Autophagic Mechanism in Anti-Cancer Immunity: Its Pros and Cons for Cancer Therapy

Abstract
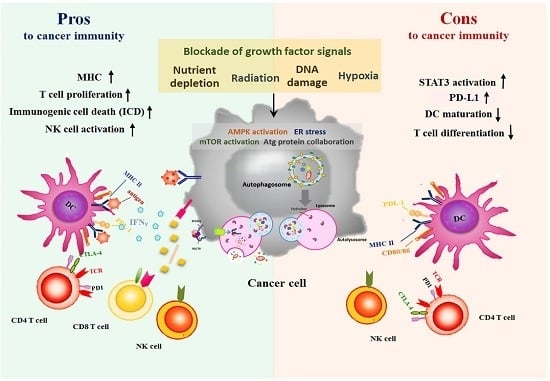
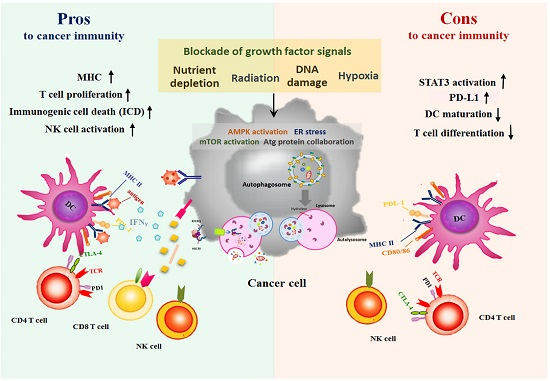
:
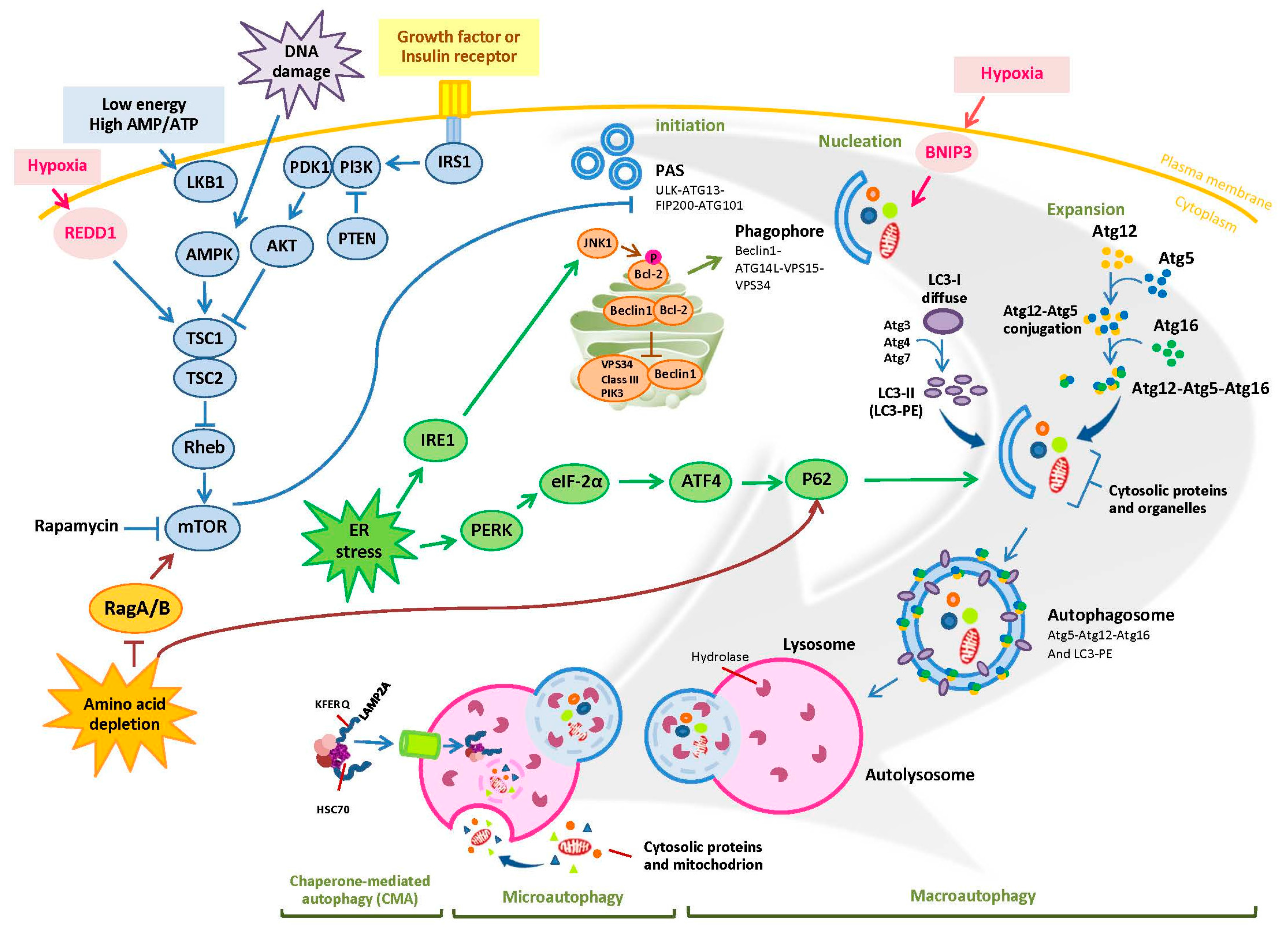
1. Overview of Autophagy
2. The Role of Autophagy in Cancer Cells: Survival or Death Signals?
2.1. Autophagy and Cancer Cell Survival
2.2. Autophagy and Cancer Cell Death
3. The Interplay between Autophagy and Tumor Microenvironment
3.1. Hypoxia Induces Autophagy in Tumor Microenvironment
3.2. The Effect of Inflammation on Autophagy in Tumor Milieu
3.3. The Impact of Autophagy on Tumor Promoting Inflammation
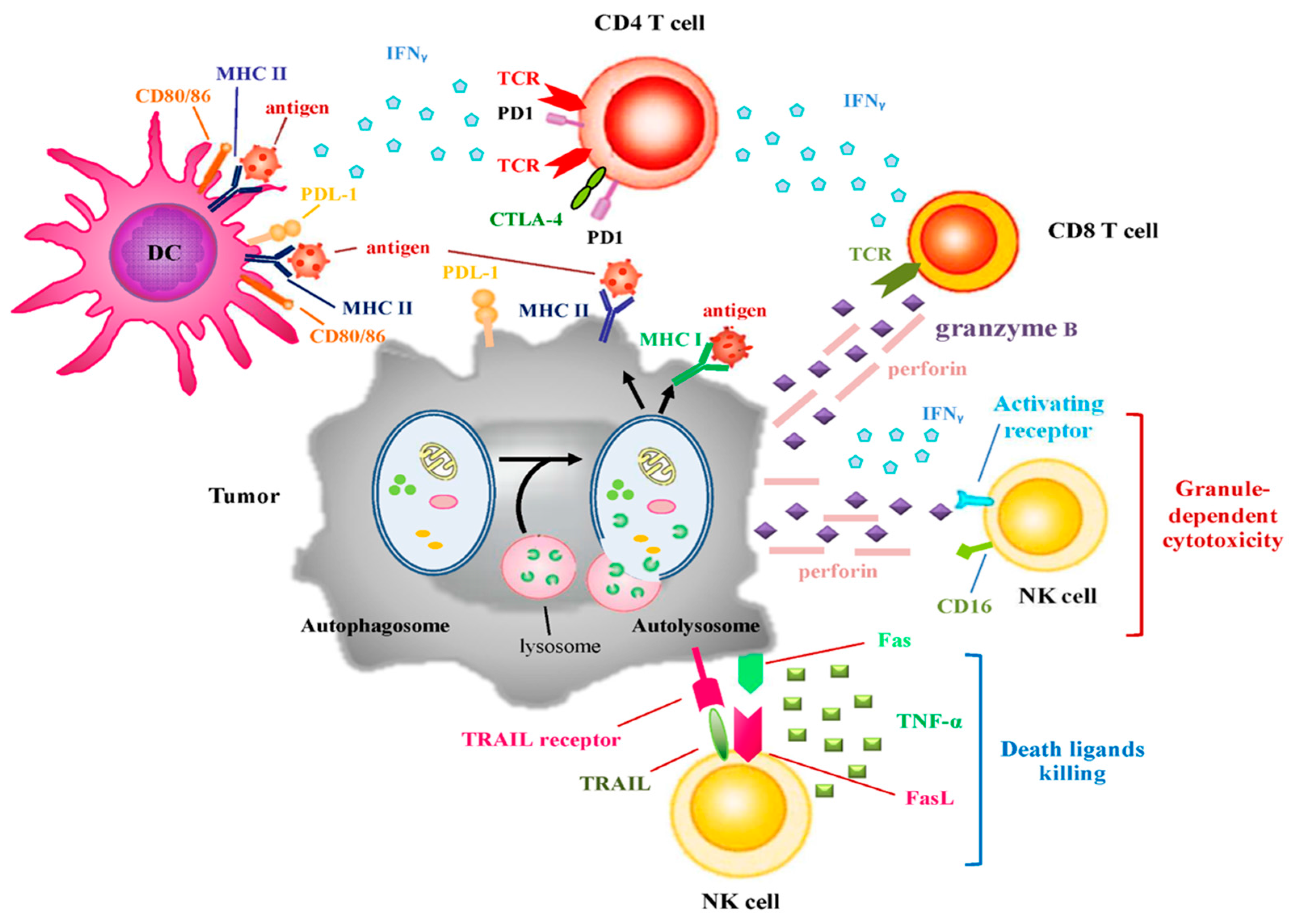
4. The Influence of Autophagy on Immune Surveillance
4.1. Autophagy and Immunogenicity
4.2. The Roles of Autophagy in Immune Cell Cytotoxicity
4.3. Autophagy and Immune Checkpoints
5. Autophagy Paradox: Activating Autophagy by Therapeutic Agents is Pros or Cons for Anti-Cancer Immunity?
5.1. Anticancer Treatments-Induced Autophagy and Immune System
5.2. Are Autophagy Antagonists Suitable to Combine with Immunotherapies?
6. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ADCC | Antibody-dependent cell-mediated cytotoxicity |
| ADI | Arginine deiminase |
| ALL | Acute lymphoblastic leukemia |
| AMPK | 5′ AMP-activated protein kinase |
| ASS1 | Argininosuccinate synthase 1 |
| ATF4 | Activating transcription factor 4 |
| Atg | Autophagy-related protein |
| ATM | Ataxia telangiectasia mutated |
| ATP | Adenosine triphosphate |
| BNIP3 | BCL2 interacting protein 3 |
| CASTOR1 | Arginine sensor for mTORC1 |
| CCL | CC chemokine ligand |
| CTLA4 | Cytotoxic T lymphocyte associated protein 4 |
| CMA | Chaperone-mediated autophagy |
| CML | Chronic myeloid leukemia |
| CQ | Chloroquine |
| CSC | Cancer stem cells |
| Cx43 | Connexin 43 |
| DAMPs | Damage-associated molecular patterns |
| DC | Dendritic cell |
| eIF-2α | α subunit of eukaryotic translation initiation factor 2 |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial to mesenchymal transition |
| ER | Endoplasmic reticulum |
| GAL-9 | Galectin-9 |
| GAP | GTPase activating protein |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| HCC | Hepatoceulluar carcinoma |
| HCQ | Hydroxy-chloroquine |
| HDAC | Histone Deacetylase |
| HER2 | Human epidermal growth factor receptor 2 |
| HIF-1α | Hypoxia-induced factor-1 alpha |
| HMGB1 | High mobility group box 1 |
| Hsc70 | Cytosolic heat shock protein of 70 Kd |
| IAP | Inhibitor of apoptosis protein |
| IBD | Inflammatory bowel disease |
| ICD | Immunogenic cell death |
| IL | Interleukin |
| IRE1 | Inositol-requiring enzyme 1 |
| IRS-1 | Insulin receptor substrate-1 |
| JNK | Jun N-terminal kinase |
| LAMP-2A | Lysosome-associated membrane protein-2A |
| LAP | LC3-associated phagocytosis |
| LKB1 | Liver kinase B1 |
| MAPK | Mitogen-activated protein kinase |
| MB | Medulloblastoma |
| MDSC | Myeloid-derived suppressor cell |
| MEFs | Mouse embryonic fibroblasts |
| MHC | Major histocompatibility complex |
| MICA/B | MHC I chain-related molecules A and B |
| MIP-1α | Macrophage inflammatory protein 1-alpha |
| mTOR | Mammalian target-of-rapamycin |
| mTORC | Mammalian target-of-rapamycin complex 1 |
| NK | Nature killer |
| NLRP3 | NOD-like receptor family, pyrin domain containing 3 |
| NSCLC | Non-small cell lung cancer |
| OVA | Ovalbumin (OVA) |
| PANX1 | Pannexin1 |
| PARK7 | Parkinsonism associated deglycase |
| PD-1 | Program death-1 |
| PD-L1/2 | Program death ligand 1/2 |
| PDK1 | Phosphoinositide-dependent kinase-1 |
| PERK | Protein kinase RNA-like endoplasmic reticulum kinase |
| PTEN | Phosphatase and tensin homologue |
| Rag | Ras-related GTPase |
| REDD1 | Rregulated in development and DNA damage responses 1 |
| Rheb | Ras homolog enriched in brain |
| RNAi | RNA interference |
| ROS | Reactive oxygen species |
| SACC | Salivary adenoid cystic carcinoma |
| SAHA | Suberoylanilide hydroxamic |
| SQSTM1 | Sequestosome 1 |
| STAT3 | Signal transducer and activator of transcription 3 |
| TAM | Tumor associated macrophage |
| TAP | Transporter associated with antigen processing |
| TCR | T cell receptor |
| TGF-β | Transforming growth factor-beta |
| TILs | Tumor infiltrating lymphocytes |
| TIM-4 | T cell immunoglobulin- and mucin-domain-containing molecule-4 |
| TSC1/2 | Tuberous sclerosis complex 1/2 |
| TRAIL | TNF-related apoptosis-inducing factor |
| Treg | Regulatory T cells |
| TRIF | Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta |
| ULBP | UL16-binding proteins |
| ULK1 | Unc-51-like kinase 1 |
| UPR | Unfolded protein response |
| ZEB1 | Zinc finger E-box-binding homeobox 1 |
| 3-MA | 3-methyladenine |
| 5-FU | 5-fluorouracil |
References
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. 2013, 8, 105–137. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, E.; Cuervo, A.M. Chaperone-mediated autophagy. Proc. Am. Thorac. Soc. 2010, 7, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Sofer, A.; Lei, K.; Johannessen, C.M.; Ellisen, L.W. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol. Cell. Biol. 2005, 25, 5834–5845. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Khaled, A.R. Homeostasis and the importance for a balance between AKT/mTOR activity and intracellular signaling. Curr. Med. Chem. 2012, 19, 3748–3762. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef] [PubMed]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Nakajima, S.; Saito, Y.; Takahashi, S.; Katoh, R.; Kitamura, M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ. 2012, 19, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of BCL-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Manning, B.D. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 2013, 15, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Chantranupong, L.; Scaria, S.M.; Saxton, R.A.; Gygi, M.P.; Shen, K.; Wyant, G.A.; Wang, T.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. The castor proteins are arginine sensors for the mTORC1 pathway. Cell 2016, 165, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Huang, Z.; Li, H.; Yu, J.; Wang, C.; Chen, S.; Meng, Q.; Cheng, Y.; Gao, X.; Li, J.; et al. Leucine deprivation increases hepatic insulin sensitivity via GCN2/mTOR/S6K1 and AMPK pathways. Diabetes 2011, 60, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Savaraj, N.; You, M.; Wu, C.; Wangpaichitr, M.; Kuo, M.T.; Feun, L.G. Arginine deprivation, autophagy, apoptosis (AAA) for the treatment of melanoma. Curr. Mol. Med. 2010, 10, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Cuervo, A.M. Autophagy and human disease: Emerging themes. Curr. Opin. Genet. Dev. 2014, 26, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wullschleger, S.; Shpiro, N.; McGuire, V.A.; Sakamoto, K.; Woods, Y.L.; McBurnie, W.; Fleming, S.; Alessi, D.R. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in pten-deficient mice. Biochem. J. 2008, 412, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Jeong, J.H.; Asara, J.M.; Yuan, Y.Y.; Granter, S.R.; Chin, L.; Cantley, L.C. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol. Cell 2009, 33, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Cai, H.; Teplova, I.; Bowman-Colin, C.; Chen, G.; Price, S.; Barnard, N.; Ganesan, S.; Karantza, V.; White, E.; et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov. 2013, 3, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, E.C.; Knebel, S.M.; Lo, W.H.; Leung, Y.C.; Cheng, P.N.; Hsueh, C.T. Deprivation of arginine by recombinant human arginase in prostate cancer cells. J. Hematol. Oncol. 2012, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Xu, Z.; Dai, S.; Qian, L.; Sun, L.; Gong, Z. Targeting autophagy to sensitive glioma to temozolomide treatment. J. Exp. Clin. Cancer Res. CR 2016, 35, 23. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, T.R.; O’Sullivan, G.C.; McKenna, S.L. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011, 7, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.M.; Li, Z.G. Blockage of cisplatin-induced autophagy sensitizes cervical cancer cells to cisplatin. Genet. Mol. Res. GMR 2015, 14, 16905–16912. [Google Scholar] [CrossRef] [PubMed]
- Del Bello, B.; Toscano, M.; Moretti, D.; Maellaro, E. Cisplatin-induced apoptosis inhibits autophagy, which acts as a pro-survival mechanism in human melanoma cells. PLoS ONE 2013, 8, e57236. [Google Scholar] [CrossRef]
- Wang, J.; Wu, G.S. Role of autophagy in cisplatin resistance in ovarian cancer cells. J. Biol. Chem. 2014, 289, 17163–17173. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Jiang, Z.F.; Ding, P.S.; Shao, L.J.; Liu, R.Y. Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci. Rep. 2015, 5, 12291. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Shao, L.J.; Jiang, Z.F.; Liu, R.Y. Gemcitabine-induced autophagy protects human lung cancer cells from apoptotic death. Lung 2016, 194, 959–966. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Vitale, I.; Younes, A.B.; Maiuri, M.C.; Kroemer, G. Evaluation of rapamycin-induced cell death. Methods Mol. Biol. 2012, 821, 125–169. [Google Scholar] [PubMed]
- Zarogoulidis, P.; Lampaki, S.; Turner, J.F.; Huang, H.; Kakolyris, S.; Syrigos, K.; Zarogoulidis, K. mTOR pathway: A current, up-to-date mini-review (review). Oncol. Lett. 2014, 8, 2367–2370. [Google Scholar] [CrossRef] [PubMed]
- Palm, W.; Park, Y.; Wright, K.; Pavlova, N.N.; Tuveson, D.A.; Thompson, C.B. The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell 2015, 162, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, S.; Comincini, S. Autophagy and ionizing radiation in tumors: The “survive or not survive” dilemma. J. Cell. Physiol. 2013, 228, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Clement, J.M.; McDermott, D.F. The high-dose aldesleukin (IL-2) “select” trial: A trial designed to prospectively validate predictive models of response to high-dose IL-2 treatment in patients with metastatic renal cell carcinoma. Clin. Genitourin. Cancer 2009, 7, E7–E9. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; De Vera, M.E.; Buchser, W.J.; Romo de Vivar Chavez, A.; Loughran, P.; Beer Stolz, D.; Basse, P.; Wang, T.; Van Houten, B.; Zeh, H.J., 3rd; et al. Inhibiting systemic autophagy during interleukin 2 immunotherapy promotes long-term tumor regression. Cancer Res. 2012, 72, 2791–2801. [Google Scholar] [CrossRef] [PubMed]
- Lomonaco, S.L.; Finniss, S.; Xiang, C.; Decarvalho, A.; Umansky, F.; Kalkanis, S.N.; Mikkelsen, T.; Brodie, C. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int. J. Cancer 2009, 125, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Xie, C. Radiation-induced autophagy promotes esophageal squamous cell carcinoma cell survival via the LKB1 pathway. Oncol. Rep. 2016, 35, 3559–3565. [Google Scholar] [CrossRef] [PubMed]
- Jo, G.H.; Bogler, O.; Chwae, Y.J.; Yoo, H.; Lee, S.H.; Park, J.B.; Kim, Y.J.; Kim, J.H.; Gwak, H.S. Radiation-induced autophagy contributes to cell death and induces apoptosis partly in malignant glioma cells. Cancer Res. Treat. 2015, 47, 221–241. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, B.; Yu, F.; Chen, Q.; Tian, Y.; Ma, S.; Liu, X. The roles of mitochondria in radiation-induced autophagic cell death in cervical cancer cells. Tumour Biol. 2016, 37, 4083–4091. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, N.; Faried, A.; Tsutsumi, S.; Kuwano, H. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Eur. J. Cancer 2010, 46, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Nawrocki, S.T.; Kahue, C.N.; Zhang, H.; Yang, C.; Chung, L.; Houghton, J.A.; Huang, P.; Giles, F.J.; Cleveland, J.L. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor saha to overcome Bcr-Abl-mediated drug resistance. Blood 2007, 110, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Pardo, R.; Lo Re, A.; Archange, C.; Ropolo, A.; Papademetrio, D.L.; Gonzalez, C.D.; Alvarez, E.M.; Iovanna, J.L.; Vaccaro, M.I. Gemcitabine induces the vmp1-mediated autophagy pathway to promote apoptotic death in human pancreatic cancer cells. Pancreatology 2010, 10, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Carracedo, A.; Salanueva, I.J.; Hernandez-Tiedra, S.; Lorente, M.; Egia, A.; Vazquez, P.; Blazquez, C.; Torres, S.; Garcia, S.; et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of er stress in human glioma cells. J. Clin. Investig. 2009, 119, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.L.; Hill, D.S.; McKee, C.S.; Hernandez-Tiedra, S.; Lorente, M.; Lopez-Valero, I.; Eleni Anagnostou, M.; Babatunde, F.; Corazzari, M.; Redfern, C.P.; et al. Exploiting cannabinoid-induced cytotoxic autophagy to drive melanoma cell death. J. Investig. Dermatol. 2015, 135, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Salazar, M.; Olea-Herrero, N.; Guzman, M.; Velasco, G.; Diaz-Laviada, I. Anti-tumoral action of cannabinoids on hepatocellular carcinoma: Role of AMPK-dependent activation of autophagy. Cell Death Differ. 2011, 18, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.F.; Lin, Y.C.; Lin, Y.H.; Tsai, T.F.; Chou, K.Y.; Chen, H.E.; Hwang, T.I. Zoledronic acid induces autophagic cell death in human prostate cancer cells. J. Urol. 2011, 185, 1490–1496. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Y.; Yang, L.Q.; Jiang, Y.; Yang, W.W.; Fu, J.; Li, S.L. Reactive oxygen species and autophagy associated apoptosis and limitation of clonogenic survival induced by zoledronic acid in salivary adenoid cystic carcinoma cell line SACC-83. PLoS ONE 2014, 9, e101207. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Shi, G.; Jin, J.; Guo, H.; Guo, X.; Luo, F.; Song, Y.; Jia, X. Dual PI3K/mTOR inhibitor NVP-BEZ235-induced apoptosis of hepatocellular carcinoma cell lines is enhanced by inhibitors of autophagy. Int. J. Mol. Med. 2013, 31, 1449–1456. [Google Scholar] [PubMed]
- Echeverry, N.; Ziltener, G.; Barbone, D.; Weder, W.; Stahel, R.A.; Broaddus, V.C.; Felley-Bosco, E. Inhibition of autophagy sensitizes malignant pleural mesothelioma cells to dual PI3K/mTOR inhibitors. Cell Death Dis. 2015, 6, e1757. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.X.; Zhao, L.; Yue, P.; Fang, G.; Tao, H.; Owonikoko, T.K.; Ramalingam, S.S.; Khuri, F.R.; Sun, S.Y. Augmentation of NVP-BEZ235’s anticancer activity against human lung cancer cells by blockage of autophagy. Cancer Biol. Ther. 2011, 12, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.W.; Shin, J.S.; Moon, J.H.; Kim, Y.S.; Lee, J.; Choi, E.K.; Ha, S.H.; Lee, D.H.; Chung, H.N.; Kim, J.E.; et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, induces cell death through alternate routes in prostate cancer cells depending on the pten genotype. Apoptosis Int. J. Program. Cell Death 2014, 19, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.F.; Lin, Y.C.; Yang, S.C.; Tsai, T.F.; Chen, H.E.; Chou, K.Y.; Hwang, T.I. Autophagy inhibition enhances RAD001-induced cytotoxicity in human bladder cancer cells. Drug Des. Dev. Ther. 2016, 10, 1501–1513. [Google Scholar] [CrossRef] [PubMed]
- Crazzolara, R.; Bradstock, K.F.; Bendall, L.J. RAD001 (everolimus) induces autophagy in acute lymphoblastic leukemia. Autophagy 2009, 5, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Subhawong, T.; Albert, J.M.; Kim, K.W.; Geng, L.; Sekhar, K.R.; Gi, Y.J.; Lu, B. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res. 2006, 66, 10040–10047. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Yang, Z.J.; Yu, C.; Sinicrope, F.A. Inhibition of mTOR kinase by AZD8055 can antagonize chemotherapy-induced cell death through autophagy induction and down-regulation of p62/sequestosome 1. J. Biol. Chem. 2011, 286, 40002–40012. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Huang, H.; Zhao, R.; Li, P.; Li, M.; Miao, H.; Chen, N.; Chen, M. AZD8055 induces cell death associated with autophagy and activation of AMPK in hepatocellular carcinoma. Oncol. Rep. 2014, 31, 649–656. [Google Scholar] [PubMed]
- Zou, Y.; Ling, Y.H.; Sironi, J.; Schwartz, E.L.; Perez-Soler, R.; Piperdi, B. The autophagy inhibitor chloroquine overcomes the innate resistance of wild-type EGFR non-small-cell lung cancer cells to erlotinib. J. Thorac. Oncol. 2013, 8, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.C.; Wu, M.Y.; Hwang, M.H.; Chang, Y.T.; Huang, H.J.; Lin, A.M.; Yang, J.C. Chloroquine enhances gefitinib cytotoxicity in gefitinib-resistant nonsmall cell lung cancer cells. PLoS ONE 2015, 10, e0119135. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fan, Z. The epidermal growth factor receptor antibody cetuximab induces autophagy in cancer cells by downregulating HIF-1α and Bcl-2 and activating the beclin 1/hVps34 complex. Cancer Res. 2010, 70, 5942–5952. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Cao, Y.; Jiang, H.; Mao, A. Autophagy facilitates the sorafenib resistance of hepatocellular carcinoma cells. West Indian Med. J. 2013, 62, 698–700. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.; Mitchell, C.; Park, M.A.; Yacoub, A.; Graf, M.; Rahmani, M.; Houghton, P.J.; Voelkel-Johnson, C.; Grant, S.; Dent, P. Sorafenib and vorinostat kill colon cancer cells by CD95-dependent and -independent mechanisms. Mol. Pharmacol. 2009, 76, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Mishima, Y.; Terui, Y.; Mishima, Y.; Taniyama, A.; Kuniyoshi, R.; Takizawa, T.; Kimura, S.; Ozawa, K.; Hatake, K. Autophagy and autophagic cell death are next targets for elimination of the resistance to tyrosine kinase inhibitors. Cancer Sci. 2008, 99, 2200–2208. [Google Scholar] [CrossRef] [PubMed]
- Shingu, T.; Fujiwara, K.; Bogler, O.; Akiyama, Y.; Moritake, K.; Shinojima, N.; Tamada, Y.; Yokoyama, T.; Kondo, S. Inhibition of autophagy at a late stage enhances imatinib-induced cytotoxicity in human malignant glioma cells. Int. J. Cancer 2009, 124, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy Levy, J.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.K.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. eLife 2017, 6, e19671. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting er stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS ONE 2009, 4, e6251. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.M.; Li, Y.Y.; Tu, C.H.; Salazar, N.; Tseng, Y.Y.; Huang, S.F.; Hsieh, L.L.; Lui, T.N. Blockade of inhibitors of apoptosis proteins in combination with conventional chemotherapy leads to synergistic antitumor activity in medulloblastoma and cancer stem-like cells. PLoS ONE 2016, 11, e0161299. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.Y.; Kim, S.W.; Park, K.C.; Yun, M. The bifunctional autophagic flux by 2-deoxyglucose to control survival or growth of prostate cancer cells. BMC Cancer 2015, 15, 623. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Kurtoglu, M.; Liu, H.; Wangpaichitr, M.; You, M.; Liu, X.; Savaraj, N.; Lampidis, T.J. 2-Deoxy-d-glucose activates autophagy via endoplasmic reticulum stress rather than ATP depletion. Cancer Chemother. Pharmacol. 2011, 67, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Bean, G.R.; Kremer, J.C.; Prudner, B.C.; Schenone, A.D.; Yao, J.C.; Schultze, M.B.; Chen, D.Y.; Tanas, M.R.; Adkins, D.R.; Bomalaski, J.; et al. A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death Dis. 2016, 7, e2406. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Coates, J.M.; Bowles, T.L.; McNerney, G.P.; Sutcliffe, J.; Jung, J.U.; Gandour-Edwards, R.; Chuang, F.Y.; Bold, R.J.; Kung, H.J. Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis. Cancer Res. 2009, 69, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.M.; Cohen, G.M.; Bampton, E.T. The in vitro cleavage of the hatg proteins by cell death proteases. Autophagy 2010, 6, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Savaraj, N.; Kuo, M.T.; Wangpaichitr, M.; Varona-Santos, J.; Wu, C.; Nguyen, D.M.; Feun, L. Trail induces autophagic protein cleavage through caspase activation in melanoma cell lines under arginine deprivation. Mol. Cell. Biochem. 2013, 374, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yu, D.D.; Yan, F.; Jing, Y.Y.; Han, Z.P.; Sun, K.; Liang, L.; Hou, J.; Wei, L.X. The role of autophagy induced by tumor microenvironment in different cells and stages of cancer. Cell Biosci. 2015, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Mayer, A. Hypoxia and anemia: Effects on tumor biology and treatment resistance. Transfusion Clinique et Biologique 2005, 12, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Mayer, A.; Hockel, M. Tumor hypoxia and malignant progression. Methods Enzymol. 2004, 381, 335–354. [Google Scholar] [PubMed]
- Chen, J.L.; Lin, H.H.; Kim, K.J.; Lin, A.; Ou, J.H.; Ann, D.K. PKCδ signaling: A dual role in regulating hypoxic stress-induced autophagy and apoptosis. Autophagy 2009, 5, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Mazure, N.M.; Pouyssegur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and Atg5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Vasseur, S.; Afzal, S.; Tardivel-Lacombe, J.; Park, D.S.; Iovanna, J.L.; Mak, T.W. DJ-1/PARK7 is an important mediator of hypoxia-induced cellular responses. Proc. Natl. Acad. Sci. USA 2009, 106, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Yao, J.; Zhuang, L.; Wang, D.; Han, J.; Lam, E.W.; Network, T.R.; Gan, B. The Foxo-BNIP3 axis exerts a unique regulation of mTORC1 and cell survival under energy stress. Oncogene 2014, 33, 3183–3194. [Google Scholar] [CrossRef] [PubMed]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Cam, H.; Easton, J.B.; High, A.; Houghton, P.J. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1α. Mol. Cell 2010, 40, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Trimmer, C.; Lin, Z.; Whitaker-Menezes, D.; Chiavarina, B.; Zhou, J.; Wang, C.; Pavlides, S.; Martinez-Cantarin, M.P.; Capozza, F.; et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NF-κB activation in the tumor stromal microenvironment. Cell Cycle 2010, 9, 3515–3533. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Yoon, J.H.; Ryu, J.H. Mitophagy: A balance regulator of NLRP3 inflammasome activation. BMB Rep. 2016, 49, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Kanayama, M.; He, Y.W.; Shinohara, M.L. The lung is protected from spontaneous inflammation by autophagy in myeloid cells. J. Immunol. 2015, 194, 5465–5471. [Google Scholar] [CrossRef] [PubMed]
- Netea-Maier, R.T.; Plantinga, T.S.; van de Veerdonk, F.L.; Smit, J.W.; Netea, M.G. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy 2016, 12, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Lock, R.; Kenific, C.M.; Leidal, A.M.; Salas, E.; Debnath, J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 2014, 4, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.K.; Wen, J.; Chen, S.; Guan, J.L. Autophagy differentially regulates distinct breast cancer stem-like cells in murine models via EGFR/Stat3 and Tgfβ/Smad signaling. Cancer Res. 2016, 76, 3397–3410. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Cretney, E.; Kelly, J.M.; Westwood, J.A.; Street, S.E.; Yagita, H.; Takeda, K.; van Dommelen, S.L.; Degli-Esposti, M.A.; Hayakawa, Y. Activation of nk cell cytotoxicity. Mol. Immunol. 2005, 42, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Waldhauer, I.; Steinle, A. Nk cells and cancer immunosurveillance. Oncogene 2008, 27, 5932–5943. [Google Scholar] [CrossRef] [PubMed]
- Warren, H.S.; Smyth, M.J. Nk cells and apoptosis. Immunol. Cell Biol. 1999, 77, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Munz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Pypaert, M.; Munz, C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Crotzer, V.L.; Blum, J.S. Autophagy and its role in MHC-mediated antigen presentation. J. Immunol. 2009, 182, 3335–3341. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W.; Reits, E.; Neefjes, J. Making sense of mass destruction: Quantitating MHC class I antigen presentation. Nat. Rev. Immunol. 2003, 3, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Vyas, J.M.; Van der Veen, A.G.; Ploegh, H.L. The known unknowns of antigen processing and presentation. Nat. Rev. Immunol. 2008, 8, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Munz, C. Antigen processing via autophagy—Not only for mhc class ii presentation anymore? Curr. Opin. Immunol. 2010, 22, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, L.X.; Yang, G.; Hao, F.; Urba, W.J.; Hu, H.M. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res. 2008, 68, 6889–6895. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lei, Z.; Lichty, B.D.; Li, D.; Zhang, G.M.; Feng, Z.H.; Wan, Y.; Huang, B. Autophagy facilitates major histocompatibility complex class I expression induced by IFN-γ in B16 melanoma cells. Cancer Immunol. Immunother. CII 2010, 59, 313–321. [Google Scholar] [CrossRef] [PubMed]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippe, R.; et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Martins, I.; Ma, Y.; Kepp, O.; Galluzzi, L.; Kroemer, G. Autophagy-dependent ATP release from dying cells via lysosomal exocytosis. Autophagy 2013, 9, 1624–1625. [Google Scholar] [CrossRef] [PubMed]
- Parekh, V.V.; Wu, L.; Boyd, K.L.; Williams, J.A.; Gaddy, J.A.; Olivares-Villagomez, D.; Cover, T.L.; Zong, W.X.; Zhang, J.; Van Kaer, L. Impaired autophagy, defective T cell homeostasis, and a wasting syndrome in mice with a T cell-specific deletion of vps34. J. Immunol. 2013, 190, 5086–5101. [Google Scholar] [CrossRef] [PubMed]
- Pua, H.H.; Guo, J.; Komatsu, M.; He, Y.W. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J. Immunol. 2009, 182, 4046–4055. [Google Scholar] [CrossRef] [PubMed]
- Willinger, T.; Flavell, R.A. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T cell homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 8670–8675. [Google Scholar] [CrossRef] [PubMed]
- Bronietzki, A.W.; Schuster, M.; Schmitz, I. Autophagy in T cell development, activation and differentiation. Immunol. Cell Biol. 2015, 93, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Pua, H.H.; Dzhagalov, I.; Chuck, M.; Mizushima, N.; He, Y.W. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp. Med. 2007, 204, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Buart, S.; Van Pelt, J.; Richon, C.; Hasmim, M.; Leleu, N.; Suchorska, W.M.; Jalil, A.; Lecluse, Y.; El Hage, F.; et al. The cooperative induction of hypoxia-inducible factor-1α and STAT3 during hypoxia induced an impairment of tumor susceptibility to CTL-mediated cell lysis. J. Immunol. 2009, 182, 3510–3521. [Google Scholar] [CrossRef] [PubMed]
- Viry, E.; Baginska, J.; Berchem, G.; Noman, M.Z.; Medves, S.; Chouaib, S.; Janji, B. Autophagic degradation of gzmb/granzyme b: A new mechanism of hypoxic tumor cell escape from natural killer cell-mediated lysis. Autophagy 2014, 10, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Tittarelli, A.; Janji, B.; Van Moer, K.; Noman, M.Z.; Chouaib, S. The selective degradation of synaptic connexin 43 protein by hypoxia-induced autophagy impairs natural killer cell-mediated tumor cell killing. J. Biol. Chem. 2015, 290, 23670–23679. [Google Scholar] [CrossRef] [PubMed]
- Alexander, W. The checkpoint immunotherapy revolution: What started as a trickle has become a flood, despite some daunting adverse effects; new drugs, indications, and combinations continue to emerge. P T Peer-Rev. J. Formul. Manag. 2016, 41, 185–191. [Google Scholar]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Lastwika, K.J.; Wilson, W., 3rd; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.A.; Gupta, H.B.; Sareddy, G.; Pandeswara, S.; Lao, S.; Yuan, B.; Drerup, J.M.; Padron, A.; Conejo-Garcia, J.; Murthy, K.; et al. Tumor-intrinsic PD-L1 signals regulate cell growth, pathogenesis, and autophagy in ovarian cancer and melanoma. Cancer Res. 2016, 76, 6964–6974. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [PubMed]
- Chacon, J.A.; Schutsky, K.; Powell, D.J. The impact of chemotherapy, radiation and epigenetic modifiers in cancer cell expression of immune inhibitory and stimulatory molecules and anti-tumor efficacy. Vaccines 2016, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Bezu, L.; Gomes-de-Silva, L.C.; Dewitte, H.; Breckpot, K.; Fucikova, J.; Spisek, R.; Galluzzi, L.; Kepp, O.; Kroemer, G. Combinatorial strategies for the induction of immunogenic cell death. Front. Immunol. 2015, 6, 187. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.D.; Pollizzi, K.N.; Heikamp, E.B.; Horton, M.R. Regulation of immune responses by mTOR. Annu. Rev. Immunol. 2012, 30, 39–68. [Google Scholar] [CrossRef] [PubMed]
- Granville, C.A.; Memmott, R.M.; Balogh, A.; Mariotti, J.; Kawabata, S.; Han, W.; Lopiccolo, J.; Foley, J.; Liewehr, D.J.; Steinberg, S.M.; et al. A central role for Foxp3+ regulatory T cells in K-Ras-driven lung tumorigenesis. PLoS ONE 2009, 4, e5061. [Google Scholar] [CrossRef] [PubMed]
- Mentlik James, A.; Cohen, A.D.; Campbell, K.S. Combination immune therapies to enhance anti-tumor responses by NK cells. Front. Immunol. 2013, 4, 481. [Google Scholar] [CrossRef] [PubMed]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600e) melanoma. Sci. Transl. Med. 2015, 7, 279ra241. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.Y.; Hopson, C.; et al. The BRAF and MEK inhibitors dabrafenib and trametinib: Effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 2015, 21, 1639–1651. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ratovitski, E.A. Phospho-ΔNp63α/Rpn13-dependent regulation of LKB1 degradation modulates autophagy in cancer cells. Aging 2010, 2, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Notte, A.; Rebucci, M.; Fransolet, M.; Roegiers, E.; Genin, M.; Tellier, C.; Watillon, K.; Fattaccioli, A.; Arnould, T.; Michiels, C. Taxol-induced unfolded protein response activation in breast cancer cells exposed to hypoxia: ATF4 activation regulates autophagy and inhibits apoptosis. Int. J. Biochem. Cell Biol. 2015, 62, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sevko, A.; Michels, T.; Vrohlings, M.; Umansky, L.; Beckhove, P.; Kato, M.; Shurin, G.V.; Shurin, M.R.; Umansky, V. Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells and chronic inflammation in the spontaneous melanoma model. J. Immunol. 2013, 190, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.C.; Wang, H.C.; Hou, Y.C.; Tung, H.L.; Chiu, T.J.; Shan, Y.S. Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol. Cancer 2015, 14, 179. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Shi, A.; Song, D.; Han, B.; Zhang, Z.; Ma, L.; Liu, D.; Fan, Z. Resistin confers resistance to doxorubicin-induced apoptosis in human breast cancer cells through autophagy induction. Am. J. Cancer Res. 2017, 7, 574–583. [Google Scholar] [PubMed]
- Zheng, J.; Liu, Q.; Yang, J.; Ren, Q.; Cao, W.; Yang, J.; Yu, Z.; Yu, F.; Wu, Y.; Shi, H.; et al. Co-culture of apoptotic breast cancer cells with immature dendritic cells: A novel approach for DC-based vaccination in breast cancer. Braz. J. Med. Biol. Res. 2012, 45, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Kinsella, T.J. BNIP3 is essential for mediating 6-thioguanine- and 5-fluorouracil-induced autophagy following DNA mismatch repair processing. Cell Res. 2010, 20, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Deng, Z.; Wang, H.; Ma, W.; Zhou, C.; Zhang, S. Repeated cycles of 5-fluorouracil chemotherapy impaired anti-tumor functions of cytotoxic T cells in a CT26 tumor-bearing mouse model. BMC Immunol. 2016, 17, 29. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [PubMed]
- Nichols, P.H.; Ward, U.; Ramsden, C.W.; Primrose, J.N. The effect of 5-fluorouracil and alpha interferon and 5-fluorouracil and leucovorin on cellular anti-tumour immune responses in patients with advanced colorectal cancer. Br. J. Cancer 1994, 70, 946–949. [Google Scholar] [CrossRef] [PubMed]
- Jazirehi, A.R.; Kurdistani, S.K.; Economou, J.S. Histone deacetylase inhibitor sensitizes apoptosis-resistant melanomas to cytotoxic human T lymphocytes through regulation of TRAIL/DR5 pathway. J. Immunol. 2014, 192, 3981–3989. [Google Scholar] [CrossRef] [PubMed]
- Banerji, V.; Gibson, S.B. Targeting metabolism and autophagy in the context of haematologic malignancies. Int. J. Cell Biol. 2012, 2012, 595976. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.X.; Wan, Y.Y.; Gong, A.H.; Ge, L.; Jin, J.; Xu, M.; Wu, C.Y. Egr-1 regulates irradiation-induced autophagy through Atg4B to promote radioresistance in hepatocellular carcinoma cells. Oncogenesis 2017, 6, e292. [Google Scholar] [CrossRef] [PubMed]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Park, B.; Yee, C.; Lee, K.M. The effect of radiation on the immune response to cancers. Int. J. Mol. Sci. 2014, 15, 927–943. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, M.L.; Li, Z.; Gao, D.M.; Cai, Y.; Chang, J.; Wang, S.P. Interferon-α-2b induces autophagy in hepatocellular carcinoma cells through Beclin1 pathway. Cancer Biol. Med. 2014, 11, 64–68. [Google Scholar] [PubMed]
- Yang, Y.Q.; Dong, W.J.; Yin, X.F.; Xu, Y.N.; Yang, Y.; Wang, J.J.; Yuan, S.J.; Xiao, J.; DeLong, J.H.; Chu, L.; et al. Interferon-related secretome from direct interaction between immune cells and tumor cells is required for upregulation of PD-L1 in tumor cells. Protein Cell 2016, 7, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor pten function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Liu, Y.; Li, Q.; Li, X.; Ning, Z.; Zhao, W.; Shi, H.; Jiang, J.; Wu, C. PD-1/PD-L1 expression in non-small-cell lung cancer and its correlation with EGFR/kras mutations. Cancer Biol. Ther. 2016, 17, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Gugnoni, M.; Sancisi, V.; Manzotti, G.; Gandolfi, G.; Ciarrocchi, A. Autophagy and epithelial-mesenchymal transition: An intricate interplay in cancer. Cell Death Dis. 2016, 7, e2520. [Google Scholar] [CrossRef] [PubMed]
- Han, M.L.; Zhao, Y.F.; Tan, C.H.; Xiong, Y.J.; Wang, W.J.; Wu, F.; Fei, Y.; Wang, L.; Liang, Z.Q. Cathepsin l upregulation-induced emt phenotype is associated with the acquisition of cisplatin or paclitaxel resistance in A549 cells. Acta Pharmacol. Sin. 2016, 37, 1606–1622. [Google Scholar] [CrossRef] [PubMed]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; Andre, F.; De Cremoux, P.; Bertheau, P.; Badoual, C.; Vielh, P.; Larsen, A.K.; et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T cell-mediated lysis. Cancer Res. 2013, 73, 2418–2427. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, S.; Saeki, H.; Nakashima, Y.; Ito, S.; Oki, E.; Morita, M.; Oda, Y.; Okano, S.; Maehara, Y. PD-L1 expression at tumor invasive front is associated with EMT and poor prognosis in esophageal squamous cell carcinoma. Cancer Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Li, M.; Zhang, Y.; Teng, F.; Han, A.; Kong, L.; Zhu, H. PD-1/PD-L1 blockades in non-small-cell lung cancer therapy. OncoTargets Ther. 2016, 9, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Langer, C.J.; Gadgeel, S.M.; Borghaei, H.; Papadimitrakopoulou, V.A.; Patnaik, A.; Powell, S.F.; Gentzler, R.D.; Martins, R.G.; Stevenson, J.P.; Jalal, S.I.; et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: A randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016, 17, 1497–1508. [Google Scholar] [CrossRef]
- Azad, A.; Yin Lim, S.; D’Costa, Z.; Jones, K.; Diana, A.; Sansom, O.J.; Kruger, P.; Liu, S.; McKenna, W.G.; Dushek, O.; et al. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol. Med. 2017, 9, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Starobinets, H.; Ye, J.; Broz, M.; Barry, K.; Goldsmith, J.; Marsh, T.; Rostker, F.; Krummel, M.; Debnath, J. Antitumor adaptive immunity remains intact following inhibition of autophagy and antimalarial treatment. J. Clin. Investig. 2016, 126, 4417–4429. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Treatment | Action | Autophagy Role | Cancer Type | Reference |
|---|---|---|---|---|
| Radiation 5-FU | DNA damage, ER stress, mTOR inhibition Thymidylate synthase inhibitor | Survival Death Survival | Glioma (CSC), Esophageal Glioma, Cervical Colon, Esophageal | [41,42] [43,44] [27,45] |
| Temozolomide | DNA damage | Survival | Glioma | [26] |
| Cisplatin | DNA damage | Survival | Esophageal, Cervical, Melanoma, Ovarian, Lung | [27,28,29,30,31] |
| SAHA | HDAC inhibitor | Survival | CML | [46] |
| Gemcitabine | DNA synthesis inhibitor | Survival | Lung | [32] |
| Death | Pancreatic | [47] | ||
| Cannabinoids | ER stress | Death | Glioma, HCC, Melanoma | [48,49,50] |
| Bisphosphonates | Farnesyl pyrophosphate synthase inhibitor, mevalonate | Death | Prostate, SACC | [51,52] |
| NVP-BEZ235 | PI3K/AKT/mTOR inhibitor | Survival | HCC, Mesothelioma, Lung | [53,54,55] |
| Death | Prostate | [56] | ||
| RAD001 (Rapamycin derivative) | mTOR inhibitor | Survival | Bladder | [57] |
| Death | Prostate, ALL | [58,59] | ||
| AZD8055 | mTORC1/mTORC2 complex inhibitor | Survival | Colon | [60] |
| Death | HCC | [61] | ||
| Erlotinib, Gefitinib | EGFR mutation inhibitor | Survival | Lung | [62,63] |
| Cetuximab | EGFR inhibitor | Survival | Lung, Epidermoid carcinoma | [64] |
| Sorafenib | Tyrosine kinase inhibitor | Survival | Colon, HCC | [65,66] |
| Imatinib | Tyrosine kinase inhibitor | Survival | Glioma, CML | [67,68] |
| Vemurafenib/Dabrafenib | BRAF (V600E) inhibitor, ER stress | Survival | Melanoma, Glioma | [69,70] |
| Trastuzumab | HER2 inhibitor | Survival | Breast | [71] |
| LCL161/LBW242 + Vincristine or Cisplatin | IAPs inhibitor + Tubulin inhibitor or DNA damage | Death | MB | [72] |
| 2-DG | Glycolysis inhibitor, ER stress | Survival | Prostate, Pancreatic, Melanoma, Breast | [73,74] |
| ADI-PEG20/Arginine deiminase, Arginase | Arginine depletion | Survival | Melanoma, Prostate, Sarcoma, | [18,75,76] |
| IL-2 | Immunotherapy | Survival | Colon, Pancreatic | [40] |
| Treatment | Induction of Autophagy | Pros to Immunity | Cons to Immunity | Reference |
|---|---|---|---|---|
| Rapamycin | mTOR inhibition | Reduced Treg influx and PD-L1 | Impaired DC maturation and T cell differentiation | [121] |
| Bortezomib | ER stress | Increased ICD, DC maturation, NK activation | Decreased MHC I | [128] |
| Cyclophosphamide | ER stress | Increased ICD, MHC, TRAIL CD8 T cells, NK, and DC activation Treg depletion | [124] | |
| Trametinib | ER stress | Increased MHC and T cell proliferation , decreased PD-L1 and immunosuppressive factors | Increased PD-L1 when becoming resistant | [70,129,130] |
| Vemurafenib | ER stress | Increased MHC and T cell proliferation, decreased PD-L1 and immunosuppressive factors | Increased PD-L1 when becoming resistant | [70,129,130] |
| Cisplatin | ATM | Increased MHC, HMGB1 | Increased PD-L1 | [124,131] |
| Taxol | UPR | Increased MHC, decreased MDSC activation | Increased PD-L1 | [124,132,133] |
| Gemcitabine | NF-κB | Increased MHC, NK activation via MICA | Increased PD-L1 | [124,134] |
| Doxorubicin | AMPK–ULK1 | Increased ICD and antigen presentation of DC | [135,136] | |
| 5-FU | BNIP3 | Increased NK and T cell cytotoxicity while combined with interferons | Increased PD-L1 | [137,138] |
| SAHA | mTOR inhibition | Increased ICD and TRAIL, DC and T cell activation | [139,140,141] | |
| Arsenic Trioxide | mTOR inhibition | Increased NK activation via MICA/B and ULBP | Increased PD-L1 | [124,142] |
| Radiation | Arg4B | Increased MHC I, IFN-β mediated cross-presentation of DC, decreased Treg activity | [143,144,145] | |
| Interferons | Beclin1 | Increased MHC, NK and T cell activation | Increased PD-L1 | [146,147] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.-Y.; Feun, L.G.; Thongkum, A.; Tu, C.-H.; Chen, S.-M.; Wangpaichitr, M.; Wu, C.; Kuo, M.T.; Savaraj, N. Autophagic Mechanism in Anti-Cancer Immunity: Its Pros and Cons for Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18061297
Li Y-Y, Feun LG, Thongkum A, Tu C-H, Chen S-M, Wangpaichitr M, Wu C, Kuo MT, Savaraj N. Autophagic Mechanism in Anti-Cancer Immunity: Its Pros and Cons for Cancer Therapy. International Journal of Molecular Sciences. 2017; 18(6):1297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18061297
Chicago/Turabian StyleLi, Ying-Ying, Lynn G. Feun, Angkana Thongkum, Chiao-Hui Tu, Shu-Mei Chen, Medhi Wangpaichitr, Chunjing Wu, Macus T. Kuo, and Niramol Savaraj. 2017. "Autophagic Mechanism in Anti-Cancer Immunity: Its Pros and Cons for Cancer Therapy" International Journal of Molecular Sciences 18, no. 6: 1297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18061297