Epigenetic Regulation of the Biosynthesis & Enzymatic Modification of Heparan Sulfate Proteoglycans: Implications for Tumorigenesis and Cancer Biomarkers

Abstract
:1. Introduction
1.1. Structure and Types of Heparan Sulfate Proteoglycans (HSPGs)
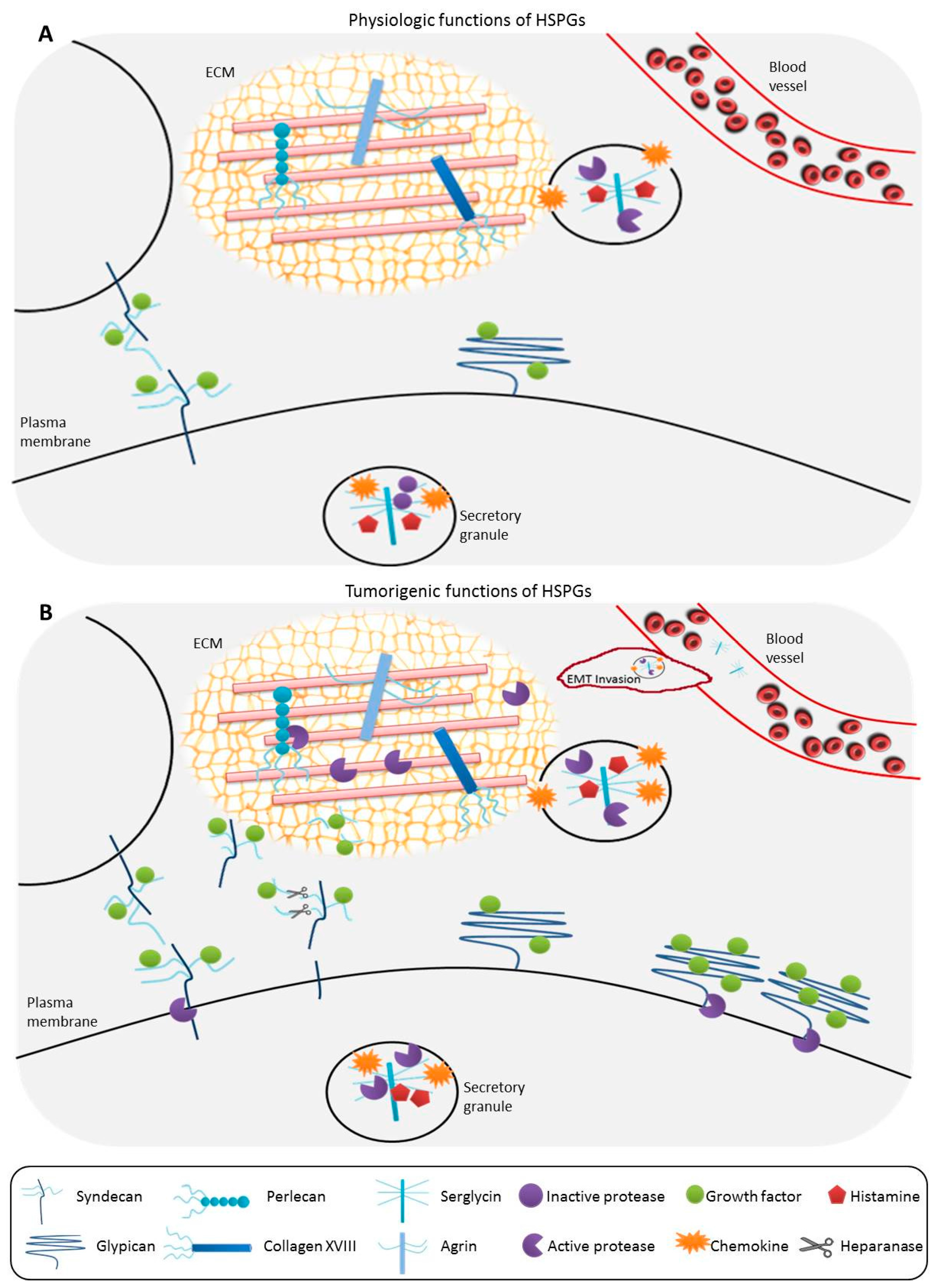
1.2. General Functions of HSPGs
1.3. Alterations of HSPGs Serving as Biomarkers in Cancer
2. Heparan Sulfate Biosynthetic Pathway
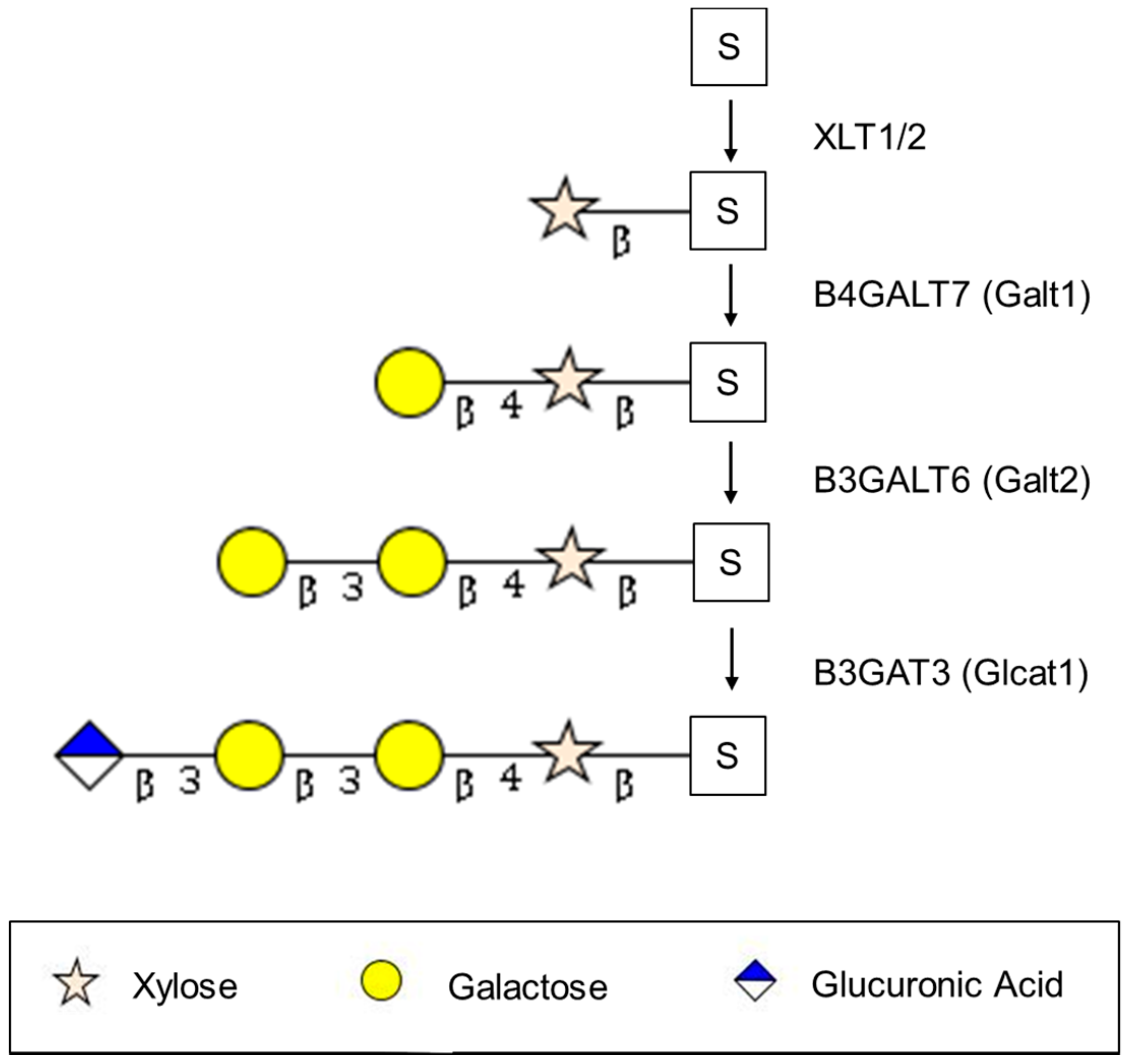
2.1. Synthesis of the Serine-Linked Tetrasaccharide Linker
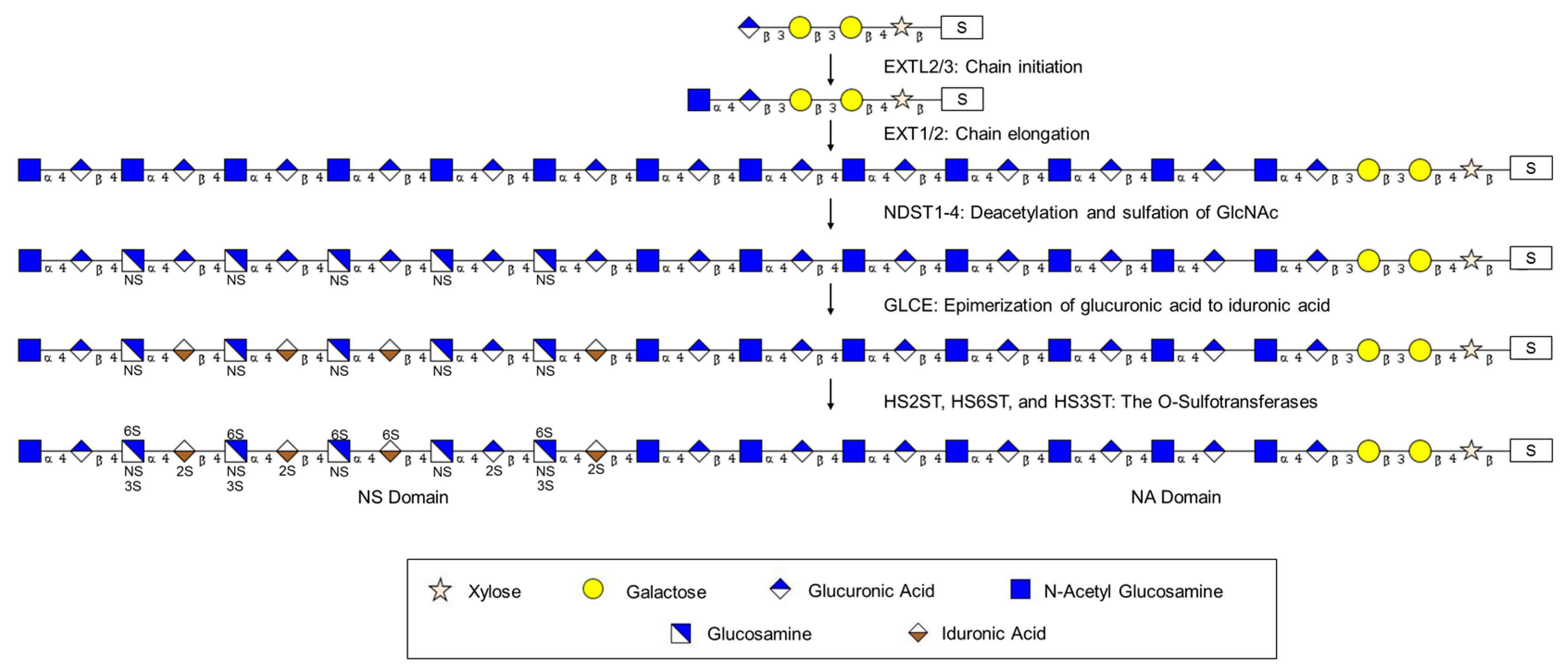
2.2. Elongation of the Tetrasaccharide Linker to Form the HS Chain: Exostosin Family
2.3. Modification of the HS Chain: Formation of Domains
2.3.1. Modification of the HS Chain: Glucosaminyl N-Deacetylase/N-Sulfotransferases (NDSTs)
2.3.2. Modification of the HS Chain: d-Glucuronyl C5-Epimerase (GLCE)
2.3.3. Modification of the HS Chain: The O-Sulfotransferases HS2ST, HS6ST, and HS3ST
2.4. Summary of Modification of the HS Chain: Complexity, Redundancy, and Protein Interactions
3. Heparan Sulfate Modification and Degradation Enzymes
3.1. SULF1 and SULF2
3.2. Heparanase
4. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
Abbreviations
| BxGALT | β 1-x galactosyltransferase |
| BxGAT | β 1-x glucuronyltransferase |
| EXT | Exostosin |
| EXTL | Exostosin-like |
| GLCE | d-glucuronyl C5-epimerase |
| GPC | Glypican |
| HS | Heparan sulfate |
| HSPE | Heparanase |
| HSPG | Heparan sulfate proteoglycan |
| HSxST | Heparan sulfate x-O-sulfotransferase |
| NA domain | N-acetylated disaccharide units |
| NS domain | N-sulfated disaccharide units |
| NDST | N-deacetylase/N-sulfotransferases |
| SULF | Sulfatase |
| XYLT | Xylosyltransferase |
References
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V. Heparan sulfate proteoglycans: Intricate molecules with intriguing functions. J. Clin. Investig. 2001, 108, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Couchman, J.R.; Pataki, C.A. An introduction to proteoglycans and their localization. J. Histochem. Cytochem. 2012, 60, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Blackhall, F.H.; Merry, C.L.; Davies, E.J.; Jayson, G.C. Heparan sulfate proteoglycans and cancer. Br. J. Cancer 2001, 85, 1094–1098. [Google Scholar] [CrossRef] [PubMed]
- Bernfield, M.; Gotte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef] [PubMed]
- Filmus, J.; Selleck, S.B. Glypicans: Proteoglycans with a surprise. J. Clin. Investig. 2001, 108, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Filmus, J.; Capurro, M.; Rast, J. Glypicans. Genome Biol. 2008, 9, 224. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.R.; Whitehouse, I.J.; Hooper, N.M. Glypican-1 mediates both prion protein lipid raft association and disease isoform formation. PLoS Pathog. 2009, 5, e1000666. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Waters, M.; Andrews, A.; Honarmandi, P.; Ebong, E.E.; Rizzo, V.; Tarbell, J.M. Fluid shear stress induces the clustering of heparan sulfate via mobility of glypican-1 in lipid rafts. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Tarbell, J.M. The adaptive remodeling of endothelial glycocalyx in response to fluid shear stress. PLoS ONE 2014, 9, e86249. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Casillas, F.; Payne, H.M.; Andres, J.L.; Massague, J. Betaglycan can act as a dual modulator of TGF-β access to signaling receptors: Mapping of ligand binding and gag attachment sites. J. Cell Biol. 1994, 124, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Bilandzic, M.; Stenvers, K.L. Betaglycan: A multifunctional accessory. Mol. Cell Endocrinol. 2011, 339, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Takashima, S.; Asano, Y.; Kato, H.; Liao, Y.; Yamazaki, S.; Tsukamoto, O.; Seguchi, O.; Yamamoto, H.; Fukushima, T.; et al. Glycosaminoglycan modification of neuropilin-1 modulates VEGFR2 signaling. EMBO J. 2006, 25, 3045–3055. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.Q.; Klagsbrun, M. Neuropilin is a mediator of angiogenesis. Cancer Met. Rev. 2000, 19, 29–37. [Google Scholar] [CrossRef]
- Soker, S.; Takashima, S.; Miao, H.Q.; Neufeld, G.; Klagsbrun, M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998, 92, 735–745. [Google Scholar] [CrossRef]
- Kolodkin, A.L.; Levengood, D.V.; Rowe, E.G.; Tai, Y.T.; Giger, R.J.; Ginty, D.D. Neuropilin is a semaphorin III receptor. Cell 1997, 90, 753–762. [Google Scholar] [CrossRef]
- Kalish, E.D.; Iida, N.; Moffat, F.L.; Bourguignon, L.Y. A new CD44v3-containing isoform is involved in tumor cell growth and migration during human breast carcinoma progression. Front. Biosci. 1999, 4, 1–8. [Google Scholar] [CrossRef]
- Wang, S.J.; Bourguignon, L.Y. Role of hyaluronan-mediated CD44 signaling in head and neck squamous cell carcinoma progression and chemoresistance. Am. J. Pathol. 2011, 178, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Wong, G.; de Heer, A.M.; Xia, W.; Bourguignon, L.Y. CD44 variant isoforms in head and neck squamous cell carcinoma progression. Laryngoscope 2009, 119, 1518–1530. [Google Scholar] [CrossRef] [PubMed]
- Jayadev, R.; Sherwood, D.R. Basement membranes. Curr. Biol. 2017, 27, R207–R211. [Google Scholar] [CrossRef] [PubMed]
- Dreyfuss, J.L.; Regatieri, C.V.; Jarrouge, T.R.; Cavalheiro, R.P.; Sampaio, L.O.; Nader, H.B. Heparan sulfate proteoglycans: Structure, protein interactions and cell signaling. An. Acad. Bras. Cienc. 2009, 81, 409–429. [Google Scholar] [CrossRef] [PubMed]
- Korpetinou, A.; Skandalis, S.S.; Labropoulou, V.T.; Smirlaki, G.; Noulas, A.; Karamanos, N.K.; Theocharis, A.D. Serglycin: At the crossroad of inflammation and malignancy. Front. Oncol. 2014, 3, 327. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.R.; Schuksz, M.; Esko, J.D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007, 446, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Olczyk, P.; Mencner, L.; Komosinska-Vassev, K. Diverse roles of heparan sulfate and heparin in wound repair. BioMed. Res. Int. 2015, 2015, 549417. [Google Scholar] [CrossRef] [PubMed]
- Celie, J.W.; Beelen, R.H.; van den Born, J. Heparan sulfate proteoglycans in extravasation: Assisting leukocyte guidance. Front. Biosci. 2009, 14, 4932–4949. [Google Scholar] [CrossRef]
- Kumar, A.V.; Katakam, S.K.; Urbanowitz, A.K.; Gotte, M. Heparan sulphate as a regulator of leukocyte recruitment in inflammation. Curr. Protein Pept. Sci. 2015, 16, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.A.; Teixeira, F.C.; Fontes, M.; Areas, A.L.; Leal, M.G.; Pavao, M.S.; Stelling, M.P. Heparan sulfate proteoglycans may promote or inhibit cancer progression by interacting with integrins and affecting cell migration. BioMed. Res. Int. 2015, 2015, 453801. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.S.; Eames, B.F. Emerging tools to study proteoglycan function during skeletal development. Methods Cell Biol. 2016, 134, 485–530. [Google Scholar] [PubMed]
- Patel, V.N.; Pineda, D.L.; Hoffman, M.P. The function of heparan sulfate during branching morphogenesis. Matrix Biol. J. Int. Soc. Matrix Biol. 2017, 57–58, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Coulson-Thomas, V.J. The role of heparan sulphate in development: The ectodermal story. Int. J. Exp. Pathol. 2016, 97, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Reine, T.M.; Vuong, T.T.; Rutkovskiy, A.; Meen, A.J.; Vaage, J.; Jenssen, T.G.; Kolset, S.O. Serglycin in quiescent and proliferating primary endothelial cells. PLoS ONE 2015, 10, e0145584. [Google Scholar] [CrossRef] [PubMed]
- Sutton, V.R.; Brennan, A.J.; Ellis, S.; Danne, J.; Thia, K.; Jenkins, M.R.; Voskoboinik, I.; Pejler, G.; Johnstone, R.W.; Andrews, D.M.; et al. Serglycin determines secretory granule repertoire and regulates natural killer cell and cytotoxic T lymphocyte cytotoxicity. FEBS J. 2016, 283, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Melo, F.R.; Vita, F.; Berent-Maoz, B.; Levi-Schaffer, F.; Zabucchi, G.; Pejler, G. Proteolytic histone modification by mast cell tryptase, a serglycin proteoglycan-dependent secretory granule protease. J. Biol. Chem. 2014, 289, 7682–7690. [Google Scholar] [CrossRef] [PubMed]
- Elfenbein, A.; Lanahan, A.; Zhou, T.X.; Yamasaki, A.; Tkachenko, E.; Matsuda, M.; Simons, M. Syndecan 4 regulates FGFR1 signaling in endothelial cells by directing macropinocytosis. Sci. Signal. 2012, 5, ra36. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Yoshida, E.; Shinkai, Y.; Yamamoto, C.; Fujiwara, Y.; Kumagai, Y.; Kaji, T. Biglycan intensifies alk5-smad2/3 signaling by TGF-β1 and downregulates syndecan-4 in cultured vascular endothelial cells. J. Cell Biochem. 2017, 118, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Yoshida, E.; Fujiwara, Y.; Yamamoto, C.; Kaji, T. Transforming growth factor-β1 modulates the expression of syndecan-4 in cultured vascular endothelial cells in a biphasic manner. J. Cell Biochem. 2016, 118, 2009–2017. [Google Scholar] [CrossRef] [PubMed]
- Obunike, J.C.; Lutz, E.P.; Li, Z.; Paka, L.; Katopodis, T.; Strickland, D.K.; Kozarsky, K.F.; Pillarisetti, S.; Goldberg, I.J. Transcytosis of lipoprotein lipase across cultured endothelial cells requires both heparan sulfate proteoglycans and the very low density lipoprotein receptor. J. Biol. Chem. 2001, 276, 8934–8941. [Google Scholar] [CrossRef] [PubMed]
- Leonova, E.I.; Galzitskaya, O.V. Role of syndecans in lipid metabolism and human diseases. Adv. Exp. Med. Biol. 2015, 855, 241–258. [Google Scholar] [PubMed]
- Christianson, H.C.; Svensson, K.J.; van Kuppevelt, T.H.; Li, J.P.; Belting, M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc. Natl. Acad. Sci. USA 2013, 110, 17380–17385. [Google Scholar] [CrossRef] [PubMed]
- Christianson, H.C.; Belting, M. Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biol. J. Int. Soc. Matrix Biol. 2014, 35, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jin, H.; Beauvais, D.M.; Rapraeger, A.C. Cytoplasmic domain interactions of syndecan-1 and syndecan-4 with α6β4 integrin mediate human epidermal growth factor receptor (HER1 and HER2)-dependent motility and survival. J. Biol. Chem. 2014, 289, 30318–30332. [Google Scholar] [CrossRef] [PubMed]
- Beauvais, D.M.; Burbach, B.J.; Rapraeger, A.C. The syndecan-1 ectodomain regulates αvβ3 integrin activity in human mammary carcinoma cells. J. Cell Biol. 2004, 167, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.M.; Bhat, R.; Correia, A.L.; Mott, J.D.; Ilan, N.; Vlodavsky, I.; Pavao, M.S.; Bissell, M. Mammary branching morphogenesis requires reciprocal signaling by heparanase and mmp-14. J. Cell Biochem. 2015, 116, 1668–1679. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Sai, J.; Richmond, A. Cell surface heparan sulfate participates in cxcl1-induced signaling. Biochemistry 2003, 42, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Li, J.P.; Spillmann, D. Heparan sulfate proteoglycans as multifunctional cell regulators: Cell surface receptors. Methods Mol. Biol. 2012, 836, 239–255. [Google Scholar] [PubMed]
- Chu, W.; Song, X.; Yang, X.; Ma, L.; Zhu, J.; He, M.; Wang, Z.; Wu, Y. Neuropilin-1 promotes epithelial-to-mesenchymal transition by stimulating nuclear factor-κb and is associated with poor prognosis in human oral squamous cell carcinoma. PLoS ONE 2014, 9, e101931. [Google Scholar] [CrossRef] [PubMed]
- Tse, B.W.; Volpert, M.; Ratther, E.; Stylianou, N.; Nouri, M.; McGowan, K.; Lehman, M.L.; McPherson, S.J.; Roshan-Moniri, M.; Butler, M.S.; et al. Neuropilin-1 is upregulated in the adaptive response of prostate tumors to androgen-targeted therapies and is prognostic of metastatic progression and patient mortality. Oncogene 2017, 36, 3417–3427. [Google Scholar] [CrossRef] [PubMed]
- Graziani, G.; Lacal, P.M. Neuropilin-1 as therapeutic target for malignant melanoma. Front. Oncol. 2015, 5, 125. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.W.; Datta, S. Role of heparan sulfate 2-O-sulfotransferase in prostate cancer cell proliferation, invasion, and growth factor signaling. Prostate Cancer 2011, 2011, 893208. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.T.; Hogue, D.; Strong, L.C.; Hansen, M.F.; Blanton, S.H.; Wagner, M. Hereditary multiple exostosis and chondrosarcoma: Linkage to chromosome II and loss of heterozygosity for EXT-linked markers on chromosomes II and 8. Am. J. Hum. Gen. 1995, 56, 1125–1131. [Google Scholar]
- Lind, T.; Tufaro, F.; McCormick, C.; Lindahl, U.; Lidholt, K. The putative tumor suppressors EXT1 and EXT2 are glycosyltransferases required for the biosynthesis of heparan sulfate. J. Biol. Chem. 1998, 273, 26265–26268. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Morimoto, K.; Shimizu, T.; Takahashi, M.; Kurosawa, H.; Shirasawa, T. Association of EXT1 and EXT2, hereditary multiple exostoses gene products, in golgi apparatus. Biochem. Biophys. Res. Commun. 2000, 268, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Jao, T.M.; Li, Y.L.; Lin, S.W.; Tzeng, S.T.; Yu, I.S.; Yen, S.J.; Tsai, M.H.; Yang, Y.C. Alteration of colonic epithelial cell differentiation in mice deficient for glucosaminyl N-deacetylase/N-sulfotransferase 4. Oncotarget 2016, 7, 84938–84950. [Google Scholar] [CrossRef] [PubMed]
- Couchman, J.R.; Gopal, S.; Lim, H.C.; Norgaard, S.; Multhaupt, H.A. Syndecans: From peripheral coreceptors to mainstream regulators of cell behaviour. Int. J. Exp. Pathol. 2015, 96, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, H.; Ding, H. Gpc-3 in hepatocellular carcinoma: Current perspectives. J. Hepatocell. Carcinoma 2016, 3, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Montalbano, M.; Georgiadis, J.; Masterson, A.L.; McGuire, J.T.; Prajapati, J.; Shirafkan, A.; Rastellini, C.; Cicalese, L. Biology and function of glypican-3 as a candidate for early cancerous transformation of hepatocytes in hepatocellular carcinoma (review). Oncol. Rep. 2017, 37, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Aydin, O.; Yildiz, L.; Baris, S.; Dundar, C.; Karagoz, F. Expression of glypican 3 in low and high grade urothelial carcinomas. Diagn. Pathol. 2015, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Wade, A.; Robinson, A.E.; Engler, J.R.; Petritsch, C.; James, C.D.; Phillips, J.J. Proteoglycans and their roles in brain cancer. FEBS J. 2013, 280, 2399–2417. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vega, I.; Garcia-Suarez, O.; Garcia, B.; Crespo, A.; Astudillo, A.; Quiros, L.M. Heparan sulfate proteoglycans undergo differential expression alterations in right sided colorectal cancer, depending on their metastatic character. BMC Cancer 2015, 15, 742. [Google Scholar] [CrossRef] [PubMed]
- Kreuger, J.; Kjellen, L. Heparan sulfate biosynthesis: Regulation and variability. J. Histochem. Cytochem. 2012, 60, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.; Gotting, C.; Schnolzer, M.; Kempf, T.; Brinkmann, T.; Kleesiek, K. First isolation of human UDP-d-Xylose: Proteoglycan core protein β-d-xylosyltransferase secreted from cultured jar choriocarcinoma cells. J. Biol. Chem. 2001, 276, 4940–4947. [Google Scholar] [CrossRef] [PubMed]
- Gotting, C.; Kuhn, J.; Zahn, R.; Brinkmann, T.; Kleesiek, K. Molecular cloning and expression of human UDP-d-Xylose:Proteoglycan core protein β-d-xylosyltransferase and its first isoform XT-II. J. Mol. Biol. 2000, 304, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, Y.; Ramakrishnan, B.; Qasba, P.K. Crystal structures of β-1,4-galactosyltransferase 7 enzyme reveal conformational changes and substrate binding. J. Biol. Chem. 2013, 288, 31963–31970. [Google Scholar] [CrossRef] [PubMed]
- Hiyama, A.; Gogate, S.S.; Gajghate, S.; Mochida, J.; Shapiro, I.M.; Risbud, M.V. Bmp-2 and TGF-β stimulate expression of β1,3-glucuronosyl transferase 1 (Glcat-1) in nucleus pulposus cells through AP1, tonebp, and Sp1: Role of mapks. J. Bone Miner. Res. 2010, 25, 1179–1190. [Google Scholar] [PubMed]
- Koike, T.; Izumikawa, T.; Sato, B.; Kitagawa, H. Identification of phosphatase that dephosphorylates xylose in the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 2014, 289, 6695–6708. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Wu, G.; Li, H.; Qian, P.; Han, J.; Pan, F.; Li, W.; Li, J.; Ji, F. Promoter methylation profiles between human lung adenocarcinoma multidrug resistant a549/cisplatin (a549/ddp) cells and its progenitor a549 cells. Biol. Pharm. Bull. 2013, 36, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.; Beck, S.; Kogner, P.; Martinsson, T.; Caren, H. Genome-wide methylation profiling identifies novel methylated genes in neuroblastoma tumors. Epigenetics 2016, 11, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Poeta, M.L.; Massi, E.; Parrella, P.; Pellegrini, P.; de Robertis, M.; Copetti, M.; Rabitti, C.; Perrone, G.; Muda, A.O.; Molinari, F.; et al. Aberrant promoter methylation of β-1,4 galactosyltransferase 1 as potential cancer-specific biomarker of colorectal tumors. Genes Chromosomes Cancer 2012, 51, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Louwagie, J.; Carvalho, B.; Terhaar Sive Droste, J.S.; Park, H.L.; Chae, Y.K.; Yamashita, K.; Liu, J.; Ostrow, K.L.; Ling, S.; et al. Promoter DNA methylation of oncostatin M receptor-β as a novel diagnostic and therapeutic marker in colon cancer. PLoS ONE 2009, 4, e6555. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Chung, T.W.; Kim, C.H.; Jeong, H.S.; Joo, M.; Youn, B.; Ha, K.T. Estrogen induced β-1,4-galactosyltransferase 1 expression regulates proliferation of human breast cancer MCF-7 cells. Biochem. Biophys. Res. Commun. 2012, 426, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Busse, M.; Feta, A.; Presto, J.; Wilen, M.; Gronning, M.; Kjellen, L.; Kusche-Gullberg, M. Contribution of EXT1, EXT2, and EXTL3 to heparan sulfate chain elongation. J. Biol. Chem. 2007, 282, 32802–32810. [Google Scholar] [CrossRef] [PubMed]
- Busse-Wicher, M.; Wicher, K.B.; Kusche-Gullberg, M. The extostosin family: Proteins with many functions. Matrix Biol. 2014, 35, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Katta, K.; Imran, T.; Busse-Wicher, M.; Gronning, M.; Czajkowski, S.; Kusche-Gullberg, M. Reduced expression of EXTL2, a member of the exostosin (EXT) family of glycosyltransferases, in human embryonic kidney 293 cells results in longer heparan sulfate chains. J. Biol. Chem. 2015, 290, 13168–13177. [Google Scholar] [CrossRef] [PubMed]
- Oud, M.M.; Tuijnenburg, P.; Hempel, M.; van Vlies, N.; Ren, Z.; Ferdinandusse, S.; Jansen, M.H.; Santer, R.; Johannsen, J.; Bacchelli, C.; et al. Mutations in EXTL3 cause neuro-immuno-skeletal dysplasia syndrome. Am. J. Hum. Gen. 2017, 100, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.B.; Pacifici, M.; Hilton, M.J. Multiple hereditary exostoses (MHE): Elucidating the pathogenesis of a rare skeletal disorder through interdisciplinary research. Connect. Tissue Res. 2014, 55, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, H.; Shimakawa, H.; Sugahara, K. The tumor suppressor EXT-like gene EXTL2 encodes an α1, 4-N-acetylhexosaminyltransferase that transfersn-acetylgalactosamine and N-acetylglucosamine to the common glycosaminoglycan-protein linkage region: The key enzyme for the chain initiation of heparan sulfate. J. Biol. Chem. 1999, 274, 13933–13937. [Google Scholar] [PubMed]
- Nadanaka, S.; Zhou, S.; Kagiyama, S.; Shoji, N.; Sugahara, K.; Sugihara, K.; Asano, M.; Kitagawa, H. EXTL2, a member of the EXT family of tumor suppressors, controls glycosaminoglycan biosynthesis in a xylose kinase-dependent manner. J. Biol. Chem. 2013, 288, 9321–9333. [Google Scholar] [CrossRef] [PubMed]
- Nadanaka, S.; Kitagawa, H. EXTL2 controls liver regeneration and aortic calcification through xylose kinase-dependent regulation of glycosaminoglycan biosynthesis. Matrix Biol. J. Int. Soc. Matrix Biol. 2014, 35, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bansal, P.; Bridge, A.J.; Poux, S.; Bougueleret, L.; Xenarios, I. Uniprotkb/swiss-prot, the manually annotated section of the uniprot knowledgebase: How to use the entry view. Methods Mol. Biol. 2016, 1374, 23–54. [Google Scholar] [PubMed]
- Duncan, G.; McCormick, C.; Tufaro, F. The link between heparan sulfate and hereditary bone disease: Finding a function for the ext family of putative tumor suppressor proteins. J. Clin. Investig. 2001, 108, 511–516. [Google Scholar] [CrossRef] [PubMed]
- McCormick, C.; Duncan, G.; Goutsos, K.T.; Tufaro, F. The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc. Natl. Acad. Sci. USA 2000, 97, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Nadanaka, S.; Shoji, N.; Tamura, J.; Kitagawa, H. Biosynthesis of heparan sulfate in EXT1-deficient cells. Biochem. J. 2010, 428, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Ludecke, H.J.; Lindow, S.; Horton, W.A.; Lee, B.; Wagner, M.J.; Horsthemke, B.; Wells, D.E. Cloning of the putative tumour suppressor gene for hereditary multiple exostoses (EXT1). Nat. Genet. 1995, 11, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Francannet, C.; Cohen-Tanugi, A.; Le Merrer, M.; Munnich, A.; Bonaventure, J.; Legeai-Mallet, L. Genotype-phenotype correlation in hereditary multiple exostoses. J. Med. Gen. 2001, 38, 430–434. [Google Scholar] [CrossRef]
- Sinha, S.; Mundy, C.; Bechtold, T.; Sgariglia, F.; Ibrahim, M.M.; Billings, P.C.; Carroll, K.; Koyama, E.; Jones, K.B.; Pacifici, M. Unsuspected osteochondroma-like outgrowths in the cranial base of hereditary multiple exostoses patients and modeling and treatment with a bmp antagonist in mice. PLoS Genet. 2017, 13, e1006742. [Google Scholar] [CrossRef] [PubMed]
- Farhan, S.M.; Wang, J.; Robinson, J.F.; Prasad, A.N.; Rupar, C.A.; Siu, V.M.; Hegele, R.A. Old gene, new phenotype: Mutations in heparan sulfate synthesis enzyme, ext2 leads to seizure and developmental disorder, no exostoses. J. Med. Gen. 2015, 52, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Volpi, S.; Yamazaki, Y.; Brauer, P.M.; van Rooijen, E.; Hayashida, A.; Slavotinek, A.; Sun Kuehn, H.; Di Rocco, M.; Rivolta, C.; Bortolomai, I.; et al. EXTL3 mutations cause skeletal dysplasia, immune deficiency, and developmental delay. J. Exp. Med. 2017, 214, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Elcioglu, N.H.; Mizumoto, S.; Wang, Z.; Noyan, B.; Albayrak, H.M.; Yamada, S.; Matsumoto, N.; Miyake, N.; Nishimura, G.; et al. Identification of biallelic EXTL3 mutations in a novel type of spondylo-epi-metaphyseal dysplasia. J. Hum. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ropero, S.; Setien, F.; Espada, J.; Fraga, M.F.; Herranz, M.; Asp, J.; Benassi, M.S.; Franchi, A.; Patino, A.; Ward, L.S.; et al. Epigenetic loss of the familial tumor-suppressor gene exostosin-1 (EXT1) disrupts heparan sulfate synthesis in cancer cells. Hum. Mol. Genet. 2004, 13, 2753–2765. [Google Scholar] [CrossRef] [PubMed]
- Khoontawad, J.; Hongsrichan, N.; Chamgramol, Y.; Pinlaor, P.; Wongkham, C.; Yongvanit, P.; Pairojkul, C.; Khuntikeo, N.; Roytrakul, S.; Boonmars, T.; et al. Increase of exostosin 1 in plasma as a potential biomarker for opisthorchiasis-associated cholangiocarcinoma. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Wang, H.; Bernfield, M.; Gallagher, J.T.; Turnbull, J.E. Cell surface syndecan-1 on distinct cell types differs in fine structure and ligand binding of its heparan sulfate chains. J. Biol. Chem. 1994, 269, 18881–18890. [Google Scholar] [PubMed]
- Suhovskih, A.V.; Domanitskaya, N.V.; Tsidulko, A.Y.; Prudnikova, T.Y.; Kashuba, V.I.; Grigorieva, E.V. Tissue-specificity of heparan sulfate biosynthetic machinery in cancer. Cell Adh. Migr. 2015, 9, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Suhovskih, A.V.; Tsidulko, A.Y.; Kutsenko, O.S.; Kovner, A.V.; Aidagulova, S.V.; Ernberg, I.; Grigorieva, E.V. Transcriptional activity of heparan sulfate biosynthetic machinery is specifically impaired in benign prostate hyperplasia and prostate cancer. Front. Oncol. 2014, 4, 79. [Google Scholar] [CrossRef] [PubMed]
- Van den Born, J.; Pikas, D.S.; Pisa, B.J.; Eriksson, I.; Kjellen, L.; Berden, J.H. Antibody-based assay for N-deacetylase activity of heparan sulfate/heparin N-deacetylase/N-sulfotransferase (NDST): Novel characteristics of NDST-1 and -2. Glycobiology 2003, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dou, W.; Xu, Y.; Pagadala, V.; Pedersen, L.C.; Liu, J. Role of deacetylase activity of N-deacetylase/N-sulfotransferase 1 in forming N-sulfated domain in heparan sulfate. J. Biol. Chem. 2015, 290, 20427–20437. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, J.; Grobe, K.; Tsujimoto, M.; Esko, J.D. Multiple isozymes of heparan sulfate/heparin glcnac N-deacetylase/glcn N-sulfotransferase. Structure and activity of the fourth member, NDST4. J. Biol. Chem. 2001, 276, 5876–5882. [Google Scholar] [CrossRef] [PubMed]
- Pikas, D.S.; Eriksson, I.; Kjellen, L. Overexpression of different isoforms of glucosaminyl N-deacetylase/N-sulfotransferase results in distinct heparan sulfate N-sulfation patterns. Biochemistry 2000, 39, 4552–4558. [Google Scholar] [CrossRef] [PubMed]
- Holmborn, K.; Ledin, J.; Smeds, E.; Eriksson, I.; Kusche-Gullberg, M.; Kjellen, L. Heparan sulfate synthesized by mouse embryonic stem cells deficient in NDST1 and NDST2 is 6-O-sulfated but contains no N-sulfate groups. J. Biol. Chem. 2004, 279, 42355–42358. [Google Scholar] [CrossRef] [PubMed]
- Humphries, D.E.; Wong, G.W.; Friend, D.S.; Gurish, M.F.; Qiu, W.T.; Huang, C.; Sharpe, A.H.; Stevens, R.L. Heparin is essential for the storage of specific granule proteases in mast cells. Nature 1999, 400, 769–772. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, E.; Pejler, G.; Ringvall, M.; Lunderius, C.; Tomasini-Johansson, B.; Kusche-Gullberg, M.; Eriksson, I.; Ledin, J.; Hellman, L.; Kjellen, L. Abnormal mast cells in mice deficient in a heparin-synthesizing enzyme. Nature 1999, 400, 773–776. [Google Scholar] [PubMed]
- Tzeng, S.T.; Tsai, M.H.; Chen, C.L.; Lee, J.X.; Jao, T.M.; Yu, S.L.; Yen, S.J.; Yang, Y.C. NDST4 is a novel candidate tumor suppressor gene at chromosome 4q26 and its genetic loss predicts adverse prognosis in colorectal cancer. PLoS ONE 2013, 8, e67040. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Menzaghi, C.; Smith, S.; Liang, L.; de Rekeneire, N.; Garcia, M.E.; Lohman, K.K.; Miljkovic, I.; Strotmeyer, E.S.; Cummings, S.R.; et al. Genome-wide association analysis identifies TYW3/CRYZ and NDST4 loci associated with circulating resistin levels. Hum. Mol. Genet. 2012, 21, 4774–4780. [Google Scholar] [CrossRef] [PubMed]
- Eicher, J.D.; Powers, N.R.; Miller, L.L.; Akshoomoff, N.; Amaral, D.G.; Bloss, C.S.; Libiger, O.; Schork, N.J.; Darst, B.F.; Casey, B.J.; et al. Genome-wide association study of shared components of reading disability and language impairment. Genes Brain Behav. 2013, 12, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Reuter, M.S.; Musante, L.; Hu, H.; Diederich, S.; Sticht, H.; Ekici, A.B.; Uebe, S.; Wienker, T.F.; Bartsch, O.; Zechner, U.; et al. NDST1 missense mutations in autosomal recessive intellectual disability. Am. J. Med. Genet. Part A 2014, 164A, 2753–2763. [Google Scholar] [CrossRef] [PubMed]
- Najmabadi, H.; Hu, H.; Garshasbi, M.; Zemojtel, T.; Abedini, S.S.; Chen, W.; Hosseini, M.; Behjati, F.; Haas, S.; Jamali, P.; et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011, 478, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Lencz, T.; Guha, S.; Liu, C.; Rosenfeld, J.; Mukherjee, S.; DeRosse, P.; John, M.; Cheng, L.; Zhang, C.; Badner, J.A.; et al. Genome-wide association study implicates NDST3 in schizophrenia and bipolar disorder. Nat. Commun. 2013, 4, 2739. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lu, W.; Wang, Z.; Ni, J.; Zhang, J.; Tang, W.; Fang, Y. A comprehensive analysis of NDST3 for schizophrenia and bipolar disorder in han chinese. Trans. Psychiatry 2016, 6, e701. [Google Scholar] [CrossRef] [PubMed]
- Deligny, A.; Dierker, T.; Dagalv, A.; Lundequist, A.; Eriksson, I.; Nairn, A.V.; Moremen, K.W.; Merry, C.L.; Kjellen, L. NDST2 (N-deacetylase/N-sulfotransferase-2) enzyme regulates heparan sulfate chain length. J. Biol. Chem. 2016, 291, 18600–18607. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Liu, R.; Xu, Y.; Liu, J. The dominating role of N-deacetylase/N-sulfotransferase 1 in forming domain structures in heparan sulfate. J. Biol. Chem. 2011, 286, 19768–19776. [Google Scholar] [CrossRef] [PubMed]
- Bui, C.; Ouzzine, M.; Talhaoui, I.; Sharp, S.; Prydz, K.; Coughtrie, M.W.; Fournel-Gigleux, S. Epigenetics: Methylation-associated repression of heparan sulfate 3-O-sulfotransferase gene expression contributes to the invasive phenotype of h-emc-ss chondrosarcoma cells. FASEB J. 2010, 24, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Su, S.; Long, J.; Mei, B.; Chen, Y. Microrna-191 targets N-deacetylase/N-sulfotransferase 1 and promotes cell growth in human gastric carcinoma cell line MGC803. Acta Biochim. Biophys. Sin. 2011, 43, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Kasza, Z.; Fredlund Fuchs, P.; Tamm, C.; Eriksson, A.S.; O’Callaghan, P.; Heindryckx, F.; Spillmann, D.; Larsson, E.; Le Jan, S.; Eriksson, I.; et al. Microrna-24 suppression of N-deacetylase/N-sulfotransferase-1 (NDST1) reduces endothelial cell responsiveness to vascular endothelial growth factor a (VEGFA). J. Biol. Chem. 2013, 288, 25956–25963. [Google Scholar] [CrossRef] [PubMed]
- He, D.X.; Gu, X.T.; Li, Y.R.; Jiang, L.; Jin, J.; Ma, X. Methylation-regulated mir-149 modulates chemoresistance by targeting glcnac N-deacetylase/N-sulfotransferase-1 in human breast cancer. FEBS J. 2014, 281, 4718–4730. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Xu, Y.; Dulaney, S.B.; Huang, X.; Liu, J. Uncovering biphasic catalytic mode of c5-epimerase in heparan sulfate biosynthesis. J. Biol. Chem. 2012, 287, 20996–21002. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Song, T.; Lindahl, U.; Li, J.P. Enzyme overexpression—An exercise toward understanding regulation of heparan sulfate biosynthesis. Sci. Rep. 2016, 6, 31242. [Google Scholar] [CrossRef] [PubMed]
- Grigorieva, E.V.; Prudnikova, T.Y.; Domanitskaya, N.V.; Mostovich, L.A.; Pavlova, T.V.; Kashuba, V.I.; Zabarovsky, E.R. d-Glucuronyl c5-epimerase suppresses small-cell lung cancer cell proliferation in vitro and tumour growth in vivo. Br. J. Cancer 2011, 105, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Prudnikova, T.Y.; Mostovich, L.A.; Domanitskaya, N.V.; Pavlova, T.V.; Kashuba, V.I.; Zabarovsky, E.R.; Grigorieva, E.V. Antiproliferative effect of d-glucuronyl c5-epimerase in human breast cancer cells. Cancer Cell Int. 2010, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, E.E.; Prudnikova, T.Y.; Zabarovsky, E.R.; Kashuba, V.I.; Grigorieva, E.V. d-Glucuronyl c5-epimerase cell type specifically affects angiogenesis pathway in different prostate cancer cells. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 3237–3245. [Google Scholar] [CrossRef] [PubMed]
- Mostovich, L.A.; Prudnikova, T.Y.; Kondratov, A.G.; Gubanova, N.V.; Kharchenko, O.A.; Kutsenko, O.S.; Vavilov, P.V.; Haraldson, K.; Kashuba, V.I.; Ernberg, I.; et al. The TCF4/β-catenin pathway and chromatin structure cooperate to regulate d-glucuronyl c5-epimerase expression in breast cancer. Epigenetics 2012, 7, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Prudnikova, T.Y.; Soulitzis, N.; Kutsenko, O.S.; Mostovich, L.A.; Haraldson, K.; Ernberg, I.; Kashuba, V.I.; Spandidos, D.A.; Zabarovsky, E.R.; Grigorieva, E.V. Heterogeneity of d-glucuronyl c5-epimerase expression and epigenetic regulation in prostate cancer. Cancer Med. 2013, 2, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Smeds, E.; Feta, A.; Kusche-Gullberg, M. Target selection of heparan sulfate hexuronic acid 2-O-sulfotransferase. Glycobiology 2010, 20, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Clegg, J.M.; Conway, C.D.; Howe, K.M.; Price, D.J.; Mason, J.O.; Turnbull, J.E.; Basson, M.A.; Pratt, T. Heparan sulfotransferases HS6ST1 and HS2ST keep erk in check for mouse corpus callosum development. J. Neurosci. 2014, 34, 2389–2401. [Google Scholar] [CrossRef] [PubMed]
- Conway, C.D.; Howe, K.M.; Nettleton, N.K.; Price, D.J.; Mason, J.O.; Pratt, T. Heparan sulfate sugar modifications mediate the functions of slits and other factors needed for mouse forebrain commissure development. J. Neurosci. 2011, 31, 1955–1970. [Google Scholar] [CrossRef] [PubMed]
- Merry, C.L.; Bullock, S.L.; Swan, D.C.; Backen, A.C.; Lyon, M.; Beddington, R.S.; Wilson, V.A.; Gallagher, J.T. The molecular phenotype of heparan sulfate in the Hs2st−/− mutant mouse. J. Biol. Chem. 2001, 276, 35429–35434. [Google Scholar] [CrossRef] [PubMed]
- Maccarana, M.; Sakura, Y.; Tawada, A.; Yoshida, K.; Lindahl, U. Domain structure of heparan sulfates from bovine organs. J. Biol. Chem. 1996, 271, 17804–17810. [Google Scholar] [CrossRef] [PubMed]
- Ai, X.; Do, A.T.; Kusche-Gullberg, M.; Lindahl, U.; Lu, K.; Emerson, C.P., Jr. Substrate specificity and domain functions of extracellular heparan sulfate 6-O-endosulfatases, QSULF1 and QSULF2. J. Biol. Chem. 2006, 281, 4969–4976. [Google Scholar] [CrossRef] [PubMed]
- Uchimura, K.; Morimoto-Tomita, M.; Rosen, S.D. Measuring the activities of the sulfs: Two novel heparin/heparan sulfate endosulfatases. Methods Enzymol. 2006, 416, 243–253. [Google Scholar] [PubMed]
- Shworak, N.W.; HajMohammadi, S.; de Agostini, A.I.; Rosenberg, R.D. Mice deficient in heparan sulfate 3-O-sulfotransferase-1: Normal hemostasis with unexpected perinatal phenotypes. Glycoconj. J. 2002, 19, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Yabe, T.; Shukla, D.; Spear, P.G.; Rosenberg, R.D.; Seeberger, P.H.; Shworak, N.W. Portable sulphotransferase domain determines sequence specificity of heparan sulphate 3-O-sulphotransferases. Biochem. J. 2001, 359, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Xu, Y.; Liu, J.; Ho, M. Epitope mapping by a wnt-blocking antibody: Evidence of the wnt binding domain in heparan sulfate. Sci. Rep. 2016, 6, 26245. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.L.; Hansen, S.U.; Barath, M.; Rushton, G.; Gardiner, J.M.; Avizienyte, E.; Jayson, G.C. Synthetic heparan sulfate oligosaccharides inhibit endothelial cell functions essential for angiogenesis. PLoS ONE 2010, 5, e11644. [Google Scholar] [CrossRef] [PubMed]
- Vijaya Kumar, A.; Salem Gassar, E.; Spillmann, D.; Stock, C.; Sen, Y.P.; Zhang, T.; Van Kuppevelt, T.H.; Hulsewig, C.; Koszlowski, E.O.; Pavao, M.S.; et al. HS3ST2 modulates breast cancer cell invasiveness via map kinase- and Tcf4 (Tcf7l2)-dependent regulation of protease and cadherin expression. Int. J. Cancer 2014, 135, 2579–2592. [Google Scholar] [CrossRef] [PubMed]
- Meneghetti, M.C.Z.; Hughes, A.J.; Rudd, T.R.; Nader, H.B.; Powell, A.K.; Yates, E.A.; Lima, M.A. Heparan sulfate and heparin interactions with proteins. J. Royal Soc. Interface 2015, 12, 0589. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Gauche, C.; Coughtrie, M.W.; Bui, C.; Gulberti, S.; Merhi-Soussi, F.; Ramalanjaona, N.; Bertin-Jung, I.; Diot, A.; Dumas, D.; et al. The heparan sulfate sulfotransferase 3-OST3A (HS3ST3A) is a novel tumor regulator and a prognostic marker in breast cancer. Oncogene 2016, 35, 5043–5055. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.A.; Kim, Y.; Hong, S.H.; Lee, J.; Cho, Y.G.; Han, J.Y.; Kim, Y.H.; Han, J.; Shim, Y.M.; Lee, Y.S.; et al. Epigenetic inactivation of heparan sulfate (glucosamine) 3-O-sulfotransferase 2 in lung cancer and its role in tumorigenesis. PLoS ONE 2013, 8, e79634. [Google Scholar] [CrossRef] [PubMed]
- Roperch, J.P.; Grandchamp, B.; Desgrandchamps, F.; Mongiat-Artus, P.; Ravery, V.; Ouzaid, I.; Roupret, M.; Phe, V.; Ciofu, C.; Tubach, F.; et al. Promoter hypermethylation of HS3ST2, SEPTIN9 and SLIT2 combined with FGFR3 mutations as a sensitive/specific urinary assay for diagnosis and surveillance in patients with low or high-risk non-muscle-invasive bladder cancer. BMC Cancer 2016, 16, 704. [Google Scholar] [CrossRef] [PubMed]
- Lamanna, W.C.; Frese, M.A.; Balleininger, M.; Dierks, T. Sulf loss influences N-, 2-O-, and 6-O-sulfation of multiple heparan sulfate proteoglycans and modulates fibroblast growth factor signaling. J. Biol. Chem. 2008, 283, 27724–27735. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Bethea, H.N.; Liu, J. Using engineered 2-O-sulfotransferase to determine the activity of heparan sulfate c5-epimerase and its mutants. J. Biol. Chem. 2010, 285, 11106–11113. [Google Scholar] [CrossRef] [PubMed]
- Presto, J.; Thuveson, M.; Carlsson, P.; Busse, M.; Wilen, M.; Eriksson, I.; Kusche-Gullberg, M.; Kjellen, L. Heparan sulfate biosynthesis enzymes EXT1 and EXT2 affect NDST1 expression and heparan sulfate sulfation. Proc. Natl Acad Sci. USA 2008, 105, 4751–4756. [Google Scholar] [CrossRef] [PubMed]
- Esko, J.D.; Selleck, S.B. Order out of chaos: Assembly of ligand binding sites in heparan sulfate. Annu Rev. Biochem 2002, 71, 435–471. [Google Scholar] [CrossRef] [PubMed]
- Hassinen, A.; Kellokumpu, S. Organizational interplay of golgi N-glycosyltransferases involves organelle microenvironment-dependent transitions between enzyme homo- and heteromers. J. Biol. Chem. 2014, 289, 26937–26948. [Google Scholar] [CrossRef] [PubMed]
- Hassinen, A.; Pujol, F.M.; Kokkonen, N.; Pieters, C.; Kihlstrom, M.; Korhonen, K.; Kellokumpu, S. Functional organization of golgi N- and O-glycosylation pathways involves pH-dependent complex formation that is impaired in cancer cells. J. Biol. Chem. 2011, 286, 38329–38340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, F.; Sheng, J. “Coding” and “decoding”: Hypothesis for the regulatory mechanism involved in heparan sulfate biosynthesis. Carbohydr. Res. 2016, 428, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Schultz, V.; Suflita, M.; Liu, X.; Zhang, X.; Yu, Y.; Li, L.; Green, D.E.; Xu, Y.; Zhang, F.; DeAngelis, P.L.; et al. Heparan sulfate domains required for fibroblast growth factor 1 and 2 signaling through fibroblast growth factor receptor 1c. J. Biol. Chem. 2017, 292, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, U.; Pakkiriswami, S.; Pillai, A.B. Sugar tags and tumorigenesis. Front. Cell Dev. Biol. 2015, 3, 69. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.K.; Bush, K.T. Growth factor-heparan sulfate “switches” regulating stages of branching morphogenesis. Pediatr. Nephrol. 2014, 29, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Poulain, F.E.; Yost, H.J. Heparan sulfate proteoglycans: A sugar code for vertebrate development? Development 2015, 142, 3456–3467. [Google Scholar] [CrossRef] [PubMed]
- Borza, D.-B. Glomerular basement membrane heparan sulfate in health and disease: A regulator of local complement activation. Matrix Biol. 2017, 57–58, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Nadanaka, S.; Purunomo, E.; Takeda, N.; Tamura, J.; Kitagawa, H. Heparan sulfate containing unsubstituted glucosamine residues: Biosynthesis and heparanase-inhibitory activity. J. Biol. Chem. 2014, 289, 15231–15243. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, N.V.; Sarkar, A.; Desai, U.R.; Mosier, P.D. Designing “high-affinity, high-specificity” glycosaminoglycan sequences through computerized modeling. Methods Mol. Biol. 2015, 1229, 289–314. [Google Scholar] [PubMed]
- Dai, Y.; Yang, Y.; MacLeod, V.; Yue, X.; Rapraeger, A.C.; Shriver, Z.; Venkataraman, G.; Sasisekharan, R.; Sanderson, R.D. Hsulf-1 and hsulf-2 are potent inhibitors of myeloma tumor growth in vivo. J. Biol. Chem. 2005, 280, 40066–40073. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Xiong, A.; Spyrou, A.; Wicher, G.; Marinescu, V.D.; Edqvist, P.D.; Zhang, L.; Essand, M.; Dimberg, A.; Smits, A.; et al. Heparanase promotes glioma progression and is inversely correlated with patient survival. Mol. Cancer Res. 2016, 14, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Higginson, J.R.; Thompson, S.M.; Santos-Silva, A.; Guimond, S.E.; Turnbull, J.E.; Barnett, S.C. Differential sulfation remodelling of heparan sulfate by extracellular 6-O-sulfatases regulates fibroblast growth factor-induced boundary formation by glial cells: Implications for glial cell transplantation. J. Neurosci. 2012, 32, 15902–15912. [Google Scholar] [CrossRef] [PubMed]
- Khurana, A.; Beleford, D.; He, X.; Chien, J.; Shridhar, V. Role of heparan sulfatases in ovarian and breast cancer. Am. J. Cancer Res. 2013, 3, 34–45. [Google Scholar] [PubMed]
- Shire, A.; Lomberk, G.; Lai, J.P.; Zou, H.; Tsuchiya, N.; Aderca, I.; Moser, C.D.; Gulaid, K.H.; Oseini, A.; Hu, C.; et al. Restoration of epigenetically silenced sulf1 expression by 5-aza-2-deoxycytidine sensitizes hepatocellular carcinoma cells to chemotherapy-induced apoptosis. Med. Epigenet. 2015, 3, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Newman, D.R.; Sannes, P.L. Hsulf-1 inhibits erk and akt signaling and decreases cell viability in vitro in human lung epithelial cells. Respirat. Res. 2012, 13, 69. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Ji, W.; Su, Y.; Xu, Y.; Yan, Y.; Shen, S.; Li, X.; Sun, B.; Qian, H.; Chen, L.; et al. Sulfatase 1 (HSULF-1) reverses basic fibroblast growth factor-stimulated signaling and inhibits growth of hepatocellular carcinoma in animal model. Oncotarget 2014, 5, 5029–5039. [Google Scholar] [CrossRef] [PubMed]
- Tessema, M.; Yingling, C.M.; Thomas, C.L.; Klinge, D.M.; Bernauer, A.M.; Liu, Y.; Dacic, S.; Siegfried, J.M.; Dahlberg, S.E.; Schiller, J.H.; et al. Sulf2 methylation is prognostic for lung cancer survival and increases sensitivity to topoisomerase-i inhibitors via induction of ISG15. Oncogene 2012, 31, 4107–4116. [Google Scholar] [CrossRef] [PubMed]
- Hur, K.; Han, T.S.; Jung, E.J.; Yu, J.; Lee, H.J.; Kim, W.H.; Goel, A.; Yang, H.K. Up-regulated expression of sulfatases (SULF1 and SULF2) as prognostic and metastasis predictive markers in human gastric cancer. J. Pathol. 2012, 228, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.P.; Yu, C.; Moser, C.D.; Aderca, I.; Han, T.; Garvey, T.D.; Murphy, L.M.; Garrity-Park, M.M.; Shridhar, V.; Adjei, A.A.; et al. Sulf1 inhibits tumor growth and potentiates the effects of histone deacetylase inhibitors in hepatocellular carcinoma. Gastroenterology 2006, 130, 2130–2144. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wei, J.; Wang, H.; Yang, Y.; Yue, G.; Wang, L.; Yu, L.; Xie, L.; Sun, X.; Bian, X.; et al. Sulf2 methylation is associated with in vitro cisplatin sensitivity and clinical efficacy for gastric cancer patients treated with a modified folfox regimen. PLoS ONE 2013, 8, e75564. [Google Scholar] [CrossRef] [PubMed]
- Takei, Y.; Takigahira, M.; Mihara, K.; Tarumi, Y.; Yanagihara, K. The metastasis-associated microrna mir-516a-3p is a novel therapeutic target for inhibiting peritoneal dissemination of human scirrhous gastric cancer. Cancer Res. 2011, 71, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Yan, Y.; Xu, C.; Ji, W.; Shen, S.; Xu, G.; Zeng, Y.; Sun, B.; Qian, H.; Chen, L.; et al. microRNA-21 suppresses PTEN and HSULF-1 expression and promotes hepatocellular carcinoma progression through AKT/ERK pathways. Cancer Lett. 2013, 337, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Yamada, S.; Toyoshima, M.; Dong, J.; Nakajima, M.; Sugahara, K. Structural recognition by recombinant human heparanase that plays critical roles in tumor metastasis. Hierarchical sulfate groups with different effects and the essential target disulfated trisaccharide sequence. J. Biol. Chem. 2002, 277, 42488–42495. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.B.; Liu, J. Unraveling the specificity of heparanase utilizing synthetic substrates. J. Biol. Chem. 2010, 285, 14504–14513. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.; Atzmon, R.; Vlodavsky, I.; Ilan, N. Heparanase processing by lysosomal/endosomal protein preparation. FEBS Lett. 2005, 579, 2334–2338. [Google Scholar] [CrossRef] [PubMed]
- Gingis-Velitski, S.; Zetser, A.; Flugelman, M.Y.; Vlodavsky, I.; Ilan, N. Heparanase induces endothelial cell migration via protein kinase b/AKT activation. J. Biol. Chem. 2004, 279, 23536–23541. [Google Scholar] [CrossRef] [PubMed]
- Zetser, A.; Bashenko, Y.; Edovitsky, E.; Levy-Adam, F.; Vlodavsky, I.; Ilan, N. Heparanase induces vascular endothelial growth factor expression: Correlation with p38 phosphorylation levels and src activation. Cancer Res. 2006, 66, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kaplan, V.; Naroditsky, I.; Zetser, A.; Ilan, N.; Vlodavsky, I.; Doweck, I. Heparanase induces VEGF c and facilitates tumor lymphangiogenesis. Int. J. Cancer 2008, 123, 2566–2573. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.C.; Yang, Y.; Ren, Y.; Nan, L.; Sanderson, R.D. Heparanase plays a dual role in driving hepatocyte growth factor (HGF) signaling by enhancing hgf expression and activity. J. Biol. Chem. 2011, 286, 6490–6499. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Brenchley, P.E.; Jayson, G.C.; Hampson, L.; Davies, J.; Hampson, I.N. Hypoxia increases heparanase-dependent tumor cell invasion, which can be inhibited by antiheparanase antibodies. Cancer Res. 2004, 64, 3928–3933. [Google Scholar] [CrossRef] [PubMed]
- Cassinelli, G.; Favini, E.; Dal Bo, L.; Tortoreto, M.; De Maglie, M.; Dagrada, G.; Pilotti, S.; Zunino, F.; Zaffaroni, N.; Lanzi, C. Antitumor efficacy of the heparan sulfate mimic roneparstat (SST0001) against sarcoma models involves multi-target inhibition of receptor tyrosine kinases. Oncotarget 2016, 7, 47848–47863. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.C.; Zhan, F.; He, J.; Barbieri, P.; Noseda, A.; Tricot, G.; Sanderson, R.D. Targeting heparanase overcomes chemoresistance and diminishes relapse in myeloma. Oncotarget 2016, 7, 1598–1607. [Google Scholar] [PubMed]
- Ogishima, T.; Shiina, H.; Breault, J.E.; Tabatabai, L.; Bassett, W.W.; Enokida, H.; Li, L.C.; Kawakami, T.; Urakami, S.; Ribeiro-Filho, L.A.; et al. Increased heparanase expression is caused by promoter hypomethylation and up-regulation of transcriptional factor early growth response-1 in human prostate cancer. Clin. Cancer Res. 2005, 11, 1028–1036. [Google Scholar] [PubMed]
- Qu, H.; Zheng, L.; Pu, J.; Mei, H.; Xiang, X.; Zhao, X.; Li, D.; Li, S.; Mao, L.; Huang, K.; et al. miRNA-558 promotes tumorigenesis and aggressiveness of neuroblastoma cells through activating the transcription of heparanase. Hum. Mol. Genet. 2015, 24, 2539–2551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sullivan, P.S.; Goodman, J.C.; Gunaratne, P.H.; Marchetti, D. microRNA-1258 suppresses breast cancer brain metastasis by targeting heparanase. Cancer Res. 2011, 71, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vega, I.; Garcia, O.; Crespo, A.; Castanon, S.; Menendez, P.; Astudillo, A.; Quiros, L.M. Specific genes involved in synthesis and editing of heparan sulfate proteoglycans show altered expression patterns in breast cancer. BMC Cancer 2013, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Gurda, D.; Handschuh, L.; Kotkowiak, W.; Jakubowski, H. Homocysteine thiolactone and N-homocysteinylated protein induce pro-atherogenic changes in gene expression in human vascular endothelial cells. Amino Acids 2015, 47, 1319–1339. [Google Scholar] [CrossRef] [PubMed]
- Okuda, H.; Kobayashi, A.; Xia, B.; Watabe, M.; Pai, S.K.; Hirota, S.; Xing, F.; Liu, W.; Pandey, P.R.; Fukuda, K.; et al. Hyaluronan synthase has 2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells with macrophages and stromal cells. Cancer Res. 2012, 72, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Mercier, I.; Casimiro, M.C.; Wang, C.; Rosenberg, A.L.; Quong, J.; Minkeu, A.; Allen, K.G.; Danilo, C.; Sotgia, F.; Bonuccelli, G.; et al. Human breast cancer-associated fibroblasts (CAFS) show caveolin-1 downregulation and RB tumor suppressor functional inactivation: Implications for the response to hormonal therapy. Cancer Biol. Ther. 2008, 7, 1212–1225. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, Z.; Liu, C.; Jin, C.; Zhang, J.; Miao, X.; Jia, L. B4galt1 gene knockdown inhibits the Hedgehog pathway and reverses multidrug resistance in the human leukemia k562/adriamycin-resistant cell line. IUBMB Life 2012, 64, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Zhu, Y.; An, H.; Wang, H.; Zhu, Y.; Fu, H.; Wang, Z.; Fu, Q.; Xu, J.; Ye, D. Increased b4galt1 expression associates with adverse outcome in patients with non-metastatic clear cell renal cell carcinoma. Oncotarget 2016, 7, 32723–32730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Y.; Xie, H.; Fu, Q.; Liu, Z.; Zhu, Y.; Xu, L.; Zhang, W.; Yang, Y.; Xu, J. β-1,4-galactosyltransferase II predicts poor prognosis of patients with non-metastatic clear-cell renal cell carcinoma. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2017, 39, 1010428317691417. [Google Scholar] [CrossRef] [PubMed]
- Lo Sasso, G.; Bovenga, F.; Murzilli, S.; Salvatore, L.; di Tullio, G.; Martelli, N.; D’Orazio, A.; Rainaldi, S.; Vacca, M.; Mangia, A.; et al. Liver x receptors inhibit proliferation of human colorectal cancer cells and growth of intestinal tumors in mice. Gastroenterology 2013, 144, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Karibe, T.; Fukui, H.; Sekikawa, A.; Shiratori, K.; Fujimori, T. EXTL3 promoter methylation down-regulates EXTL3 and heparan sulphate expression in mucinous colorectal cancers. J. Pathol. 2008, 216, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Bret, C.; Hose, D.; Reme, T.; Sprynski, A.C.; Mahtouk, K.; Schved, J.F.; Quittet, P.; Rossi, J.F.; Goldschmidt, H.; Klein, B. Expression of genes encoding for proteins involved in heparan sulphate and chondroitin sulphate chain synthesis and modification in normal and malignant plasma cells. Br. J. Haematol. 2009, 145, 350–368. [Google Scholar] [CrossRef] [PubMed]
- Okolicsanyi, R.K.; van Wijnen, A.J.; Cool, S.M.; Stein, G.S.; Griffiths, L.R.; Haupt, L.M. Heparan sulfate proteoglycans and human breast cancer epithelial cell tumorigenicity. J. Cell Biochem. 2014, 115, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Paschall, A.V.; Liu, K. Epigenetic regulation of apoptosis and cell cycle regulatory genes in human colon carcinoma cells. Genom. Data 2015, 5, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Li, Q.; Peng, Y.B.; Li, J.; Ding, K.; Chen, L.J.; Shao, C.H.; Zhang, L.J.; Li, P. Silencing of HHS6ST2 inhibits progression of pancreatic cancer through inhibition of notch signalling. Biochem. J. 2011, 436, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Iravani, O.; Bay, B.H.; Yip, G.W. Silencing HS6ST3 inhibits growth and progression of breast cancer cells through suppressing IGF1R and inducing XAF1. Exp. Cell Res. 2017, 350, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.L.; Rushton, G.; Jayson, G.C.; Avizienyte, E. Ovarian cancer cell heparan sulfate 6-O-sulfotransferases regulate an angiogenic program induced by heparin-binding epidermal growth factor (EGF)-like growth factor/egf receptor signaling. J. Biol. Chem. 2014, 289, 10488–10501. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.; Vergoten, G.; Colomb, F.; Bobowski, M.; Steenackers, A.; Carpentier, M.; Allain, F.; Delannoy, P.; Julien, S. Over-sulfated glycosaminoglycans are alternative selectin ligands: Insights into molecular interactions and possible role in breast cancer metastasis. Clin. Exp. Met. 2013, 30, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Gai, X.; Han, S.; Moser, C.D.; Hu, C.; Shire, A.M.; Floyd, R.A.; Roberts, L.R. The human sulfatase 2 inhibitor 2,4-disulfonylphenyl-TERT-butylnitrone (OKN-007) has an antitumor effect in hepatocellular carcinoma mediated via suppression of TGFB1/SMAD2 and Hedgehog/GLI1 signaling. Genes. Chromosomes Cancer 2013, 52, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.M.; Hosono-Fukao, T.; Tang, R.; Sugaya, N.; van Kuppevelt, T.H.; Jenniskens, G.J.; Kimata, K.; Rosen, S.D.; Uchimura, K. Direct detection of HSULF-1 and HSULF-2 activities on extracellular heparan sulfate and their inhibition by PI-88. Glycobiology 2010, 20, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.P.; Sandhu, D.S.; Yu, C.; Moser, C.D.; Hu, C.; Shire, A.M.; Aderca, I.; Murphy, L.M.; Adjei, A.A.; Sanderson, S.; et al. Sulfatase 2 protects hepatocellular carcinoma cells against apoptosis induced by the pi3k inhibitor ly294002 and erk and jnk kinase inhibitors. Liver Int. 2010, 30, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Elkin, M.; Cohen, I.; Zcharia, E.; Orgel, A.; Guatta-Rangini, Z.; Peretz, T.; Vlodavsky, I.; Kleinman, H.K. Regulation of heparanase gene expression by estrogen in breast cancer. Cancer Res. 2003, 63, 8821–8826. [Google Scholar] [PubMed]
- Jiang, G.; Zheng, L.; Pu, J.; Mei, H.; Zhao, J.; Huang, K.; Zeng, F.; Tong, Q. Small RNAs targeting transcription start site induce heparanase silencing through interference with transcription initiation in human cancer cells. PLoS ONE 2012, 7, e31379. [Google Scholar] [CrossRef] [PubMed]
- Levy-Adam, F.; Feld, S.; Cohen-Kaplan, V.; Shteingauz, A.; Gross, M.; Arvatz, G.; Naroditsky, I.; Ilan, N.; Doweck, I.; Vlodavsky, I. Heparanase 2 interacts with heparan sulfate with high affinity and inhibits heparanase activity. J. Biol. Chem. 2010, 285, 28010–28019. [Google Scholar] [CrossRef] [PubMed]
- Debeb, B.G.; Lacerda, L.; Xu, W.; Larson, R.; Solley, T.; Atkinson, R.; Sulman, E.P.; Ueno, N.T.; Krishnamurthy, S.; Reuben, J.M.; et al. Histone deacetylase inhibitors stimulate dedifferentiation of human breast cancer cells through WNT/β-catenin signaling. Stem. Cells 2012, 30, 2366–2377. [Google Scholar] [CrossRef] [PubMed]
- Slingerland, M.; Guchelaar, H.J.; Gelderblom, H. Histone deacetylase inhibitors: An overview of the clinical studies in solid tumors. Anti-Cancer Drugs 2014, 25, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Hameetman, L.; David, G.; Yavas, A.; White, S.J.; Taminiau, A.H.; Cleton-Jansen, A.M.; Hogendoorn, P.C.; Bovee, J.V. Decreased ext expression and intracellular accumulation of heparan sulphate proteoglycan in osteochondromas and peripheral chondrosarcomas. J. Pathol. 2007, 211, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Lyon, M.; Paraskeva, C.; Turnbull, J.E.; Deakin, J.A.; Gallagher, J.T. Heparan sulfate undergoes specific structural changes during the progression from human colon adenoma to carcinoma in vitro. J. Biol. Chem. 1998, 273, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Munkley, J.; Elliott, D.J. Hallmarks of glycosylation in cancer. Oncotarget 2016, 7, 35478–35489. [Google Scholar] [PubMed]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Pal, K.; Yadav, J.; Tang, H.; Partyka, K.; Kletter, D.; Hsueh, P.; Ensink, E.; Kc, B.; Hostetter, G.; et al. Upregulation of glycans containing 3’ fucose in a subset of pancreatic cancers uncovered using fusion-tagged lectins. J. Proteome Res. 2015, 14, 2594–2605. [Google Scholar] [CrossRef] [PubMed]
- Fry, S.A.; Afrough, B.; Lomax-Browne, H.J.; Timms, J.F.; Velentzis, L.S.; Leathem, A.J. Lectin microarray profiling of metastatic breast cancers. Glycobiology 2011, 21, 1060–1070. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Lu, W.; Li, X.; Yang, G.; Guo, J.; Yu, H.; Li, Z.; Guan, F. Altered N-glycan expression profile in epithelial-to-mesenchymal transition of nmumg cells revealed by an integrated strategy using mass spectrometry and glycogene and lectin microarray analysis. J. Proteom. Res. 2014, 13, 2783–2795. [Google Scholar] [CrossRef] [PubMed]
- Patsos, G.; Hebbe-Viton, V.; Robbe-Masselot, C.; Masselot, D.; San Martin, R.; Greenwood, R.; Paraskeva, C.; Klein, A.; Graessmann, M.; Michalski, J.C.; et al. O-glycan inhibitors generate aryl-glycans, induce apoptosis and lead to growth inhibition in colorectal cancer cell lines. Glycobiology 2009, 19, 382–398. [Google Scholar] [CrossRef] [PubMed]
- Costa, J. Glycoconjugates from extracellular vesicles: Structures, functions and emerging potential as cancer biomarkers. Biochim. Biophys. Acta 2017, 1868, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Echevarria, J.; Royo, F.; Pazos, R.; Salazar, L.; Falcon-Perez, J.M.; Reichardt, N.C. Microarray-based identification of lectins for the purification of human urinary extracellular vesicles directly from urine samples. Chembiochem 2014, 15, 1621–1626. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.; Gomes-Alves, P.; Carvalho, S.B.; Peixoto, C.; Alves, P.M.; Altevogt, P.; Costa, J. Extracellular vesicles from ovarian carcinoma cells display specific glycosignatures. Biomolecules 2015, 5, 1741–1761. [Google Scholar] [CrossRef] [PubMed]
- Ceroni, A.; Dell, A.; Haslam, S.M. The glycanbuilder: A fast, intuitive and flexible software tool for building and displaying glycan structures. Source Code Biol. Med. 2007, 2, 3. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Major Function | Expression Change | Possible Therapeutic Targeting | Type(s) of Cancer | References |
|---|---|---|---|---|---|
| XYLT1/2 | Addition of xylose to a serine on a core HSPG to initiate HS chain synthesis | Up | shRNA targeting of XYLT1; DNA methylating agents | Breast cancer/bone metastasis; breast cancer associate fibroblasts; multidrug resistance | [66,179,180] |
| B4GALT1 | Formation of the β 4 xyl-gal linkage | Varied | 5-Aza-dC treatment; estrogen receptor blockers | Colon cancer; breast cancer | [68,69,70,181,182,183] |
| B3GALT6 | Formation of the β 3 gal-gal linkage | Up | Liver X receptor agonists | Colon cancer | [184] |
| B3GAT3 | Catalyzes the β 3 glc-gal linkage | Up | DNA methylating agent | Multidrug resistance | [66] |
| EXT1/2 | Catalyzes the addition of both α-d-glucoronate (GlcA) and GlcNAc during HS chain elongation | Varied | 5-Aza-dc treatment | Osteochondromas, cholangiocarcinoma, leukemia | [89,90] |
| EXTL1/2/3 | Adds the required N-acetyl-d-Glucosamine (GlcNAc) for elongation of the HS chain | Down | 5-Aza-dc treatment; siRNA | Colon cancer | [185] |
| NDST1-4 | Replaces the N-acetyl groups (GlcNAc) with N-sulfate groups (GlcNS) on a glucosamine residue | Varied | 5-Aza-dc treatment; miRNA interference | Colon cancer (NDST4); breast cancer | [101,112,113,177] |
| GLCE | Converts glucuronic acid (GlcA) to its epimer iduronic acid | Varied | Cancer-type dependent; ectopic overexpression improves breast and lung cancer prognosis, while overexpression is associated with increased aggressiveness in prostate cancer | Breast cancer; lung cancer; prostate cancer | [93,116,117,118,119,120] |
| HS2ST1 | Mediates 2-O-sulfation of both l-iduronyl and d-glucuronyl residues within the maturing HS | Up | Heparin treatment; histone methyltransferase inhibitor | Breast cancer; multiple myeloma | [186,187,188] |
| HS6ST1-3 | Catalyzes the transfer of sulfate from 3-Phosphoadenosine 5-Phosphosulfate (PAPS) to position 6 of the N-sulfoglucosamine residue (GlcNS) of heparan sulfate | Up | HS6ST inhibitors and HS mimetics | Ovarian cancer; breast cancer; pancreatic cancer | [59,189,190,191] |
| HS3ST1-6 | Utilizes 3-phospho-5-adenylyl sulfate (PAPS) to catalyze the transfer of a sulfo group to position 3 of glucosamine residues in heparan | Down | 5-Aza-dc treatment | Breast cancer; invasive ductal carcinomas; chondrosarcoma | [59,110,134,135,136,177,192] |
| SULF1 | Selectively removes 6-O-sulfate groups from HS chains | Varied | HS mimetic (PI-88); 5-Aza-dc treatment; miRNA interference | Multiple cancers | [193,194,195] |
| SULF2 | Selectively remove 6-O-sulfate groups from heparan sulfate | Up | Sulf inhibitors (OKN-007); proteasome inhibitors (bortezomib); HS mimetic (PI-88) | Multiple cancers | [154,193,194] |
| HPSE | Cleaves heparan sulfate proteoglycans to permit cell movement through remodeling of the extracellular matrix | Up | Roneparstat; miRNA interference; estrogen receptor antagonists | Multiple myeloma; brain cancer; breast cancer; colon cancer | [173,174,175,176,196,197] |
| HPSE2 | Binds heparin and heparan sulfate with high affinity, but lacks heparanase activity | Down | Prognostic biomarker as elevated HPSE2 is correlated to improved outcomes | Breast cancers; head and neck cancers | [177,198] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hull, E.E.; Montgomery, M.R.; Leyva, K.J. Epigenetic Regulation of the Biosynthesis & Enzymatic Modification of Heparan Sulfate Proteoglycans: Implications for Tumorigenesis and Cancer Biomarkers. Int. J. Mol. Sci. 2017, 18, 1361. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18071361
Hull EE, Montgomery MR, Leyva KJ. Epigenetic Regulation of the Biosynthesis & Enzymatic Modification of Heparan Sulfate Proteoglycans: Implications for Tumorigenesis and Cancer Biomarkers. International Journal of Molecular Sciences. 2017; 18(7):1361. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18071361
Chicago/Turabian StyleHull, Elizabeth E., McKale R. Montgomery, and Kathryn J. Leyva. 2017. "Epigenetic Regulation of the Biosynthesis & Enzymatic Modification of Heparan Sulfate Proteoglycans: Implications for Tumorigenesis and Cancer Biomarkers" International Journal of Molecular Sciences 18, no. 7: 1361. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18071361