochem. Physiol. 1995, 28, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Iverson, S.L.; Hu, L.Q.; Vukomanoci, V.; Bolton, J.L. The influence of the p-alkyl substituent on the isomerization of o-quinones to p-quinone methides: Potential bioactivation mechanism for catechols. Chem. Res. Toxicol. 1995, 8, 109–113. [Google Scholar] [CrossRef]
- Bolton, J.L.; Wu, H.W.; Hu, L.Q. Mechanism of isomerization of 4-propyl-o-quinone to its tautomeric p-quinone methide. Chem. Res. Toxicol. 1996, 9, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Musson, D.G.; Karashima, D.; Rubiero, H.; Melmon, K.L.; Cheng, A.; Castagnoli, N., Jr. Synthetic and preliminary hemodynamic and whole animal toxicity studies on (R,S)-, (R)-, and (S)-2-methyl-3-(2,4,5-trihydroxyphenyl)alanine. J. Med. Chem. 1980, 23, 1318–1323. [Google Scholar] [CrossRef] [PubMed]
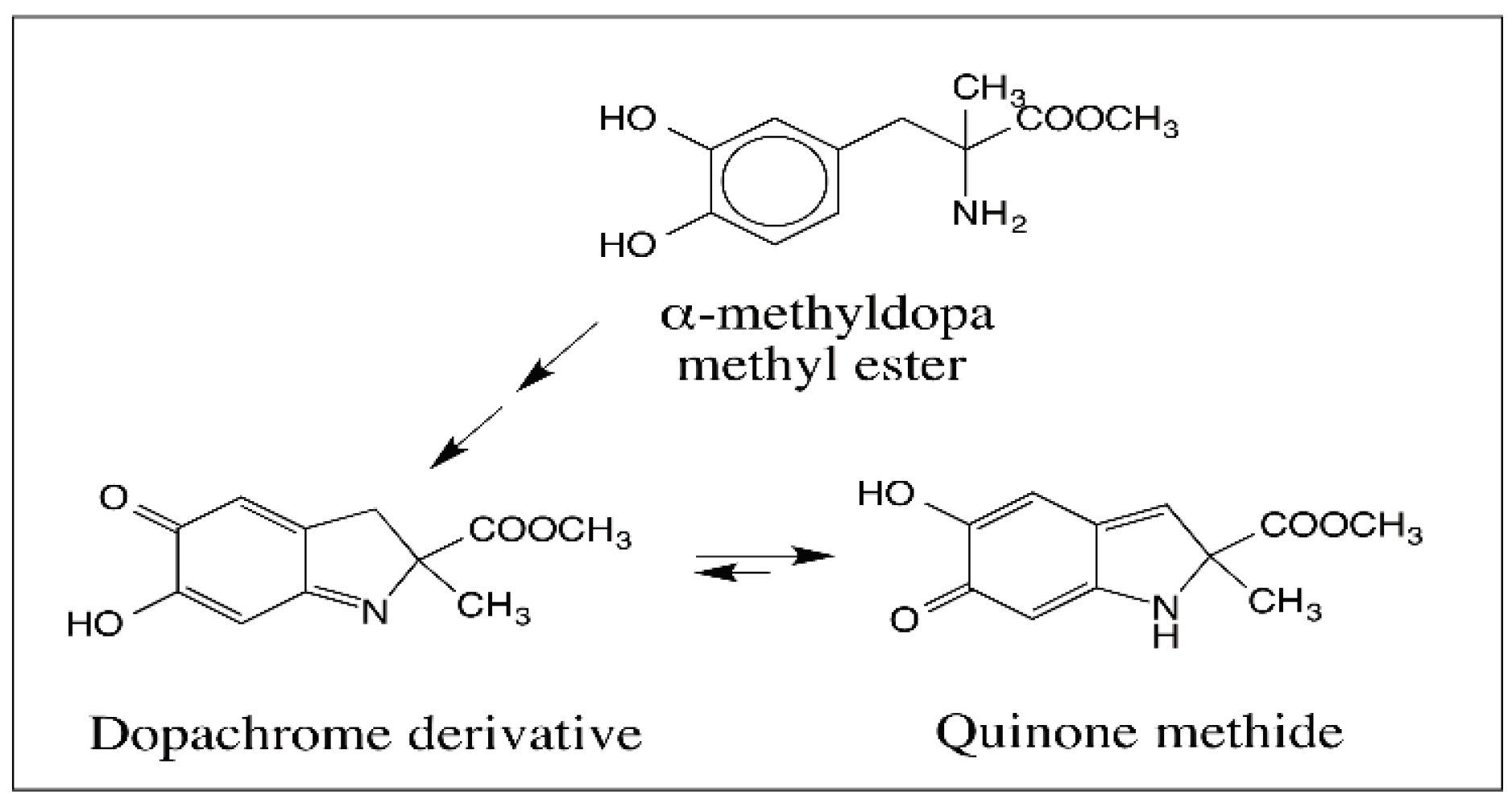
- Sugumaran, M.; Dali, H.; Semensi, V. Formation of a stable quinone methide during tyrosinase catalyzed oxidation of α-methyldopa methyl ester and its implication in melanin biosynthesis. Bioorg. Chem. 1990, 18, 144–153. [Google Scholar] [CrossRef]
- Crescenzi, O.; Costantini, C.; Prota, G. Evidence for the intermediacy of quinone-methides in the rearrangement of aminochromes to 5,6-dihydroxyindoles. Tetrahedron Lett. 1990, 31, 6095–6096. [Google Scholar] [CrossRef]
- Costantini, C.; Crescenzi, O.; Prota, G. Mechanism of the rearrangement of dopachrome to 5,6-dihydroxyindole. Tetrahedron Lett. 1991, 32, 3849–3850. [Google Scholar] [CrossRef]
- Palumbo, A.; d’Ischia, M.; Misuraca, G.; Prota, G. Effect of metal ions on the rearrangement of dopachrome. Biochim. Biophys. Acta 1987, 925, 203–209. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Ito, S. Preparation of eumelanin related metabolites, 5,6-dihydroxyindole, 5,6-dihydroxyindole-2-carboxylic acid, and their O-methyl derivatives. Anal. Biochem. 1988, 170, 335–340. [Google Scholar] [CrossRef]
- Palumbo, A.; d’Ischia, M.; Misuraca, G.; Prota, G. A new look at the rearrangement of adrenochrome under biomimetic conditions. Biochim. Biophys. Acta 1989, 990, 297–302. [Google Scholar] [CrossRef]
- Manini, P.; Panzella, L.; Napolitano, A.; d’Ischia, M. Oxidation chemistry of norepinephrine: Partitioning of the o-quinone between competing cyclization and chain breakdown pathways and their roles in melanin formation. Chem. Res. Toxicol. 2007, 20, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- Kroesche, C.; Peter, M.G. Detection of melanochromes by MALDI-TOF mass spectrometry. Tetrahedron 1996, 52, 3947–3952. [Google Scholar] [CrossRef]
- Al-Kazwini, A.T.; O’Neill, P.; Cundall, R.B.; Admas, G.E.; Junino, A.; Maignan, J. Direct observation of the reaction of the quinone methide from 5,6-dihydroxyindole with the nucleophilic azide ion. Tetrahedron Lett. 1992, 33, 3045–3048. [Google Scholar] [CrossRef]
- Al-Kazwini, A.T.; O’Neill, P.; Admas, G.E.; Cundall, R.B.; Lang, G.; Junino, A. Reactions of indolic radicals produced upon one-electron oxidation of 5,6-dihydroxyindole and its N(1)-methylated analogue. J. Chem. Soc. Perkin Trans. II 1991. [Google Scholar] [CrossRef]
- Napolitano, A.; Corradini, M.G.; Prota, G. A reinvestigation of the structure of melanochrome. Tetrahedron Lett. 1985, 26, 2805–2808. [Google Scholar] [CrossRef]
- d’Ischia, M.; Napolitano, A.; Tsiakas, K.; Prota, G. New intermediates in the oxidative polymerization of 5,6-dihydroxyindole to melanin promoted by the peroxidase/H2O2 system. Tetrahedron 1990, 46, 5789–5796. [Google Scholar] [CrossRef]
- Pezzella, A.; Panzella, L.; Crescenzi, O.; Napolitano, A.; Navaratman, S.; Edge, R.; Land, E.J.; Barone, V.; d’Ischia, M. Short-lived quinonoid species from 5,6-dihydroxyindole dimers en route to eumelanin polymers: Integrated chemical, pulse radiolytic, and quantum mechanical investigation. J. Am. Chem. Soc. 2006, 128, 15490–15498. [Google Scholar] [CrossRef] [PubMed]
- Panzella, L.; Pezzella, A.; Napolitano, A.; d’Ischia, M. The first 5,6-dihydroxyindole tetramer by oxidation of 5,5′,6,6′-tetrahydroxy-2,4′-biindolyl and an unexpected issue of positional reactivity en route to eumelanin-related polymers. Tetrahedron Lett. 2007, 9, 1411–1414. [Google Scholar] [CrossRef] [PubMed]
- Pezzella, A.; Panzella, L.; Natangelo, A.; Arzillo, M.; Napolitano, A.; d’Ischia, M. 5,6-Dihydroxyindole tetramers with “anomalous” interunit bonding patterns by oxidative coupling of 5,5′,6,6′-Tetrahydroxy-2,7′-biindolyl: Emerging complexities on the way toward an improved model of eumelanin buildup. J. Org. Chem. 2007, 72, 9225–9230. [Google Scholar] [CrossRef] [PubMed]
- Okunda, H.; Wakamatsu, K.; Ito, S.; Sota, T. Possible oxidative polymerization mechanism of 5,6-dihydroxyindole from ab Initio calculations. J. Phys. Chem. A 2008, 112, 11213–11222. [Google Scholar] [CrossRef] [PubMed]
- Pezzella, A.; Napolitano, A.; d’Ischia, M.; Prota, G. Oxidative polymerization of 5,6-dihydroxy indole-2-carboxylic acid: A new insight. Tetrahedron 1996, 23, 7913–7920. [Google Scholar] [CrossRef]
- Pezzella, A.; Vogna, D.; Prota, G. Atropoisomeric melanin intermediates by oxidation of the melanogenic precursor 5,6-dihydroxyindole-2-carboxylic acid under biomimetic conditions. Tetrahedron 2002, 58, 3681–3687. [Google Scholar] [CrossRef]
- D’ischia, M.; Napolitano, A.; Pezzella, A. 5,6-Dihydroxyindole chemistry: Unexplored opportunities beyond eumelanin. Eur. J. Org. Chem. 2011, 2011, 5501–5516. [Google Scholar] [CrossRef]
- Saul, S.J.; Sugumaran, M. Characterization of a new enzyme system that desaturates the side chain of N-acetyldopamine. FEBS Lett. 1989, 251, 69–73. [Google Scholar] [CrossRef]
- Saul, S.J.; Sugumaran, M. N-Acetyldopamine quinone methide/1,2-dehydro-N-acetyldopamine tautomerase—A new enzyme involved in sclerotization of insect cuticle. FEBS Lett. 1989, 255, 340–344. [Google Scholar] [CrossRef]
- Ricketts, D.; Sugumaran, M. 1,2-dehydro-N-β-alanyldopamine as a new intermediate in insect cuticular sclerotization. J. Biol. Chem. 1994, 269, 22217–22221. [Google Scholar] [PubMed]
- Sugumaran, M.; Dali, H.; Semensi, V.; Hennigan, B. Tyrosinase catalyzed unusual oxidative dimerization of 1,2-dehydro-N-acetyldopamine. J. Biol. Chem. 1987, 262, 10546–10549. [Google Scholar] [PubMed]
- Sugumaran, M.; Semensi, V.; Kalyanaraman, B.; Bruce, J.M.; Land, E.J. Evidence for the formation of a quinone methide during the oxidation of the insect cuticular sclerotizing precursor, 1.2-dehydro-N-acetyldopamine. J. Biol. Chem. 1992, 267, 10355–10361. [Google Scholar] [PubMed]
- Sugumaran, M. Oxidation chemistry of 1,2-dehydro-N-acetyldopamines: Direct evidence for the formation of 1,2-dehydro-N-acetyldopamine quinone. Arch. Biochem. Biophys. 2000, 378, 404–419. [Google Scholar] [CrossRef] [PubMed]
- Abebe, A.; Zheng, D.; Evans, J.; Sugumaran, M. Reexamination of the mechanisms of oxidative transformation of the insect cuticular sclerotizing precursor, 1,2-dehydro-N-acetyldopamine. Insect Biochem. Mol. Biol. 2010, 40, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M.; Robinson, W. Structure, biosynthesis and possible function of tunichromes and related compounds. Comp. Biochem. Physiol. B 2012, 163, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M.; Robinson, W.E. Bioactive dehydrotyrosyl and dehydrodopyl compounds of marine origin. Mar. Drugs 2010, 8, 2906–2935. [Google Scholar] [CrossRef] [PubMed]
- Abebe, A.; Zheng, D.; Evans, J.; Sugumaran, M. Novel post-translational oligomerization of peptidyl dehydrodopa model compound, 1,2-dehydro-N-acetyldopa methyl ester. Bioorg. Chem. 2016, 66, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, A.; Crescenzi, C.; Prota, G. Copolymerization of 5,6-dihydroxyindole and 5,6-dihydroxy indole-2-carboxylic acid in melanogenesis. Isolation of a cross coupling product. Tetrahedron Lett. 1993, 34, 885–888. [Google Scholar] [CrossRef]
- Napolitano, A.; Pezzella, A.; Prota, G.; Seraglia, R.; Traldi, P. A reassessment of the structure of 5,6-dihydroxyindole-2-carboxylic acid melanins by matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun. Mass Spectrom. 1998, 10, 204–208. [Google Scholar] [CrossRef]
- Bertazzo, A.; Costa, C.V.L.; Allegri, G.; Schiavolin, M.; Favretto, D.; Traldi, P. Enzymatic oligomerization of tyrosine by tyrosinase and peroxidase studied by matrix assisted laser desorption/ionization mass spectrometry. Rapid Commun. Mass Spectrom. 1999, 13, 542–547. [Google Scholar] [CrossRef]
- Cooksey, C.J.; Garratt, P.J.; Land, E.J.; Pavel, S.; Ramsden, C.A.; Riley, P.A.; Smit, N.P.M. Evidence of the indirect formation of the catecholic intermediate substrate responsible for the autoactivation kinetics of tyrosinase. J. Biol. Chem. 1997, 272, 26226–26235. [Google Scholar] [CrossRef] [PubMed]











© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugumaran, M. Reactivities of Quinone Methides versus o-Quinones in Catecholamine Metabolism and Eumelanin Biosynthesis. Int. J. Mol. Sci. 2016, 17, 1576. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091576
Sugumaran M. Reactivities of Quinone Methides versus o-Quinones in Catecholamine Metabolism and Eumelanin Biosynthesis. International Journal of Molecular Sciences. 2016; 17(9):1576. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091576
Chicago/Turabian StyleSugumaran, Manickam. 2016. "Reactivities of Quinone Methides versus o-Quinones in Catecholamine Metabolism and Eumelanin Biosynthesis" International Journal of Molecular Sciences 17, no. 9: 1576. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091576