
Microglial Function across the Spectrum of Age and Gender

Abstract
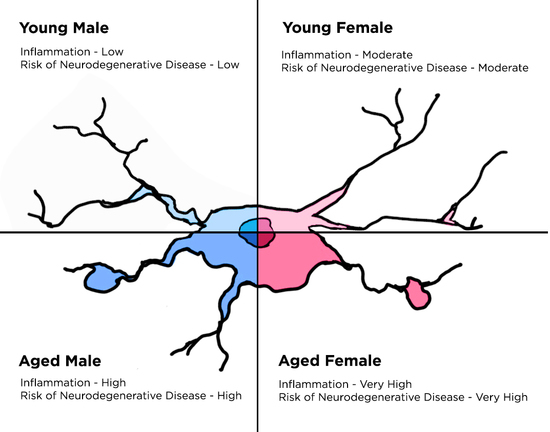
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
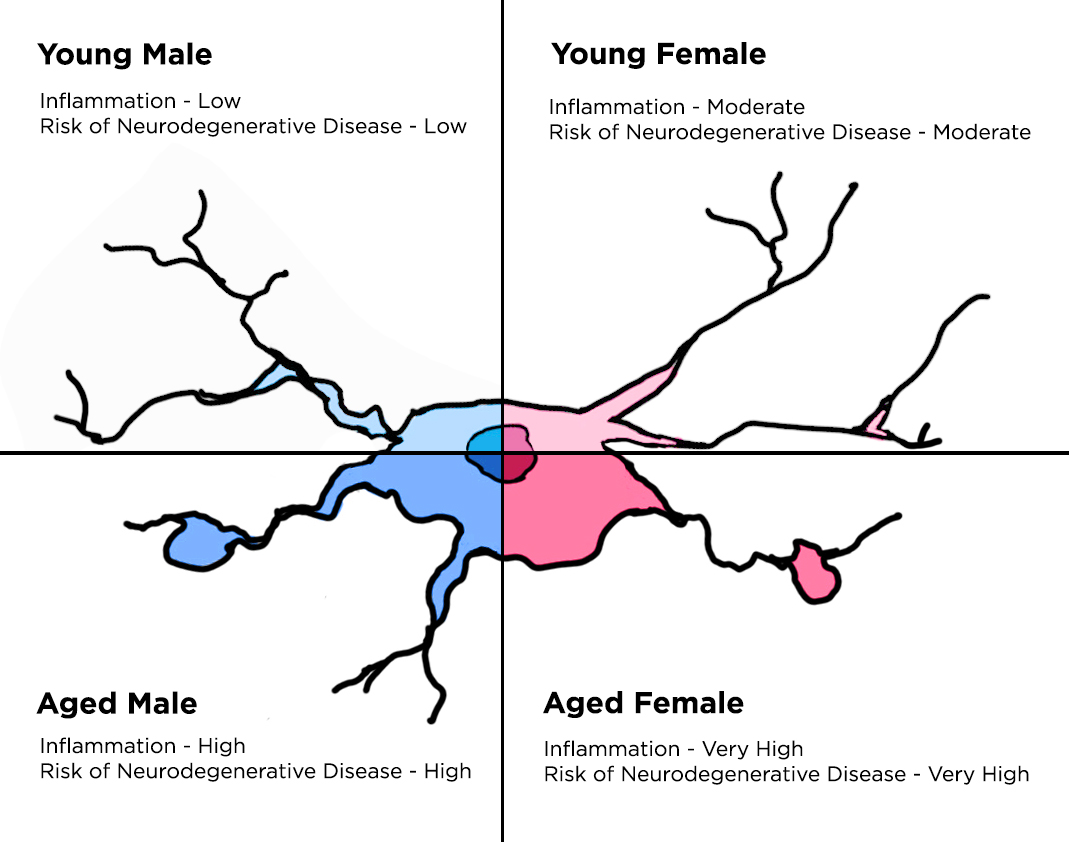
2. Microglial Changes throughout Aging
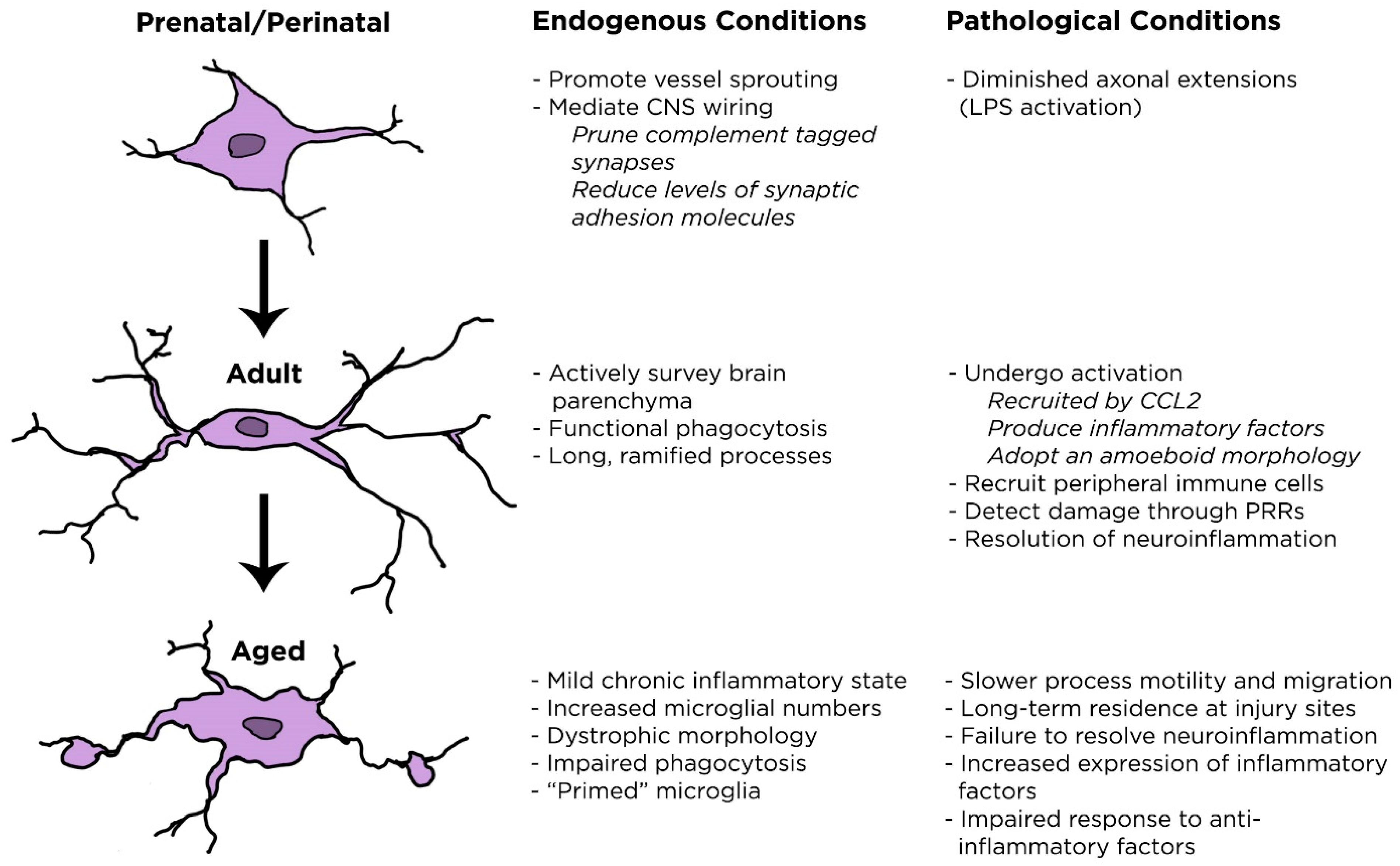
2.1. Microglia in the Developing Brain
2.2. Microglia in the Adult Brain
2.3. Microglia in the Aged Brain
3. Microglial Differences between Genders
3.1. Sexual Differentiation of the Brain
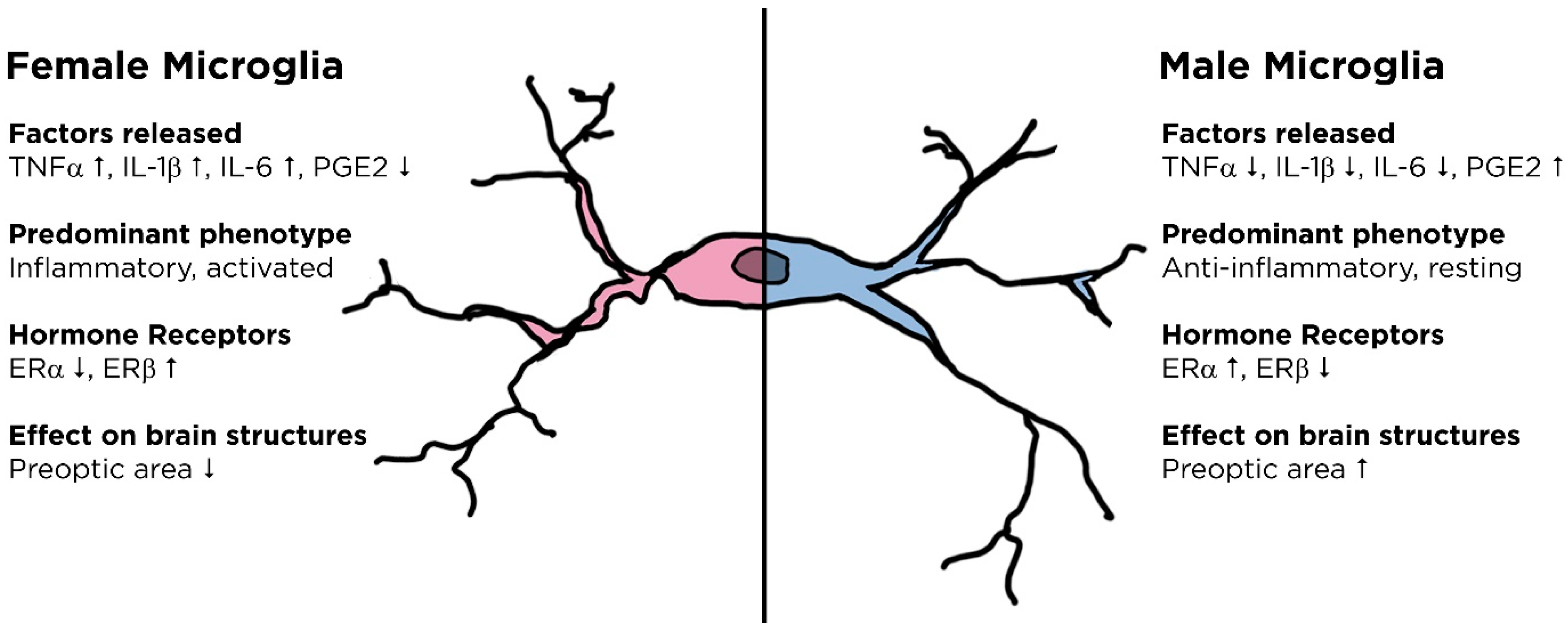
3.2. Gender Differences in Microglial Function
3.3. Hormonal Regulation of Neuroimmune Function
4. Implications in Neurodegenerative Diseases
5. Conclusions
Acknowledgements
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ATP | adenosine triphosphate |
| BBB | Blood-brain barrier |
| CNS | Central nervous system |
| CSF1R | Colony stimulating factor 1 receptor |
| DHT | Dihydrotestosterone |
| ER | Estrogen receptor |
| HSP | Heat shock protein |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| MCP1 | Monocyte chemoattractant protein 1 |
| MHC | Major histocompatibility complex |
| NO | Nitric oxide |
| NOD | Nucleotide-binding oligomerization domain-like |
| PGE2 | Prostaglandin E2 |
| POA | Preoptic area |
| PRR | Pattern recognition receptor |
| ROS | Reactive oxygen species |
| TGFβ | Transforming growth factor beta |
| TNFα | Tumor necrosis factor alpha |
References
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, D.; Hanisch, U.K. Microglia. Metab. Brain Dis. 2004, 19, 393–411. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.K.; Girvin, A.M.; Miller, S.D. Direct activation of innate and antigen-presenting functions of microglia following infection with Theiler’s virus. J. Virol. 2001, 75, 9780–9789. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Akgul, G.; Wollmuth, L.P.; Tsirka, S.E. Microglia actively regulate the number of functional synapses. PLoS ONE 2013, 8, e56293. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Varol, C.; Landsman, L.; Fogg, D.K.; Greenshtein, L.; Gildor, B.; Margalit, R.; Kalchenko, V.; Geissmann, F.; Jung, S. Monocytes give rise to mucosal, but not splenic, conventional dendritic cells. J. Exp. Med. 2007, 204, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Brew, B.J. Microglia, macrophages, perivascular macrophages, and pericytes: A review of function and identification. J. Leukoc. Biol. 2004, 75, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Alliot, F.; Godin, I.; Pessac, B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res. Dev. Brain Res. 1999, 117, 145–152. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Checchin, D.; Sennlaub, F.; Levavasseur, E.; Leduc, M.; Chemtob, S. Potential role of microglia in retinal blood vessel formation. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3595–3602. [Google Scholar] [CrossRef] [PubMed]
- Fantin, A.; Vieira, J.M.; Gestri, G.; Denti, L.; Schwarz, Q.; Prykhozhij, S.; Peri, F.; Wilson, S.W.; Ruhrberg, C. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 2010, 116, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Rymo, S.F.; Gerhardt, H.; Wolfhagen Sand, F.; Lang, R.; Uv, A.; Betsholtz, C. A two-way communication between microglial cells and angiogenic sprouts regulates angiogenesis in aortic ring cultures. PLoS ONE 2011, 6, e15846. [Google Scholar] [CrossRef] [PubMed]
- Vela, J.M.; Dalmau, I.; Gonzalez, B.; Castellano, B. Morphology and distribution of microglial cells in the young and adult mouse cerebellum. J. Comp. Neurol. 1995, 361, 602–616. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Croom, D.; Hida, H.; Kirov, S.A. Capillary blood flow around microglial somata determines dynamics of microglial processes in ischemic conditions. Glia 2011, 59, 1744–1753. [Google Scholar] [CrossRef] [PubMed]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [PubMed]
- Chechik, G.; Meilijson, I.; Ruppin, E. Synaptic pruning in development: A computational account. Neural Comput. 1998, 10, 1759–1777. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.E.; Lowery, R.L.; Majewska, A.K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010, 8, e1000527. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Jin, X.; Parada, I.; Pesic, A.; Stevens, B.; Barres, B.; Prince, D.A. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc. Natl. Acad Sci. USA 2010, 107, 7975–7980. [Google Scholar] [CrossRef] [PubMed]
- Squarzoni, P.; Oller, G.; Hoeffel, G.; Pont-Lezica, L.; Rostaing, P.; Low, D.; Bessis, A.; Ginhoux, F.; Garel, S. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 2014, 8, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Carson, M.J.; Doose, J.M.; Melchior, B.; Schmid, C.D.; Ploix, C.C. CNS immune privilege: Hiding in plain sight. Immunol. Rev. 2006, 213, 48–65. [Google Scholar] [CrossRef] [PubMed]
- Erblich, B.; Zhu, L.; Etgen, A.M.; Dobrenis, K.; Pollard, J.W. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS ONE 2011, 6, e26317. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, K.; Zhu, L.; Pollard, J.W. Conditional deletion of the colony stimulating factor-1 receptor (c-fms proto-oncogene) in mice. Genesis 2006, 44, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, E.B.; McNulty, J.A.; Castro, A.J.; Fox, L.M.; Zimmer, J.; Finsen, B. Enriched immune-environment of blood-brain barrier deficient areas of normal adult rats. J. Neuroimmunol. 1997, 76, 117–131. [Google Scholar] [CrossRef]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Raivich, G.; Banati, R. Brain microglia and blood-derived macrophages: Molecular profiles and functional roles in multiple sclerosis and animal models of autoimmune demyelinating disease. Brain Res. Brain Res. Rev. 2004, 46, 261–281. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Hanisch, U.K. Microglia as a source and target of cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.C.; Hu, S.; Molitor, T.W.; Shaskan, E.G.; Peterson, P.K. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J. Immunol. 1992, 149, 2736–2741. [Google Scholar] [PubMed]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 2009, 29, 13435–13444. [Google Scholar] [CrossRef] [PubMed]
- Lo, D.; Feng, L.; Li, L.; Carson, M.J.; Crowley, M.; Pauza, M.; Nguyen, A.; Reilly, C.R. Integrating innate and adaptive immunity in the whole animal. Immunol. Rev. 1999, 169, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Coyle, P.K. Dissecting the immune component of neurologic disorders: A grand challenge for the 21st century. Front. Neurol. 2011, 2, 37. [Google Scholar] [CrossRef] [PubMed]
- Hanke, M.L.; Kielian, T. Toll-like receptors in health and disease in the brain: Mechanisms and therapeutic potential. Clin. Sci. 2011, 121, 367–387. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.S. “Recruitment signals” from apoptotic cells: Invitation to a quiet meal. Cell 2003, 113, 817–820. [Google Scholar] [CrossRef]
- Sivagnanam, V.; Zhu, X.; Schlichter, L.C. Dominance of E. coli phagocytosis over LPS in the inflammatory response of microglia. J. Neuroimmunol. 2010, 227, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Caso, J.R.; Pradillo, J.M.; Hurtado, O.; Leza, J.C.; Moro, M.A.; Lizasoain, I. Toll-like receptor 4 is involved in subacute stress-induced neuroinflammation and in the worsening of experimental stroke. Stroke 2008, 39, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Lehnardt, S.; Lehmann, S.; Kaul, D.; Tschimmel, K.; Hoffmann, O.; Cho, S.; Krueger, C.; Nitsch, R.; Meisel, A.; Weber, J.R. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J. Neuroimmunol. 2007, 190, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Lehnardt, S.; Schott, E.; Trimbuch, T.; Laubisch, D.; Krueger, C.; Wulczyn, G.; Nitsch, R.; Weber, J.R. A vicious cycle involving release of heat shock protein 60 from injured cells and activation of toll-like receptor 4 mediates neurodegeneration in the CNS. J. Neurosci. 2008, 28, 2320–2331. [Google Scholar] [CrossRef] [PubMed]
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007, 30, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shi, X.Q.; Echeverry, S.; Mogil, J.S.; de Koninck, Y.; Rivest, S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J. Neurosci. 2007, 27, 12396–12406. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Tsirka, S.E. The CCL2-CCR2 system affects the progression and clearance of intracerebral hemorrhage. Glia 2012, 60, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, J.J.; Zhou, C.; Gravanis, I.; Rogove, A.D.; Wu, Y.P.; Bogenhagen, D.F.; Tsirka, S.E. Proteolytic activation of monocyte chemoattractant protein-1 by plasmin underlies excitotoxic neurodegeneration in mice. J. Neurosci. 2007, 27, 1738–1745. [Google Scholar] [CrossRef] [PubMed]
- Castellano, B.; Bosch-Queralt, M.; Almolda, B.; Villacampa, N.; Gonzalez, B. Purine Signaling and Microglial Wrapping. Adv. Exp. Med. Biol. 2016, 949, 147–165. [Google Scholar] [PubMed]
- Verkhratsky, A.; Krishtal, O.A.; Burnstock, G. Purinoceptors on neuroglia. Mol. Neurobiol. 2009, 39, 190–208. [Google Scholar] [CrossRef] [PubMed]
- Crain, J.M.; Nikodemova, M.; Watters, J.J. Expression of P2 nucleotide receptors varies with age and sex in murine brain microglia. J. Neuroinflamm. 2009, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Bernardino, L.; Balosso, S.; Ravizza, T.; Marchi, N.; Ku, G.; Randle, J.C.; Malva, J.O.; Vezzani, A. Inflammatory events in hippocampal slice cultures prime neuronal susceptibility to excitotoxic injury: A crucial role of P2X7 receptor-mediated IL-1beta release. J. Neurochem. 2008, 106, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Hide, I.; Tanaka, M.; Inoue, A.; Nakajima, K.; Kohsaka, S.; Inoue, K.; Nakata, Y. Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J. Neurochem. 2000, 75, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Parvathenani, L.K.; Tertyshnikova, S.; Greco, C.R.; Roberts, S.B.; Robertson, B.; Posmantur, R. P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer’s disease. J. Biol. Chem. 2003, 278, 13309–13317. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Hide, I.; Ido, K.; Kohsaka, S.; Inoue, K.; Nakata, Y. Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J. Neurosci. 2004, 24, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Drachman, D.A. Aging of the brain, entropy, and Alzheimer disease. Neurology 2006, 67, 1340–1352. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic microglia in the aging human brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.M.; Johnson, R.W. An age-related decline in interleukin-10 may contribute to the increased expression of interleukin-6 in brain of aged mice. Neuroimmunomodulation 2001, 9, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.D.; Huang, M.H.; Albert, M.; Harris, T.; Rowe, J.W.; Seeman, T.E. Interleukin-6 and risk of cognitive decline: MacArthur studies of successful aging. Neurology 2002, 59, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Droge, W.; Schipper, H.M. Oxidative stress and aberrant signaling in aging and cognitive decline. Aging Cell 2007, 6, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Mouton, P.R.; Long, J.M.; Lei, D.L.; Howard, V.; Jucker, M.; Calhoun, M.E.; Ingram, D.K. Age and gender effects on microglia and astrocyte numbers in brains of mice. Brain Res. 2002, 956, 30–35. [Google Scholar] [CrossRef]
- Damani, M.R.; Zhao, L.; Fontainhas, A.M.; Amaral, J.; Fariss, R.N.; Wong, W.T. Age-related alterations in the dynamic behavior of microglia. Aging Cell 2011, 10, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.E.; Zettel, M.L.; Ison, J.R.; Allen, P.D.; Majewska, A.K. Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia 2012, 60, 541–558. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Xue, Q.S. The Brain’s Aging Immune System. Aging Dis. 2010, 1, 254–261. [Google Scholar] [PubMed]
- Sierra, A.; Gottfried-Blackmore, A.C.; McEwen, B.S.; Bulloch, K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 2007, 55, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Barrientos, R.M.; Biedenkapp, J.C.; Rudy, J.W.; Watkins, L.R.; Maier, S.F. mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol. Aging 2006, 27, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Letiembre, M.; Hao, W.; Liu, Y.; Walter, S.; Mihaljevic, I.; Rivest, S.; Hartmann, T.; Fassbender, K. Innate immune receptor expression in normal brain aging. Neuroscience 2007, 146, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.M.; Johnson, R.W. Increased interleukin-6 expression by microglia from brain of aged mice. J. Neuroimmunol. 1999, 93, 139–148. [Google Scholar] [CrossRef]
- Sheng, J.G.; Mrak, R.E.; Griffin, W.S. Enlarged and phagocytic, but not primed, interleukin-1 alpha-immunoreactive microglia increase with age in normal human brain. Acta Neuropathol. 1998, 95, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Norden, D.M.; Godbout, J.P. Review: Microglia of the aged brain: Primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 2013, 39, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Godbout, J.P.; Chen, J.; Abraham, J.; Richwine, A.F.; Berg, B.M.; Kelley, K.W.; Johnson, R.W. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 2005, 19, 1329–1331. [Google Scholar] [PubMed]
- Chen, J.; Buchanan, J.B.; Sparkman, N.L.; Godbout, J.P.; Freund, G.G.; Johnson, R.W. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav. Immun. 2008, 22, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, R.M.; Frank, M.G.; Hein, A.M.; Higgins, E.A.; Watkins, L.R.; Rudy, J.W.; Maier, S.F. Time course of hippocampal IL-1 beta and memory consolidation impairments in aging rats following peripheral infection. Brain Behav. Immun. 2009, 23, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, P.N.; Lovell-Badge, R. SRY and sex determination in mammals. Annu. Rev. Genet. 1993, 27, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Reyes, F.I.; Winter, J.S.; Faiman, C. Studies on human sexual development. I. Fetal gonadal and adrenal sex steroids. J. Clin. Endocrinol. Metab. 1973, 37, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Phoenix, C.H.; Goy, R.W.; Gerall, A.A.; Young, W.C. Organizing action of prenatally administered testosterone propionate on the tissues mediating mating behavior in the female guinea pig. Endocrinology 1959, 65, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Toran-Allerand, C.D. Sex steroids and the development of the newborn mouse hypothalamus and preoptic area in vitro: Implications for sexual differentiation. Brain Res. 1976, 106, 407–412. [Google Scholar] [CrossRef]
- Morris, J.A.; Jordan, C.L.; Breedlove, S.M. Sexual differentiation of the vertebrate nervous system. Nat. Neurosci. 2004, 7, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Amateau, S.K.; McCarthy, M.M. A novel mechanism of dendritic spine plasticity involving estradiol induction of prostaglandin-E2. J. Neurosci. 2002, 22, 8586–8596. [Google Scholar] [PubMed]
- Davis, E.C.; Popper, P.; Gorski, R.A. The role of apoptosis in sexual differentiation of the rat sexually dimorphic nucleus of the preoptic area. Brain Res. 1996, 734, 10–18. [Google Scholar] [CrossRef]
- De Vries, G.J.; Rissman, E.F.; Simerly, R.B.; Yang, L.Y.; Scordalakes, E.M.; Auger, C.J.; Swain, A.; Lovell-Badge, R.; Burgoyne, P.S.; Arnold, A.P. A model system for study of sex chromosome effects on sexually dimorphic neural and behavioral traits. J. Neurosci. 2002, 22, 9005–9014. [Google Scholar] [PubMed]
- Gatewood, J.D.; Wills, A.; Shetty, S.; Xu, J.; Arnold, A.P.; Burgoyne, P.S.; Rissman, E.F. Sex chromosome complement and gonadal sex influence aggressive and parental behaviors in mice. J. Neurosci. 2006, 26, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, I.; Lleo, A.; Gershwin, M.E.; Invernizzi, P. The X chromosome and immune associated genes. J. Autoimmun. 2012, 38, J187–J192. [Google Scholar] [CrossRef] [PubMed]
- Voskuhl, R. Sex differences in autoimmune diseases. Biol. Sex. Differ. 2011, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Smith-Bouvier, D.L.; Divekar, A.A.; Sasidhar, M.; Du, S.; Tiwari-Woodruff, S.K.; King, J.K.; Arnold, A.P.; Singh, R.R.; Voskuhl, R.R. A role for sex chromosome complement in the female bias in autoimmune disease. J. Exp. Med. 2008, 205, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Sholar, P.W.; Bilbo, S.D. Sex differences in microglial colonization of the developing rat brain. J. Neurochem. 2012, 120, 948–963. [Google Scholar] [CrossRef] [PubMed]
- Amateau, S.K.; McCarthy, M.M. Induction of PGE2 by estradiol mediates developmental masculinization of sex behavior. Nat. Neurosci. 2004, 7, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Lenz, K.M.; McCarthy, M.M. Organized for sex-steroid hormones and the developing hypothalamus. Eur. J. Neurosci. 2010, 32, 2096–2104. [Google Scholar] [CrossRef] [PubMed]
- Lenz, K.M.; Nugent, B.M.; Haliyur, R.; McCarthy, M.M. Microglia are essential to masculinization of brain and behavior. J. Neurosci. 2013, 33, 2761–2772. [Google Scholar] [CrossRef] [PubMed]
- Crain, J.M.; Nikodemova, M.; Watters, J.J. Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J. Neurosci. Res. 2013, 91, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Ohsawa, K.; Inoue, K.; Kohsaka, S. Purinergic receptors in microglia: Functional modal shifts of microglia mediated by P2 and P1 receptors. Glia 2013, 61, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Acaz-Fonseca, E.; Duran, J.C.; Carrero, P.; Garcia-Segura, L.M.; Arevalo, M.A. Sex differences in glia reactivity after cortical brain injury. Glia 2015, 63, 1966–1981. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.E.; Brautigam, V.M.; Watters, J.J. Estrogen modulates microglial inflammatory mediator production via interactions with estrogen receptor beta. Endocrinology 2004, 145, 5021–5032. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Gottfried-Blackmore, A.; Milner, T.A.; McEwen, B.S.; Bulloch, K. Steroid hormone receptor expression and function in microglia. Glia 2008, 56, 659–674. [Google Scholar] [CrossRef] [PubMed]
- Vegeto, E.; Bonincontro, C.; Pollio, G.; Sala, A.; Viappiani, S.; Nardi, F.; Brusadelli, A.; Viviani, B.; Ciana, P.; Maggi, A. Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J. Neurosci. 2001, 21, 1809–1818. [Google Scholar] [PubMed]
- Crain, J.M.; Watters, J.J. Estrogen and P2 Purinergic Receptor Systems in Microglia: Therapeutic Targets for Neuroprotection. Open Drug Discov. J. 2010, 2, 148–167. [Google Scholar] [PubMed]
- Crain, J.M.; Watters, J.J. Microglial P2 Purinergic Receptor and Immunomodulatory Gene Transcripts Vary By Region, Sex, and Age in the Healthy Mouse CNS. Transcr. Open Access 2015, 3, 124. [Google Scholar] [CrossRef] [PubMed]
- Loram, L.C.; Sholar, P.W.; Taylor, F.R.; Wiesler, J.L.; Babb, J.A.; Strand, K.A.; Berkelhammer, D.; Day, H.E.; Maier, S.F.; Watkins, L.R. Sex and estradiol influence glial pro-inflammatory responses to lipopolysaccharide in rats. Psychoneuroendocrinology 2012, 37, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Uylings, H.B.; de Brabander, J.M. Neuronal changes in normal human aging and Alzheimer’s disease. Brain Cogn. 2002, 49, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Katzman, R. Alzheimer’s disease as an age-dependent disorder. Ciba Found. Symp. 1988, 134, 69–85. [Google Scholar] [PubMed]
- Payami, H.; Zareparsi, S.; Montee, K.R.; Sexton, G.J.; Kaye, J.A.; Bird, T.D.; Yu, C.E.; Wijsman, E.M.; Heston, L.L.; Litt, M.; et al. Gender difference in apolipoprotein E-associated risk for familial Alzheimer disease: A possible clue to the higher incidence of Alzheimer disease in women. Am. J. Hum. Genet. 1996, 58, 803–811. [Google Scholar] [PubMed]
- Griffin, W.S.; Mrak, R.E. Interleukin-1 in the genesis and progression of and risk for development of neuronal degeneration in Alzheimer’s disease. J. Leukoc. Biol. 2002, 72, 233–238. [Google Scholar] [PubMed]
- Sala, G.; Galimberti, G.; Canevari, C.; Raggi, M.E.; Isella, V.; Facheris, M.; Appollonio, I.; Ferrarese, C. Peripheral cytokine release in Alzheimer patients: Correlation with disease severity. Neurobiol. Aging 2003, 24, 909–914. [Google Scholar] [CrossRef]
- Etminan, M.; Gill, S.; Samii, A. Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer’s disease: Systematic review and meta-analysis of observational studies. BMJ 2003, 327, 128. [Google Scholar] [CrossRef] [PubMed]
- Koenigsknecht-Talboo, J.; Landreth, G.E. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J. Neurosci. 2005, 25, 8240–8249. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Xu, F.; Previti, M.L.; Davis, J.; Grande, A.M.; Robinson, J.K.; Van Nostrand, W.E. Minocycline reduces microglial activation and improves behavioral deficits in a transgenic model of cerebral microvascular amyloid. J. Neurosci. 2007, 27, 3057–3063. [Google Scholar] [CrossRef] [PubMed]
- Floden, A.M.; Combs, C.K. Microglia demonstrate age-dependent interaction with amyloid-beta fibrils. J. Alzheimers Dis. 2011, 25, 279–293. [Google Scholar] [PubMed]
- Abiega, O.; Beccari, S.; Diaz-Aparicio, I.; Nadjar, A.; Laye, S.; Leyrolle, Q.; Gomez-Nicola, D.; Domercq, M.; Perez-Samartin, A.; Sanchez-Zafra, V.; et al. Neuronal Hyperactivity Disturbs ATP Microgradients, Impairs Microglial Motility, and Reduces Phagocytic Receptor Expression Triggering Apoptosis/Microglial Phagocytosis Uncoupling. PLoS Biol. 2016, 14, e1002466. [Google Scholar] [CrossRef] [PubMed]
- Ngo, S.T.; Steyn, F.J.; McCombe, P.A. Gender differences in autoimmune disease. Front. Neuroendocrinol. 2014, 35, 347–369. [Google Scholar] [CrossRef] [PubMed]
- Dotson, A.L.; Offner, H. Sex differences in the immune response to experimental stroke: Implications for translational research. J. Neurosci. Res. 2017, 95, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.A.; Choudhury, K.R.; Rathakrishnan, B.G.; Marks, D.M.; Petrella, J.R.; Doraiswamy, P.M.; Alzheimer’s Disease Neuroimaging Initiative. Marked gender differences in progression of mild cognitive impairment over 8 years. Alzheimers Dement. 2015, 1, 103–110. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nissen, J.C. Microglial Function across the Spectrum of Age and Gender. Int. J. Mol. Sci. 2017, 18, 561. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18030561
Nissen JC. Microglial Function across the Spectrum of Age and Gender. International Journal of Molecular Sciences. 2017; 18(3):561. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18030561
Chicago/Turabian StyleNissen, Jillian C. 2017. "Microglial Function across the Spectrum of Age and Gender" International Journal of Molecular Sciences 18, no. 3: 561. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18030561