Targeting Tumor Adaption to Chronic Hypoxia: Implications for Drug Resistance, and How It Can Be Overcome

Graduate School of Analytical Science and Technology (GRAST), Chungnam National University, Daejeon 34134, Korea
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(9), 1854; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18091854
Submission received: 21 July 2017
/
Revised: 21 August 2017
/
Accepted: 22 August 2017
/
Published: 25 August 2017
(This article belongs to the Special Issue Adaptation to Chronic Hypoxia: The Last Word Has Not yet Been Said)
Abstract
:The rapid and uncontrolled proliferation of tumors limits the availability of oxygen and nutrients supplied from the tumor vasculature, thus exposing them to low oxygen environments. Thus, diminished oxygen availability, or hypoxia, is the most common microenvironment feature of nearly all solid tumors. All living cells have the ability to sense changes in oxygen tension and adapt to this stress to preserve survival. Likewise, cancer cells adapt to chronic hypoxic stress via several mechanisms, including promotion of angiogenic factor production, metabolic shift to consume less oxygen, and reduction of apoptotic potential. Adaptation of tumor cells to hypoxia is believed to be the main driver for selection of more invasive and therapy-resistant cancer phenotypes. In this review, we discuss molecular mechanisms by which tumor cells adapt to hypoxia, with a specific focus on hypoxia-inducible factor (HIF) transcription factor. We further discuss the current understandings on hypoxia-mediated drug resistance and strategies to overcome it.
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Tumor cells require a constant supply of oxygen. In small tumors with a diameter less than 1 mm, oxygen can be supplied by diffusion from blood vessels. However, as the tumor grows, cells distant from the blood vessel are exposed to limited oxygen availability, leading to the formation of necrotic tumor tissue. A fraction of cells which survive this hypoxic stress become problematic, since they exhibit a more invasive phenotype and are refractory to cancer therapies [1,2,3]. Thus, targeting hypoxia is becoming an exciting area garnering significant attention in the efforts to overcome cancer drug resistance. In this review, we discuss our recent understandings on molecular mechanisms by which hypoxic tumor cells adapt to hypoxic stress, with a focus on hypoxia-inducible factor (HIF) transcription factor and current therapeutic strategies to attenuate hypoxia-mediated drug resistance.
1.1. Hypoxia-Inducible Factor (HIF)
1.1.1. HIF Regulation in Hypoxia
The HIF family of transcription factor plays a pivotal role in the adaptation of cancer cells to hypoxia. The HIF transcription factor functions as a heterodimer, composed of one of two oxygen-labile α subunits (HIF-1α, -2α) and an oxygen-insensitive β subunit. HIF-1α is expressed ubiquitously, whereas HIF-α is expressed only in particular cell types such as hepatocytes and endothelial cells [4]. The stability of HIF-α family proteins are tightly regulated by oxygen status. Under normoxic conditions, a set of enzymes called HIF prolyl hydroxylase domain family proteins (PHDs) hydroxylates two critical proline residues in the HIF-α subunits. On hydroxylation of the critical proline residues, the von-Hippel Lindau (VHL) tumor suppressor E3 ligase recognizes and ubiquitinates HIF-α, leading to rapid proteosomal degradation. In addition to the proline residues, HIF-α subunits contain a critical arginine residue in the C-terminal transactivation domain. Factor inhibiting HIF (FIH) hydroxylates HIF-α at these arginine residues, leading to decreased transcriptional activity via disruption of its binding with transcriptional coactivator p300/CREB-binding protein (CBP). Since both PHD and FIH require oxygen as a substrate, the hydroxylation of HIF-α is impaired under hypoxic conditions. This leads to the stabilization and translocation of HIF-α to the nucleus where it heterodimerizes with HIF-1β. The HIF-α/β heterodimer complex binds to the hypoxia-responsive elements (HRE) located in the promoter region of HIF target genes. Through this mechanism, HIF transcription factors promote the expression of target genes involved in angiogenesis, metabolic adaptation, migration, and suppression of apoptosis [5,6,7] (Figure 1).
1.1.2. HIF Expression in Tumors
The robust growth of tumor cells creates a relatively large distance between a portion of the tumor area and the tumor vasculature. Due to limited diffusion of oxygen from the blood vessels (which has been measured to be around 150 µm [8,9]), some tumor cells are inevitably exposed to hypoxic stress. The resulting limited supply of oxygen to tumor cells leads to the formation of centric regions of necrotic cells, which are frequently observed upon histological examination of human solid tumors [10]. This makes hypoxia one of the most common features within the microenvironment of solid tumors, and hypoxia responsive HIF-1α level is found to be particularly elevated in many human solid tumors including colon, gastric, lung, and prostate cancers [11]. Although hematological malignancies are not considered solid tumors, recent studies revealed the unique hypoxic environment of bone marrow promoting maintenance of hematological cancer stem cells. These studies indicate that hypoxia–HIF axis could play a pivotal role in the development of hematological malignancies and drug resistance as well [12,13,14]. HIF-1α overexpression is also mediated by aberrant activation of oncogenic signaling providing a mechanism for adaptation to hypoxia by tumor cells, in addition to hypoxia-mediated mechanisms. V-Src, but not c-Src, promotes HIF-1 expression and its target gene vascular endothelial growth factor (VEGF) expression [15]. HIF-1 expression is induced by activation of the oncogenic receptor tyrosine kinases, including epidermal growth factor receptor (EGFR), fibroblast growth factor receptor (FGFR), and insulin-like growth factors 1/2 receptor (IGF1/2R) [16]. Of note, HIF-1 is required for the expression of IGF2 and IGF-binding proteins (IGFBP)-2 and -3, suggesting that HIF-1 promotes the autocrine growth factor loop [16].
1.2. Mechanism of Tumor Adaptation to Hypoxia, and Its Implication in Drug Resistance
1.2.1. Hypoxia-Induced Autophagy
Hypoxia-induced autophagy is now recognized as a part of adaptive mechanisms promoting cell survival. Autophagy is an evolutionally conserved cellular process that clears old or damaged cellular components. Although all cells undergo basal autophagy under normal conditions, autophagy is induced by stresses such as nutrient starvation, metabolic stresses, and hypoxia in order to maintain metabolic homeostasis [17]. Since such stresses are common in nutrient deficient solid tumors, autophagy is recognized as a key regulator of cellular viability in cancer cells. The precise role of autophagy in cancer development is still elusive; however, increasing evidence indicates that autophagy is associated with poor outcome in multiple cancers as well as with therapy resistance, indicating the cytoprotective role of autophagy protecting cancer cells [18,19,20,21,22]. Ablation of autophagy augments the efficacy of chemotherapeutic reagents [18,23,24] as well as targeted reagents in imatinib-treated chronic myeloid leukemia [25] and vemurafenib-treated BRAF mutant melanoma [26]. The cytoprotective role of autophagy is presumably mediated by the recycling of ATP and cellular breakdown products to maintain cellular biosynthesis and survival [27]. Given its importance in drug resistance, autophagy could be involved in the failure of cancer therapies associated with hypoxia, and targeting autophagy could therefore be an attractive option to improve therapy outcomes. Like other hypoxic responses, HIF transcription factor plays a key role in hypoxia-induced autophagy induction. Bellot and colleagues reported that the HIF-1α target genes, BNIP3 and BNIP3L, are responsible for the induction of autophagy under hypoxic conditions via disruption of the Bcl-2:Beclin1 complex, and HIF-mediated autophagy is a survival mechanism involved in tumor progression [28]. Attenuation of HIF-1 expression inhibited hypoxia-induced autophagy and potentiated the efficacy of cytotoxic treatment under hypoxia, supporting the probable role of HIF in hypoxia-induced drug resistance [29]. Of note, it has been reported that HIF-1 specifically promotes hypoxia-induced autophagy of mitochondria, namely mitophagy, leading to the downregulation of oxidative phosphorylation during metabolic adaption of cancer cells to hypoxia. Hypoxia-induced mitophagy prevents the accumulation of reactive oxygen species (ROS), thereby promoting survival, via induction of HIF-dependent transcription of the BNIP3 gene [30]. However, this notion needs to be supported by future studies, wherein the following points need to be clarified: (i) the physiological role of hypoxia-induced mitophagy in the context of drug resistance; and (ii) the importance of HIF-induced BNIP3 and/or BNIP3L in hypoxia-induced drug resistance. While physiological hypoxia-induced autophagy is recognized as a survival mechanism, autophagy associated with severe hypoxic condition (O2 level less than 0.1%), namely anoxia, could lead to a different outcome. Anoxic condition is often accompanied by the drastic restriction of nutrients (e.g., amino acids, glucose), leading to inactivation of the mammalian target of rapamycin (mTOR) pathway as a result of AMP-activated protein kinase (AMPK) activation [31]. Impaired mTOR activity is linked to induction of autophagy, and this HIF-independent autophagy induction under severe hypoxia is reportedly associated with autophagic cell death, rather than cytoprotection [32]. Thus, targeting hypoxia should be carefully considered when treating tumors which are likely to be exposed to severe hypoxic condition.
1.2.2. Regulation of Cell Death under Hypoxia
Under hypoxic conditions, the non-adapted cancer cells undergo apoptosis, which provides for the strong selection of cells that survive cancer therapy. Hypoxia induces apoptosis, which is dependent on HIF-1 and p53-dependent mechanisms [33]. HIF-1 reportedly induces transcription of the Bcl2-family proteins, BNIP3 and NIX, promoting apoptosis under hypoxic condition [34,35,36]. BNIP3 is a pro-apoptotic mitochondrial protein through interaction with E1B19K and Bcl2 [37]. Interestingly, BNIP3-mediated cell death is independent of cytochrome C release and caspase activation. Rather, it involves increased plasma and mitochondrial membrane permeability, leading to mitochondrial damage and mitophagy. This is followed by loss of mitochondrial electrochemical potential and increased production of reactive oxygen species (ROS), which is a typical phenotype of aponecrosis [38,39]. However, other studies report that HIF-induced BNIP3 is poorly apoptotic, thereby the proapoptotic role of BNIP3 in hypoxia is still controversial [28,40]. Hypoxia also induces mitochondrial pathway apoptosis via increasing the p53 activity. Under severe hypoxic conditions, where the oxygen levels fall below 0.2%, p53 protein is phosphorylated and accumulated via the ataxia telangiectasia and Rad3-related protein (ATR) kinase signaling pathway [41]. Also, several reports indicate that HIF-1α stabilizes p53 to induce apoptosis [42,43,44,45].
In contrast, other studies indicated that hypoxia promotes anti-apoptotic pathways under certain conditions, such as DNA damage stress. Hypoxia attenuates the expression of pro-apoptotic proteins including Bax and Bid in a HIF-1 independent manner, which contributes to chemoresistance in colon cancer cells [46]. In the context of non-small cell lung cancer (NSCLC), the expression of the anti-apoptotic protein survivin is positively correlated with HIF-1α, and promoter activity for survivin expression is impaired by mutating the HIF-1α binding site, thus indicating that hypoxia promotes survival signaling via HIF-1α-survivin axis [47]. Moreover, the hypoxia protects mammary epithelial cells from anoikis-induced cell death by blocking the expression of pro-apoptotic proteins Bim and Bmf [48]. Survivin is reportedly a mediator of doxorubicin resistance in breast cancer [49], suggesting hypoxia-induced survivin expression could promote chemoresistance in at least some human solid tumors. Here, one importance question is raised; what is the molecular mechanism differentiating apoptosis-resistant and -sensitive cells under hypoxic stress? Dong and colleagues reported that hypoxic induction of the inhibitor of apoptosis protein-2 (IAP-2) promotes the survival of cells under hypoxic stress [50,51], raising the possibility that cells overexpressing IAP family proteins are resistant to hypoxia-induced apoptosis. As IAP family proteins have been implicated in the development of cancer development [52,53], it is worth further investigating the precise role of IAPs in the hypoxic adaptation of cancer cells, and, furthermore, in the development of drug resistance. Overall, the effect of hypoxia and subsequent activation of HIF-1 transcription factor in the determination of cell fate is multifaceted and context dependent (e.g., related to tumor type and oxygen concentration). It is plausible that hypoxia-induced cell death drives the selection of a drug-resistant population within the tumor.
1.3. Strategies to Improve Therapy
1.3.1. Targeting HIF-1 Directly
HIF-1 is a central mediator for the adaptation of cancer cells to hypoxia, via the aforementioned mechanisms. The genetic deletion of HIF-1 in endothelial cells (ECs) disrupts hypoxia-induced EC behavior, leading to the profound inhibition of tumor formation [54]. Several studies have reported that suppression of HIF-1 via RNAi impairs tumor progression. The in vivo repression of HIF-1 using RNAi resulted in tumor regression associated with increased necrosis [55]. Suppression of HIF-1 by RNAi, antisense oligonucleotide, and the dominant negative form of HIF-1 showed anti-tumor effects in various preclinical models including pancreatic cancer, tongue squamous cell carcinoma, and gastric cancer [56,57,58]. Thus, targeting the HIF-1 activation has emerged as an attractive therapeutic strategy to inhibit tumor progression, and to potentially overcome drug resistance. However, although the genetic modulation of HIF-1 expression (e.g., via RNAi) is not yet generally applicable in the clinic, small molecule inhibitors targeting HIF-1 could be an attractive way to overcome the adversity of HIF-1. HIF-1 chemical inhibitors could be divided into two groups: (i) agents modulating HIF-1 transcriptional activity; and (ii) agents modulating the HIF-1 expression. For transcriptional activation, HIF-1 forms a protein complex with transcriptional coactivator p300. Through high-throughput screening, Kung and colleagues identified chetomin as a disruptor of the HIF:p300 binding. Chetomin disrupts the structure of CH1 domain of p300, which is required for its association with HIF-1, thereby leading to the attenuation of hypoxia-inducible transcription and the inhibition of tumor growth in the xenograft prostate cancer model [59]. Echinomycin, a cycle peptide with anti-microbial properties, reportedly binds to the HIF-1 recognition sequence 5′-CGTG-3′, and inhibits the DNA-binding and transcriptional activation of HIF-1 [60]. However, a previous phase II clinical study showed that echinomycin has minimal anti-tumor activity associated with severe side effects, limiting its application to the clinic [61]. It has been reported that several compounds are able to reduce HIF-1 levels. Geldanamycin and its analog 17-AAG are inhibitors of molecular chaperon heat shock protein 90 (Hsp90), which is required for HIF-1 protein stability [62]. Geldanamycin induces the degradation of HIF-1, accompanied by the reduction of HIF-1 transcription activity in kidney and prostate cancer cells [63]. Several clinical trials have evaluated 17-AAG in combination with kinase inhibitor drugs, including Raf inhibitor sorefenib and human epidermal growth factor receptor 2 (HER2) inhibitor trastuzumab, in VHL mutant kidney cancer and HER2-positive breast cancers, respectively [64,65]. Bortezomib, a proteasome inhibitor, is reported to inhibit tumor adaptation to hypoxia via blocking of hypoxic activation of HIF-1 and induction of its target genes, including VEGF and erythropoietin (EPO) [66]. Since inhibition of chaperone or proteasome could be highly non-specific and disrupt normal tissue functions, efforts have been made to discover more specific HIF-1 inhibitors. It is reported that PX-478 suppresses the constitutive and hypoxia-induced levels of HIF-1 in cancer cells, and exhibits an antitumor effect in human tumor xenograft model [67]. NSC-134754 was discovered from a compound screening to inhibit both HIF-1 protein levels and its activity induced by hypoxia, and subsequent induction of HIF-1 target gene expression [68,69].
1.3.2. Targeting HIF-1 Signaling
In addition to targeting HIF-1, another method to reverse the adverse effects of HIF-1 would be: (i) to block signaling pathways leading to HIF-1 accumulation and activation; and (ii) to target the consequence(s) of HIF-1 activation. Studies reported hypoxia-independent induction of HIF driven by activation of kinase signaling, which could provide potential therapeutic strategies to inhibit HIF activation. The aberrant activation of PI3K-AKT-mTOR signaling induces the overexpression of HIF-1 in cancer [70], and the dual PI3K/mTOR inhibitor, NVP-BEZ235, suppresses hypoxia-induced HIF-1 expression and enhances apoptosis of cancer cells under hypoxic stress [71]. Resveratrol, a natural compound known to be anti-tumorigenic, reportedly decreases HIF-1 and its target gene (VEGF) by inhibiting PI3K-AKT and mitogen-activated protein kinase (MAPK) activation [72]. In addition, the Ras inhibitor trans-farnesylthiosalicylic acid (FTS) exhibits a profound antitumor effect in glioblastoma cells by repressing the Ras-signaling induced HIF-1 expression [73]. Fara-A, a nucleotide analog, inhibits HIF-1 expression, and blocks the VEGF transcription via the inhibition of PI3K-AKT signaling in ovarian cancer cells [74]. Moreover, the activation of receptor tyrosine kinase (RTK) is linked to HIF induction, enhancing tumor adaptation to hypoxia. Multiple RTK signaling, which includes VEGFR, platelet-derived growth factor receptor (PDGFR), and EGFR, increase the HIF-1 and HIF-2 expression, and therefore the pharmacological inhibition of these RTKs using tyrosine kinase inhibitors (TKIs) could abrogate hypoxia-induced HIF accumulation in the context of neuroblastoma cells [75]. Interestingly, some studies report that hypoxia can promote oncogenic RTK signaling, indicating a bidirectional crosstalk between oncogenic (RTK) signaling and HIF. Hypoxia-induced HIF promotes mRNA expression of an oncogenic RTK c-Met, amplifies HGF signaling, thereby promoting invasive growth of cancer under hypoxic condition [76]. This suggests c-Met is an important mediator of tumor progression under hypoxia, and this notion was supported by a study reporting that pharmacological inhibition of c-Met abrogates hypoxia-induced invasion in a mouse lung cancer xenograft model [77]. Furthermore, c-MET inhibitor PHA665753 sensitizes gastric cancer cells to radiotherapy [78]. As hypoxia is believed to be one of main drivers of radiotherapy resistance [79], it is reasonable to mention that targeting c-Met could be an attractive way to inhibit tumor progression as well as to overcome radiotherapy resistance. Hypoxia is also linked to overexpression of the ERBB family of RTKs, whose aberrant expression is closely associated with multiple human cancers [80], and with drug resistance to tyrosine kinase inhibitors (TKIs) targeting ERBB proteins. EGFR/ERBB1 expression can be induced in response to hypoxia via the HIF-2α dependent increase of EGFR mRNA translation, providing a non-mutational activation of EGFR expression in human tumors [81]. Thus, in HER2/ERBB2-driven breast cancer, hypoxia-induced HIF-1 promotes HER2 inhibitor lapatinib resistance via inhibiting MAPK phosphatase dual-specificity phosphatase (DUSP2) expression, thereby compensating the loss of MAPK activity in lapatinib-treated cells [82]. These studies indicate that HIF-1 can be induced by aberrant activation of oncogenic RTKs, and hypoxia-induced HIF activation promotes RTK expression, indicating the possible existence of a positive feedback loop. Hence, a combinatory approach that attacks this loop could be effective, especially in cancers driven by oncogenic RTK activation. Finally, VEGF is a well-established HIF-1 target gene associated with tumor progression via the promotion of hypoxia-induced angiogenesis. The efficacy of anti-VEGF monoclonal antibody bevacizumab (Avastin) has been evaluated in multiple clinical studies, showing good clinical effects when combined with chemotherapy in breast, non-small cell lung cancer, renal cell carcinoma, pancreatic cancer, and sarcoma, whereas the effect of monotherapy is marginal [83,84,85].
1.3.3. Targeting Hypoxia-Induced Autophagy
Given the importance of autophagy in tumor adaption to hypoxia and its implication in hypoxia-associated drug resistance, targeting tumor specific autophagy would be an attractive way to improve cancer therapy. The key step in autophagy is the fusion of autophagosomes with acidic lysosome, where the autophagosome is degraded and recycled back to the cytosol [86]. The increased glycolysis in a hypoxic tumor results in tumor acidity, which is closely linked to tumor progression and drug resistance [87,88]. Hence, acidic organelles such as lysosomes play a key role in processes under hypoxia, and agents that inhibit endosomal acidification, including an antimalarial drug chloroquine and proton pump inhibitors (PPIs), are proposed as possible anticancer strategies. A known PPI, pantoprazole, inhibits autophagy possibly through inhibiting the acidification of endosomes and then autophagosome fusion [89]. Tan and colleagues reported that autophagy is a mechanism of docetaxel resistance, and inhibiting autophagy by pantoprazole enhanced the efficacy of docetaxel [90,91]. Several other studies indicate that PPIs such as omeprazole and esomeprazole also overcome chemoresistance via inhibition of autophagy [92,93,94].
1.3.4. Targeting Hypoxia-HIF to Improve Immunotherapy
Recent evidence indicates that chronic tumor hypoxia is linked to tumor maintenance via the suppression of T lymphocytes, thus targeting hypoxia-HIF axis has emerged as a novel therapeutic approach to improve immunotherapy. HIF-dependent expression of CD39/CD73 ectoenzymes is responsible for the accumulation of extracellular adenosine in tumor microenvironment. The resulting activation of adenosine receptors (A2AR/A2BR) elevates cAMP level in T cells leading to inhibition of anti-tumor T cell function, providing tumor permissive tumor microenvironment [95,96]. Hatfield and colleagues showed that exposing tumor bearing mice to hyperoxia (60% oxygen) decreased intratumoral hypoxia and concentration of extracellular adenosine via decreasing CD39/CD73 expression in tumor, reversing hypoxia-adenosinergic immunosuppression [97,98]. These studies indicate that the suppression of HIF signaling could target cancer cells as well as relieve hypoxia-mediated immunosuppressive mechanisms (Figure 2).
2. Summary
Despite recent advances in anti-cancer therapies, cancer still remains the leading cause of death worldwide. Drug resistance is the primary cause for cancer therapy failure, and its underlying mechanisms involve pharmacokinetic resistance, tumor cell intrinsic resistance, and factors associated with tumor microenvironment. The relatively overlooked importance of microenvironment-induced drug resistance has recently been recognized. Hypoxia is a common feature of the microenvironment of many solid tumors and hematological malignancies, and increasing evidence indicates that it promotes tumorigenesis, and, furthermore, confers drug resistance by altering the tumor cell physiology in regard to the reduction of apoptotic potential, induction of cytoprotective autophagy, and immunosuppressive tumor microenvironment. Since the HIF-1 transcription factor is a central player in the hypoxic adaption of tumor cells, therapeutic strategies targeting HIF-1 itself or signaling pathways up and downstream of HIF are garnering significant attention in the effort to overcome hypoxia-induced drug resistance. Targeting tumor specific autophagy and receptor tyrosine kinase activation associated with hypoxia/HIF could be another attractive therapeutic option, however, challenges still remain. First, drugs targeting HIF lack specificity (e.g., HSP90 inhibitor) or mechanism of action, limiting clinical application of those drugs. Chemical library screenings could lead to the discovery of more specific HIF inhibitors in the near future. Second, targeting the kinase signaling network associated with HIF could lead to acquired drug resistance, allowing reactivation of HIF. The occurrence of this bypass signaling is a common feature in kinase inhibitor therapy, thus rational drug combination to abrogate cancer cells’ adaptive responses should also be considered. Third, it is important to note that the degree and significance of hypoxic adaptation to cancer therapy varies for each patient. Thus, in the era of personalized medicine, tumors need to be screened for hypoxia markers such as HIF-1 and biochemical markers for autophagy in order to achieve more specific targeted therapies to overcome hypoxia-mediated drug resistance. Overall, given the important role of hypoxia-induced drug resistance, therapeutic strategies hijacking hypoxia adaptation should be considered in combination with conventional cancer therapies.
Acknowledgments
This work is supported by research fund of Chungnam National University in 2017 to Jae-Young Kim, and 2012M3A9C6050087, 2016M3A9E1918329, and 2015R1D1A1A01058257, Chungnam National University to Joo-Yong Lee.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Brown, L.M.; Cowen, R.L.; Debray, C.; Eustace, A.; Erler, J.T.; Sheppard, F.C.; Parker, C.A.; Stratford, I.J.; Williams, K.J. Reversing hypoxic cell chemoresistance in vitro using genetic and small molecule approaches targeting hypoxia inducible factor-1. Mol. Pharmacol. 2006, 69, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A. Hypoxia and drug resistance. Cancer Metastasis Rev. 1994, 13, 139–168. [Google Scholar] [CrossRef] [PubMed]
- Tatum, J.L.; Kelloff, G.J.; Gillies, R.J.; Arbeit, J.M.; Brown, J.M.; Chao, K.S.; Chapman, J.D.; Eckelman, W.C.; Fyles, A.W.; Giaccia, A.J.; et al. Hypoxia: Importance in tumor biology, noninvasive measurement by imaging, and value of its measurement in the management of cancer therapy. Inte. J. Radiat. Biol. 2006, 82, 699–757. [Google Scholar] [CrossRef] [PubMed]
- Wiesener, M.S.; Jurgensen, J.S.; Rosenberger, C.; Scholze, C.K.; Horstrup, J.H.; Warnecke, C.; Mandriota, S.; Bechmann, I.; Frei, U.A.; Pugh, C.W.; et al. Widespread hypoxia-inducible expression of HIF-2α in distinct cell populations of different organs. FASEB J. 2003, 17, 271–273. [Google Scholar] [PubMed]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol.Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; Hahnfeldt, P.; Hlatky, L. Cancer: Looking outside the genome. Nat. Rev. Mol. Cell Biol. 2000, 1, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin. Radiat. Oncol. 2004, 14, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Brahimi-Horn, M.C.; Chiche, J.; Pouyssegur, J. Hypoxia and cancer. J. Mol. Med. 2007, 85, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; de Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar] [PubMed]
- Irigoyen, M.; Garcia-Ruiz, J.C.; Berra, E. The hypoxia signalling pathway in haematological malignancies. Oncotarget 2017, 8, 36832–36844. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Thall, P.F.; Yi, C.A.; Borthakur, G.; Coveler, A.; Bueso-Ramos, C.; Benito, J.; Konoplev, S.; Gu, Y.; Ravandi, F.; et al. Phase I/II study of the hypoxia-activated prodrug PR104 in refractory/relapsed acute myeloid leukemia and acute lymphoblastic leukemia. Haematologica 2015, 100, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.O.; Mortensen, B.T.; Hodgkiss, R.J.; Iversen, P.O.; Christensen, I.J.; Helledie, N.; Larsen, J.K. Increased cellular hypoxia and reduced proliferation of both normal and leukaemic cells during progression of acute myeloid leukaemia in rats. Cell Prolif. 2000, 33, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.H.; Agani, F.; Passaniti, A.; Semenza, G.L. V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: Involvement of HIF-1 in tumor progression. Cancer Res. 1997, 57, 5328–5335. [Google Scholar] [PubMed]
- Feldser, D.; Agani, F.; Iyer, N.V.; Pak, B.; Ferreira, G.; Semenza, G.L. Reciprocal positive regulation of hypoxia-inducible factor 1α and insulin-like growth factor 2. Cancer Res. 1999, 59, 3915–3918. [Google Scholar] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, N.; Faried, A.; Tsutsumi, S.; Takeuchi, T.; Kuwano, H. Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann. Surg. Oncol. 2009, 16, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Karpathiou, G.; Sivridis, E.; Koukourakis, M.I.; Mikroulis, D.; Bouros, D.; Froudarakis, M.E.; Giatromanolaki, A. Light-chain 3A autophagic activity and prognostic significance in non-small cell lung carcinomas. Chest 2011, 140, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Sivridis, E.; Koukourakis, M.I.; Mendrinos, S.E.; Karpouzis, A.; Fiska, A.; Kouskoukis, C.; Giatromanolaki, A. Beclin-1 and LC3A expression in cutaneous malignant melanomas: A biphasic survival pattern for beclin-1. Melanoma Res. 2011, 21, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Thomas, S.; Golden, E.B.; Hofman, F.M.; Chen, T.C.; Petasis, N.A.; Schonthal, A.H.; Louie, S.G. Inhibition of autophagy and induction of breast cancer cell death by mefloquine, an antimalarial agent. Cancer Lett. 2012, 326, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Rehman, S.K.; Zhang, W.; Wen, A.; Yao, L.; Zhang, J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim. Biophys. Acta 2010, 1806, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, S.M.; Moretti, L.; Varki, V.; Lu, B. Progress in the unraveling of the endoplasmic reticulum stress/autophagy pathway and cancer: Implications for future therapeutic approaches. Drug Resist. Updates 2010, 13, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, Y.; Liu, Q.; Wang, J. Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells. Med. Oncol. 2011, 28, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R.; et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in philadelphia chromosome-positive cells, including primary cml stem cells. J. Clin. Investig. 2009, 119, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting er stress-induced autophagy overcomes braf inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.W.; Su, Y.; Zhu, H.; Cao, J.; Ding, W.J.; Zhao, Y.C.; He, Q.J.; Yang, B. HIF-1α-dependent autophagy protects Hela cells from fenretinide (4-HPR)-induced apoptosis in hypoxia. Pharmacol. Res. 2010, 62, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol.Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Sakamoto, K. Ampk: A key sensor of fuel and energy status in skeletal muscle. Physiology 2006, 21, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Mazure, N.M.; Pouyssegur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Dor, Y.; Herbert, J.M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 9082–9087. [Google Scholar] [CrossRef] [PubMed]
- Kilic, M.; Kasperczyk, H.; Fulda, S.; Debatin, K.M. Role of hypoxia inducible factor-1α in modulation of apoptosis resistance. Oncogene 2007, 26, 2027–2038. [Google Scholar] [CrossRef] [PubMed]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar] [PubMed]
- Boyd, J.M.; Malstrom, S.; Subramanian, T.; Venkatesh, L.K.; Schaeper, U.; Elangovan, B.; D′Sa-Eipper, C.; Chinnadurai, G. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell 1994, 79, 341–351. [Google Scholar] [CrossRef]
- Vande Velde, C.; Cizeau, J.; Dubik, D.; Alimonti, J.; Brown, T.; Israels, S.; Hakem, R.; Greenberg, A.H. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol. Cell. Biol. 2000, 20, 5454–5468. [Google Scholar] [CrossRef] [PubMed]
- Formigli, L.; Papucci, L.; Tani, A.; Schiavone, N.; Tempestini, A.; Orlandini, G.E.; Capaccioli, S.; Orlandini, S.Z. Aponecrosis: Morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J. Cell. Physiol. 2000, 182, 41–49. [Google Scholar] [CrossRef]
- Papandreou, I.; Krishna, C.; Kaper, F.; Cai, D.; Giaccia, A.J.; Denko, N.C. Anoxia is necessary for tumor cell toxicity caused by a low-oxygen environment. Cancer Res. 2005, 65, 3171–3178. [Google Scholar] [CrossRef] [PubMed]
- Hammond, E.M.; Denko, N.C.; Dorie, M.J.; Abraham, R.T.; Giaccia, A.J. Hypoxia links atr and p53 through replication arrest. Mol. Cell. Biol. 2002, 22, 1834–1843. [Google Scholar] [CrossRef] [PubMed]
- Hammond, E.M.; Giaccia, A.J. The role of p53 in hypoxia-induced apoptosis. Biochem. Biophys. Res. Commun. 2005, 331, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Tomida, A.; Tsuruo, T. Dephosphorylated hypoxia-inducible factor 1α as a mediator of p53-dependent apoptosis during hypoxia. Oncogene 2001, 20, 5779–5788. [Google Scholar] [CrossRef] [PubMed]
- An, W.G.; Kanekal, M.; Simon, M.C.; Maltepe, E.; Blagosklonny, M.V.; Neckers, L.M. Stabilization of wild-type p53 by hypoxia-inducible factor 1α. Nature 1998, 392, 405–408. [Google Scholar] [PubMed]
- Blagosklonny, M.V.; An, W.G.; Romanova, L.Y.; Trepel, J.; Fojo, T.; Neckers, L. p53 inhibits hypoxia-inducible factor-stimulated transcription. J. Biol. Chem. 1998, 273, 11995–11998. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Cawthorne, C.J.; Williams, K.J.; Koritzinsky, M.; Wouters, B.G.; Wilson, C.; Miller, C.; Demonacos, C.; Stratford, I.J.; Dive, C. Hypoxia-mediated down-regulation of bid and bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol. Cell. Biol. 2004, 24, 2875–2889. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Q.; Zhao, C.L.; Li, W. Effect of hypoxia-inducible factor-1α on transcription of survivin in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2009, 28, 29. [Google Scholar] [CrossRef] [PubMed]
- Whelan, K.A.; Caldwell, S.A.; Shahriari, K.S.; Jackson, S.R.; Franchetti, L.D.; Johannes, G.J.; Reginato, M.J. Hypoxia suppression of Bim and Bmf blocks anoikis and luminal clearing during mammary morphogenesis. Mol. Biol. Cell 2010, 21, 3829–3837. [Google Scholar] [CrossRef] [PubMed]
- Faversani, A.; Vaira, V.; Moro, G.P.; Tosi, D.; Lopergolo, A.; Schultz, D.C.; Rivadeneira, D.; Altieri, D.C.; Bosari, S. Survivin family proteins as novel molecular determinants of doxorubicin resistance in organotypic human breast tumors. Breast Cancer Res. 2014, 16, R55. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Venkatachalam, M.A.; Wang, J.; Patel, Y.; Saikumar, P.; Semenza, G.L.; Force, T.; Nishiyama, J. Up-regulation of apoptosis inhibitory protein IAP-2 by hypoxia HIF-1-independent mechanisms. J. Biol. Chem. 2001, 276, 18702–18709. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Nishiyama, J.; Yi, X.; Venkatachalam, M.A.; Denton, M.; Gu, S.; Li, S.; Qiang, M. Gene promoter of apoptosis inhibitory protein IAP2: Identification of enhancer elements and activation by severe hypoxia. Biochem. J. 2002, 364, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, G.S.; Duckett, C.S. Iap proteins: Blocking the road to death′s door. Nat.Rev. Mol. Cell Biol. 2002, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- LaCasse, E.C.; Baird, S.; Korneluk, R.G.; MacKenzie, A.E. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene 1998, 17, 3247–3259. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Wang, L.; Esko, J.; Giordano, F.J.; Huang, Y.; Gerber, H.P.; Ferrara, N.; Johnson, R.S. Loss of HIF-1α in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell 2004, 6, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lin, X.; Staver, M.; Shoemaker, A.; Semizarov, D.; Fesik, S.W.; Shen, Y. Evaluating hypoxia-inducible factor-1α as a cancer therapeutic target via inducible RNA interference in vivo. Cancer Res. 2005, 65, 7249–7258. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Nagao, M.; Yamada, Y.; Narikiyo, M.; Ueno, M.; Miyagishi, M.; Taira, K.; Nakajima, Y. Small interfering RNA expression vector targeting hypoxia-inducible factor 1α inhibits tumor growth in hepatobiliary and pancreatic cancers. Cancer Gene Ther. 2006, 13, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, Z.F.; Rao, J.Y.; Sato, J.D.; Brown, J.; Messadi, D.V.; Le, A.D. Treatment with siRNA and antisense oligonucleotides targeted to HIF-1α induced apoptosis in human tongue squamous cell carcinomas. Int. J. Cancer 2004, 111, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Stoeltzing, O.; McCarty, M.F.; Wey, J.S.; Fan, F.; Liu, W.; Belcheva, A.; Bucana, C.D.; Semenza, G.L.; Ellis, L.M. Role of hypoxia-inducible factor 1α in gastric cancer cell growth, angiogenesis, and vessel maturation. J. Natl. Cancer Inst. 2004, 96, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Kung, A.L.; Zabludoff, S.D.; France, D.S.; Freedman, S.J.; Tanner, E.A.; Vieira, A.; Cornell-Kennon, S.; Lee, J.; Wang, B.; Wang, J.; et al. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell 2004, 6, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Park, E.J.; Stephen, A.G.; Calvani, M.; Cardellina, J.H.; Monks, A.; Fisher, R.J.; Shoemaker, R.H.; Melillo, G. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005, 65, 9047–9055. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J.; Vogelzang, N.J.; Kilton, L.J.; Leibach, S.J.; Rademaker, A.W.; French, S.; Benson, A.B. A phase ii clinical trial of echinomycin in metastatic soft tissue sarcoma. Investig. New Drugs 1995, 13, 171–174. [Google Scholar] [CrossRef]
- Minet, E.; Mottet, D.; Michel, G.; Roland, I.; Raes, M.; Remacle, J.; Michiels, C. Hypoxia-induced activation of HIF-1: Role of HIF-1α-Hsp90 interaction. FEBS Lett. 1999, 460, 251–256. [Google Scholar] [CrossRef]
- Mabjeesh, N.J.; Post, D.E.; Willard, M.T.; Kaur, B.; Van Meir, E.G.; Simons, J.W.; Zhong, H. Geldanamycin induces degradation of hypoxia-inducible factor 1α protein via the proteosome pathway in prostate cancer cells. Cancer Res. 2002, 62, 2478–2482. [Google Scholar] [PubMed]
- Vaishampayan, U.N.; Burger, A.M.; Sausville, E.A.; Heilbrun, L.K.; Li, J.; Horiba, M.N.; Egorin, M.J.; Ivy, P.; Pacey, S.; Lorusso, P.M. Safety, efficacy, pharmacokinetics, and pharmacodynamics of the combination of sorafenib and tanespimycin. Clin. Cancer Res. 2010, 16, 3795–3804. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Stopeck, A.; Linden, H.; Solit, D.; Chandarlapaty, S.; Rosen, N.; D′Andrea, G.; Dickler, M.; Moynahan, M.E.; Sugarman, S.; et al. Hsp90 inhibition is effective in breast cancer: A phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin. Cancer Res. 2011, 17, 5132–5139. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Chun, Y.S.; Lee, D.S.; Huang, L.E.; Park, J.W. Bortezomib inhibits tumor adaptation to hypoxia by stimulating the FIH-mediated repression of hypoxia-inducible factor-1. Blood 2008, 111, 3131–3136. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.; Williams, R.; Kirkpatrick, L.; Paine-Murrieta, G.; Powis, G. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1α. Mol. Cancer Ther. 2004, 3, 233–244. [Google Scholar] [PubMed]
- Chau, N.M.; Rogers, P.; Aherne, W.; Carroll, V.; Collins, I.; McDonald, E.; Workman, P.; Ashcroft, M. Identification of novel small molecule inhibitors of hypoxia-inducible factor-1 that differentially block hypoxia-inducible factor-1 activity and hypoxia-inducible factor-1α induction in response to hypoxic stress and growth factors. Cancer Res. 2005, 65, 4918–4928. [Google Scholar] [CrossRef] [PubMed]
- Carroll, V.A.; Ashcroft, M. Role of hypoxia-inducible factor (HIF)-1α versus HIF-2α in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-i, or loss of von hippel-lindau function: Implications for targeting the hif pathway. Cancer Res. 2006, 66, 6264–6270. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Karar, J.; Cerniglia, G.J.; Lindsten, T.; Koumenis, C.; Maity, A. Dual PI3K/mtor inhibitor NVP-BEZ235 suppresses hypoxia-inducible factor (HIF)-1α expression by blocking protein translation and increases cell death under hypoxia. Cancer Biol. Ther. 2012, 13, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Fang, J.; Xia, C.; Shi, X.; Jiang, B.H. Trans-3,4,5′-trihydroxystibene inhibits hypoxia-inducible factor 1α and vascular endothelial growth factor expression in human ovarian cancer cells. Clin. Cancer Res. 2004, 10, 5253–5263. [Google Scholar] [CrossRef] [PubMed]
- Blum, R.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Kloog, Y. Ras inhibition in glioblastoma down-regulates hypoxia-inducible factor-1α, causing glycolysis shutdown and cell death. Cancer Res. 2005, 65, 999–1006. [Google Scholar] [PubMed]
- Fang, J.; Cao, Z.; Chen, Y.C.; Reed, E.; Jiang, B.H. 9-β-d-Arabinofuranosyl-2-fluoroadenine inhibits expression of vascular endothelial growth factor through hypoxia-inducible factor-1 in human ovarian cancer cells. Mol. Pharmacol. 2004, 66, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.B.; Zage, P.E.; Zeng, L.; Xu, L.; Cascone, T.; Wu, H.K.; Saigal, B.; Zweidler-McKay, P.A.; Heymach, J.V. Multiple receptor tyrosine kinases regulate HIF-1α and HIF-2α in normoxia and hypoxia in neuroblastoma: Implications for antiangiogenic mechanisms of multikinase inhibitors. Oncogene 2010, 29, 2938–2949. [Google Scholar] [CrossRef] [PubMed]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef]
- Puri, N.; Khramtsov, A.; Ahmed, S.; Nallasura, V.; Hetzel, J.T.; Jagadeeswaran, R.; Karczmar, G.; Salgia, R. A selective small molecule inhibitor of c-MET, PHA665752, inhibits tumorigenicity and angiogenesis in mouse lung cancer xenografts. Cancer Res. 2007, 67, 3529–3534. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, Y.; Mukohara, T.; Tomioka, H.; Ekyalongo, R.C.; Kataoka, Y.; Inui, Y.; Kawamori, Y.; Toyoda, M.; Kiyota, N.; Fujiwara, Y.; et al. Excessive MET signaling causes acquired resistance and addiction to MET inhibitors in the MKN45 gastric cancer cell line. Investig. New Drugs 2013, 31, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Paget, J.T.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. Erbb receptors and cancer: The complexity of targeted inhibitors. Nature Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Franovic, A.; Gunaratnam, L.; Smith, K.; Robert, I.; Patten, D.; Lee, S. Translational up-regulation of the EGFR by tumor hypoxia provides a nonmutational explanation for its overexpression in human cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 13092–13097. [Google Scholar] [CrossRef] [PubMed]
- Karakashev, S.V.; Reginato, M.J. Hypoxia/HIF1A induces lapatinib resistance in ERBB2-positive breast cancer cells via regulation of DUSP2. Oncotarget 2015, 6, 1967–1980. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.I.; Fehrenbacher, L.; Hainsworth, J.D.; Heim, W.; Berlin, J.; Holmgren, E.; Hambleton, J.; Novotny, W.F.; Kabbinavar, F. Bevacizumab in combination with fluorouracil and leucovorin: An active regimen for first-line metastatic colorectal cancer. J. Clin. Oncol. 2005, 23, 3502–3508. [Google Scholar] [CrossRef] [PubMed]
- Shih, T.; Lindley, C. Bevacizumab: An angiogenesis inhibitor for the treatment of solid malignancies. Clin. Ther. 2006, 28, 1779–1802. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, G.; Rangaswami, H.; Jain, S.; Kundu, G.C. Hypoxia regulates cross-talk between Syk and Lck leading to breast cancer progression and angiogenesis. J. Biol. Chem. 2006, 281, 11322–11331. [Google Scholar] [CrossRef] [PubMed]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Subarsky, P.; Hill, R.P. The hypoxic tumour microenvironment and metastatic progression. Clin. Exp. Metastasis 2003, 20, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Udelnow, A.; Kreyes, A.; Ellinger, S.; Landfester, K.; Walther, P.; Klapperstueck, T.; Wohlrab, J.; Henne-Bruns, D.; Knippschild, U.; Wurl, P. Omeprazole inhibits proliferation and modulates autophagy in pancreatic cancer cells. PLoS ONE 2011, 6, e20143. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Joshua, A.M.; Saggar, J.K.; Yu, M.; Wang, M.; Kanga, N.; Zhang, J.Y.; Chen, X.; Wouters, B.G.; Tannock, I.F. Effect of pantoprazole to enhance activity of docetaxel against human tumour xenografts by inhibiting autophagy. Br. J. Cancer 2015, 112, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Saggar, J.K.; Yu, M.; Wang, M.; Tannock, I.F. Mechanisms of drug resistance related to the microenvironment of solid tumors and possible strategies to inhibit them. Cancer J. 2015, 21, 254–262. [Google Scholar] [CrossRef] [PubMed]
- De Milito, A.; Iessi, E.; Logozzi, M.; Lozupone, F.; Spada, M.; Marino, M.L.; Federici, C.; Perdicchio, M.; Matarrese, P.; Lugini, L.; et al. Proton pump inhibitors induce apoptosis of human B-cell tumors through a caspase-independent mechanism involving reactive oxygen species. Cancer Res. 2007, 67, 5408–5417. [Google Scholar] [CrossRef] [PubMed]
- Yeo, M.; Kim, D.K.; Kim, Y.B.; Oh, T.Y.; Lee, J.E.; Cho, S.W.; Kim, H.C.; Hahm, K.B. Selective induction of apoptosis with proton pump inhibitor in gastric cancer cells. Clin. Cancer Res. 2004, 10, 8687–8696. [Google Scholar] [CrossRef] [PubMed]
- De Milito, A.; Canese, R.; Marino, M.L.; Borghi, M.; Iero, M.; Villa, A.; Venturi, G.; Lozupone, F.; Iessi, E.; Logozzi, M.; et al. Ph-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. Int. J. Cancer 2010, 127, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Mittal, D.; Stagg, J.; Smyth, M.J. Targeting cancer-derived adenosine: New therapeutic approaches. Cancer Discov. 2014, 4, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Page, D.B.; Wolchok, J.D. Immune modulation for cancer therapy. Br. J. Cancer 2014, 111, 2214–2219. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Belikoff, B.; Schreiber, T.H.; Sethumadhavan, S.; Abbott, R.; Philbrook, P.; Thayer, M.; Shujia, D.; et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1α-dependent and extracellular adenosine-mediated tumor protection. J. Mol. Med. 2014, 92, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
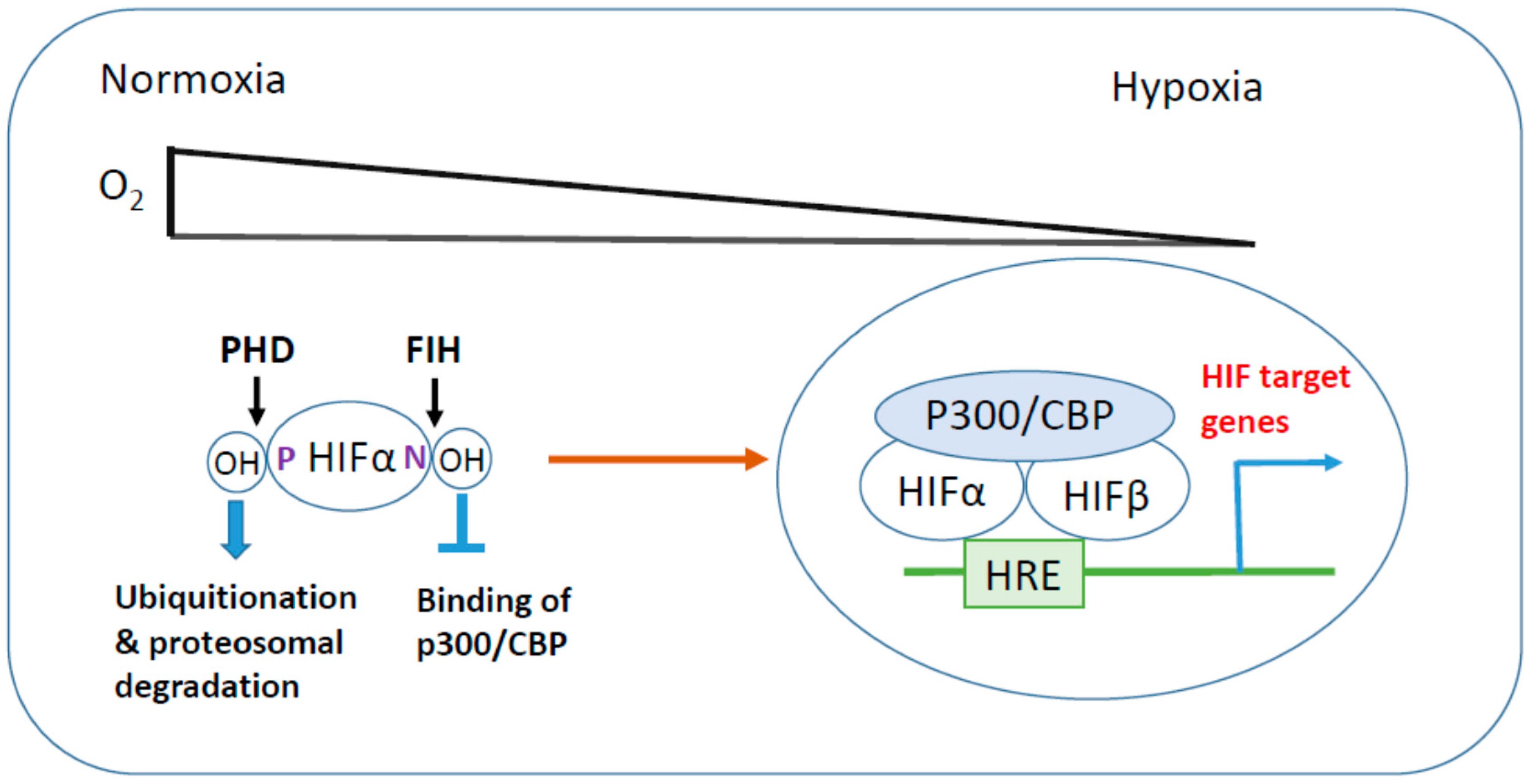
Figure 1.
Hypoxia-inducible factor (HIF) activation in hypoxic stress. Under normoxic conditions, HIF-α is hydroxylated at its critical proline (P) and arginine (N) residues by prolyl hydroxylase domain (PHD) and factor inhibiting HIF (FIH), respectively, leading to proteosomal degradation and suppression of transcriptional activity. In response to hypoxic stress, inhibitory hydroxylations of HIF-α are reduced, then HIF-α is stabilized and translocates to the nucleus where it heterodimerizes with HIF-β. HIF-α/β dimer associates with transcriptional coactivator p300/CBP and binds to hypoxia response element (HRE) to induce HIF target gene expression.
Figure 1.
Hypoxia-inducible factor (HIF) activation in hypoxic stress. Under normoxic conditions, HIF-α is hydroxylated at its critical proline (P) and arginine (N) residues by prolyl hydroxylase domain (PHD) and factor inhibiting HIF (FIH), respectively, leading to proteosomal degradation and suppression of transcriptional activity. In response to hypoxic stress, inhibitory hydroxylations of HIF-α are reduced, then HIF-α is stabilized and translocates to the nucleus where it heterodimerizes with HIF-β. HIF-α/β dimer associates with transcriptional coactivator p300/CBP and binds to hypoxia response element (HRE) to induce HIF target gene expression.

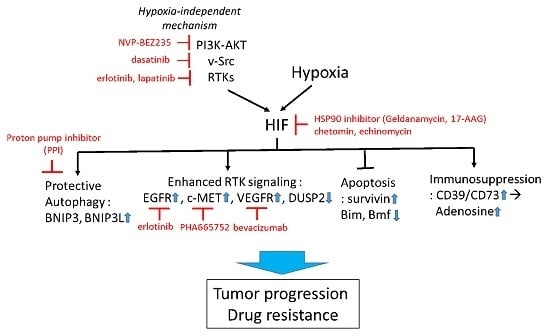
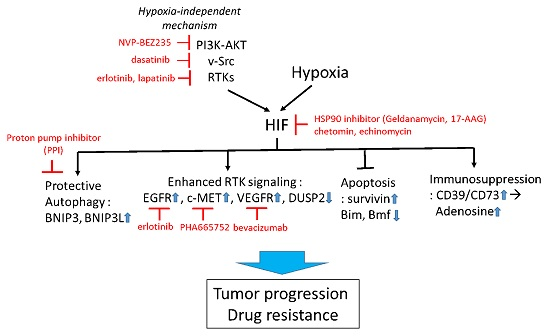
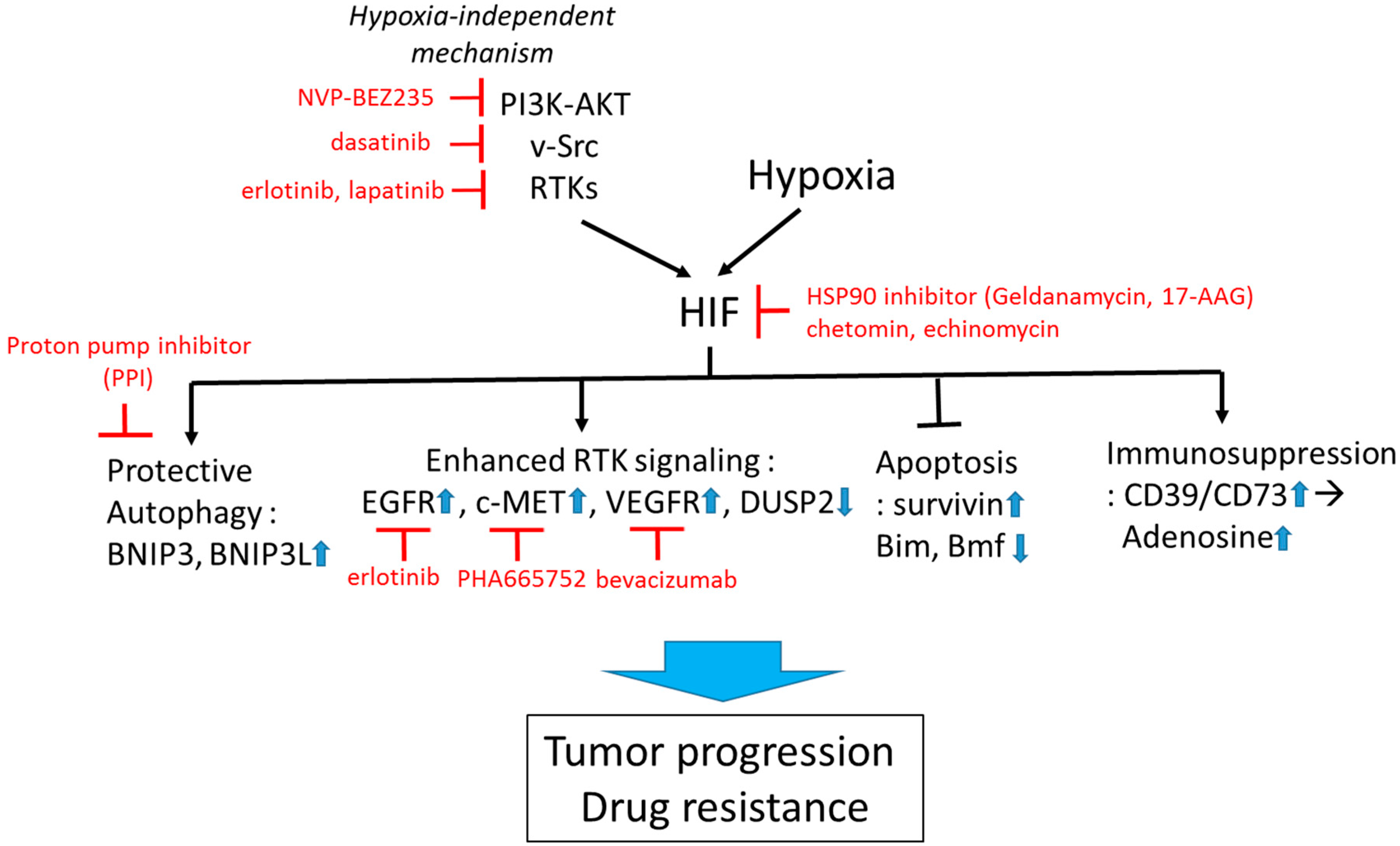
Figure 2.
Targeting HIF to overcome hypoxia-associated drug resistance. HIF can be induced by both hypoxia-dependent and -independent mechanisms. Active HIF in tumor cells promotes drug resistance via upregulation of cytoprotective autophagy, receptor tyrosine kinase (RTK) signaling, suppression of apoptosis, and promotion of immunosuppressive tumor microenvironment. Examples of pharmacological approaches targeting HIF-regulating and -regulated mechanisms are shown in red. Small blue arrows indicate the direction of changes of protein abundance or activity after HIF activation.
Figure 2.
Targeting HIF to overcome hypoxia-associated drug resistance. HIF can be induced by both hypoxia-dependent and -independent mechanisms. Active HIF in tumor cells promotes drug resistance via upregulation of cytoprotective autophagy, receptor tyrosine kinase (RTK) signaling, suppression of apoptosis, and promotion of immunosuppressive tumor microenvironment. Examples of pharmacological approaches targeting HIF-regulating and -regulated mechanisms are shown in red. Small blue arrows indicate the direction of changes of protein abundance or activity after HIF activation.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kim, J.-Y.; Lee, J.-Y. Targeting Tumor Adaption to Chronic Hypoxia: Implications for Drug Resistance, and How It Can Be Overcome. Int. J. Mol. Sci. 2017, 18, 1854. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18091854
AMA Style
Kim J-Y, Lee J-Y. Targeting Tumor Adaption to Chronic Hypoxia: Implications for Drug Resistance, and How It Can Be Overcome. International Journal of Molecular Sciences. 2017; 18(9):1854. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18091854
Chicago/Turabian StyleKim, Jae-Young, and Joo-Yong Lee. 2017. "Targeting Tumor Adaption to Chronic Hypoxia: Implications for Drug Resistance, and How It Can Be Overcome" International Journal of Molecular Sciences 18, no. 9: 1854. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18091854
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.