Protective Effects of Protocatechuic Acid on Seizure-Induced Neuronal Death

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
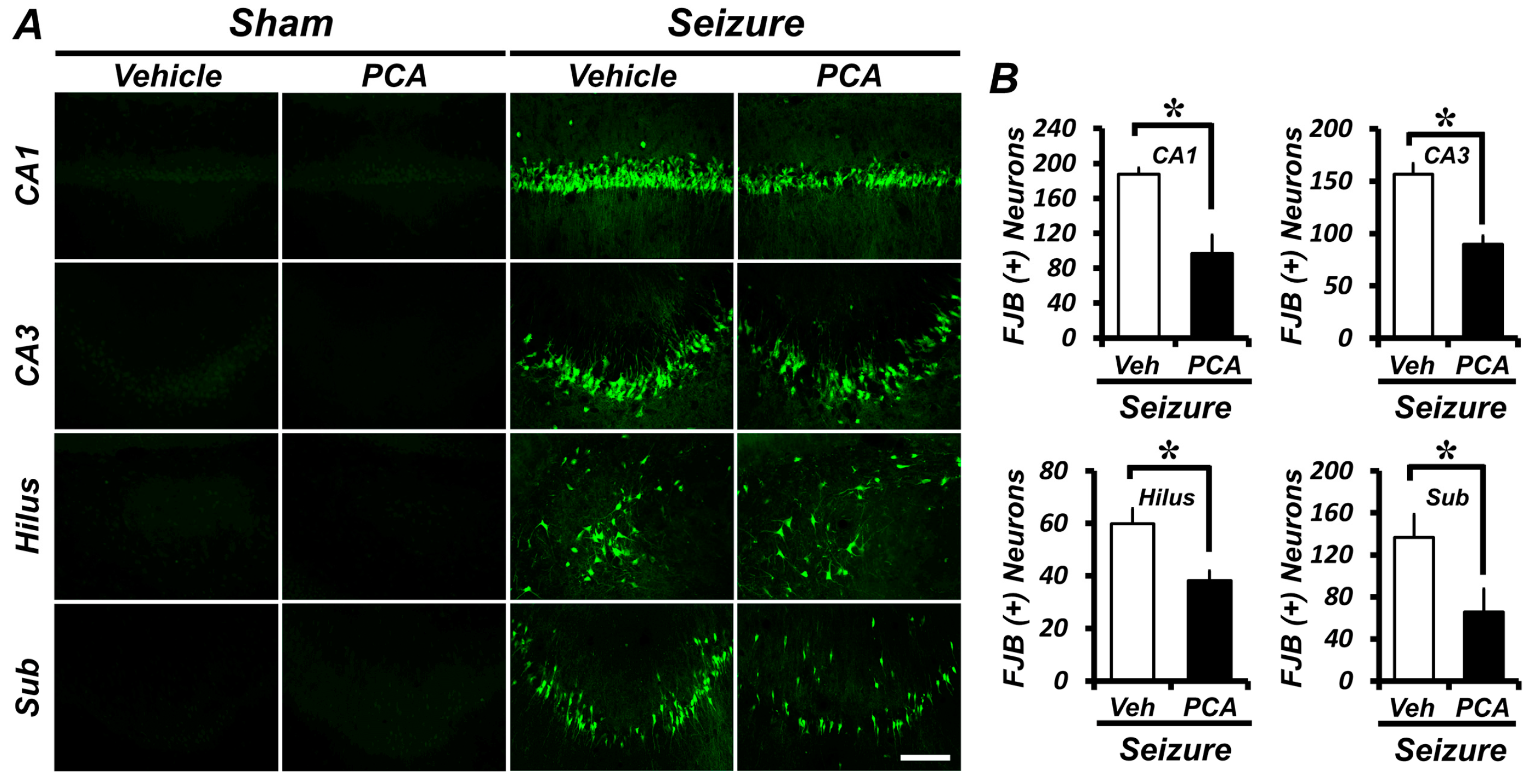
2.1. Protocatechuic Acid (PCA) Treatment Reduces Seizure-Induced Hippocampal Neuronal Death
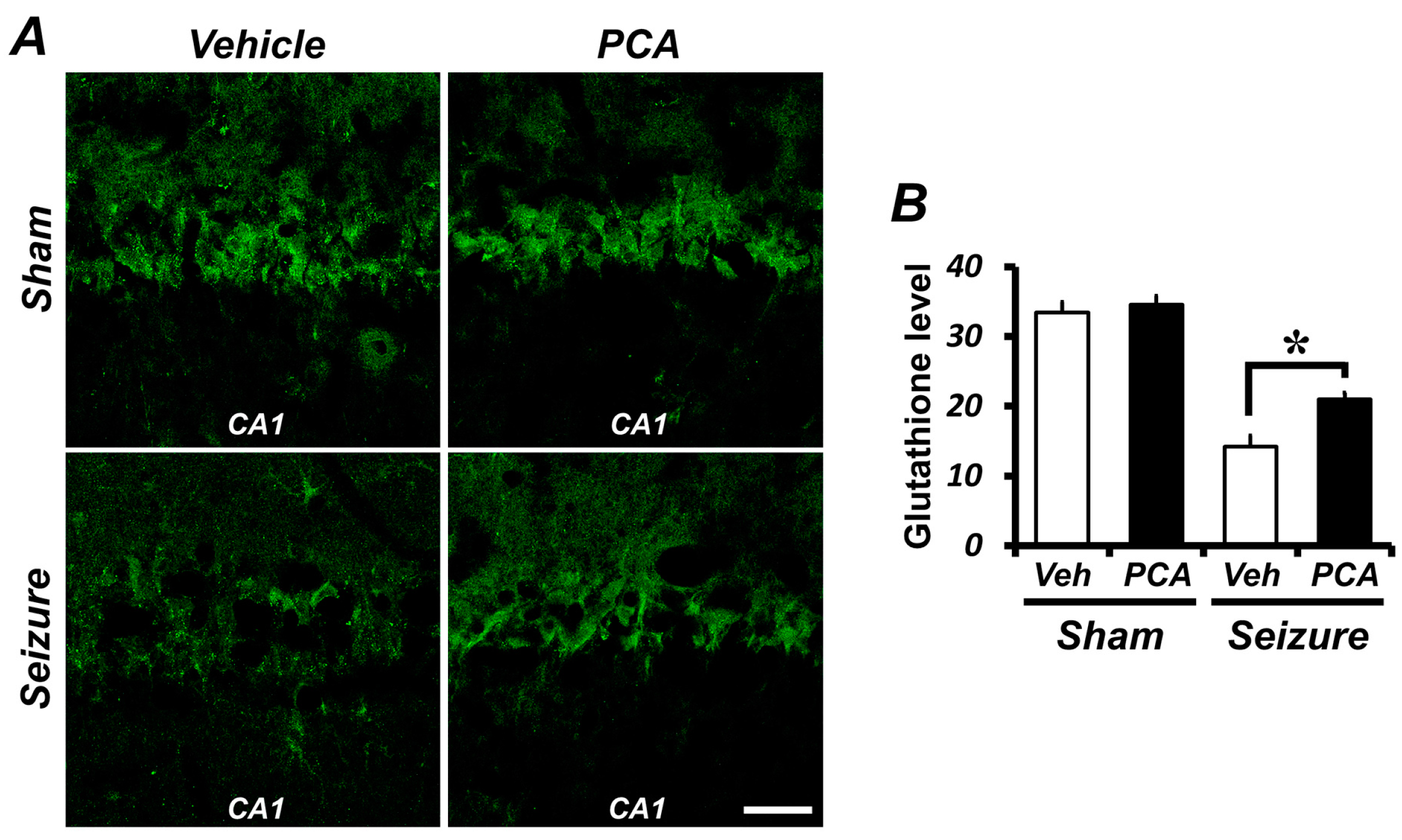
2.2. PCA Treatment Preserves Neuronal Glutathione (GSH) Loss in Hippocampal Neurons
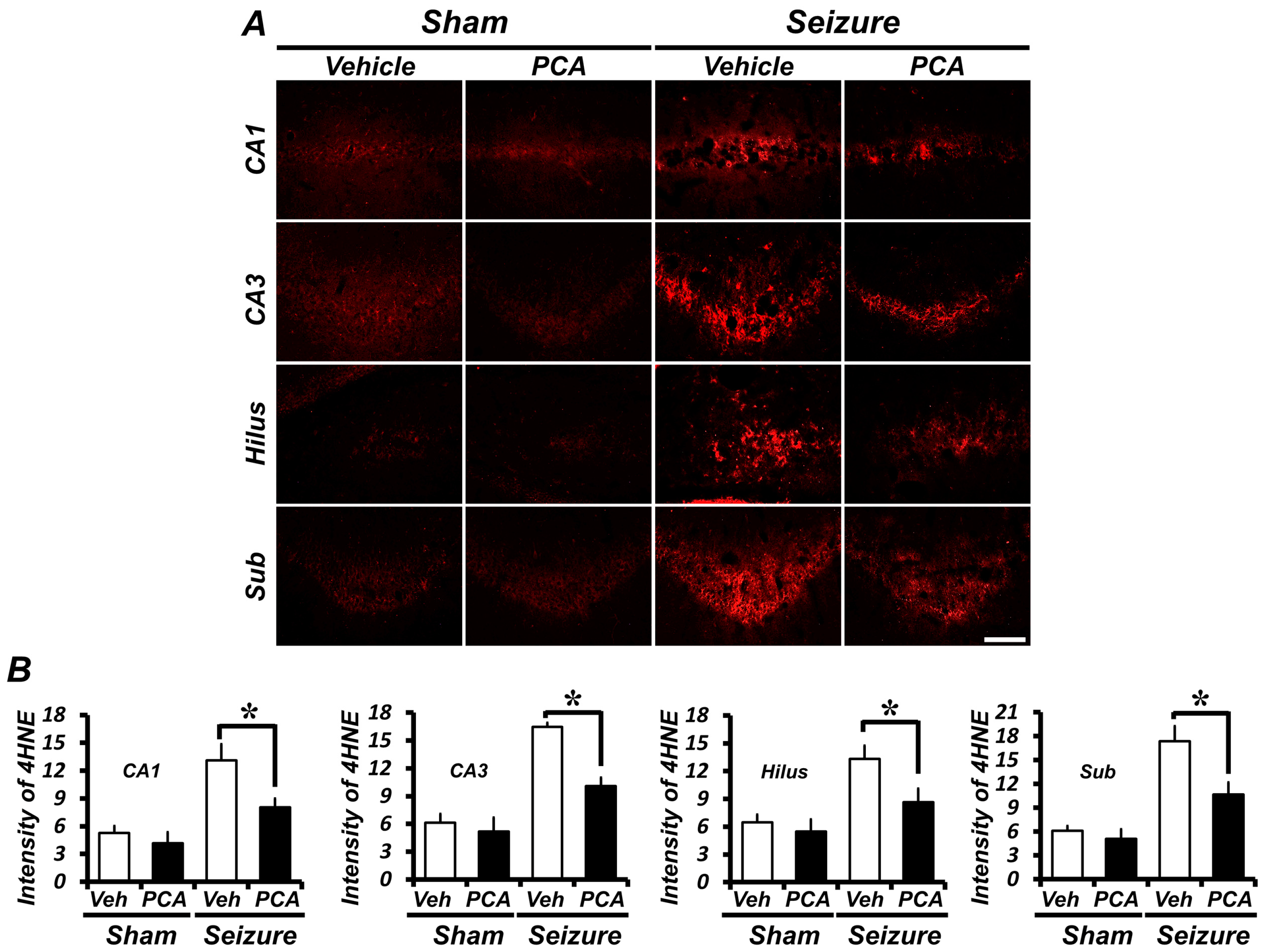
2.3. PCA Treatment Reduces Seizure-Induced Oxidative Injury in the Hippocampus
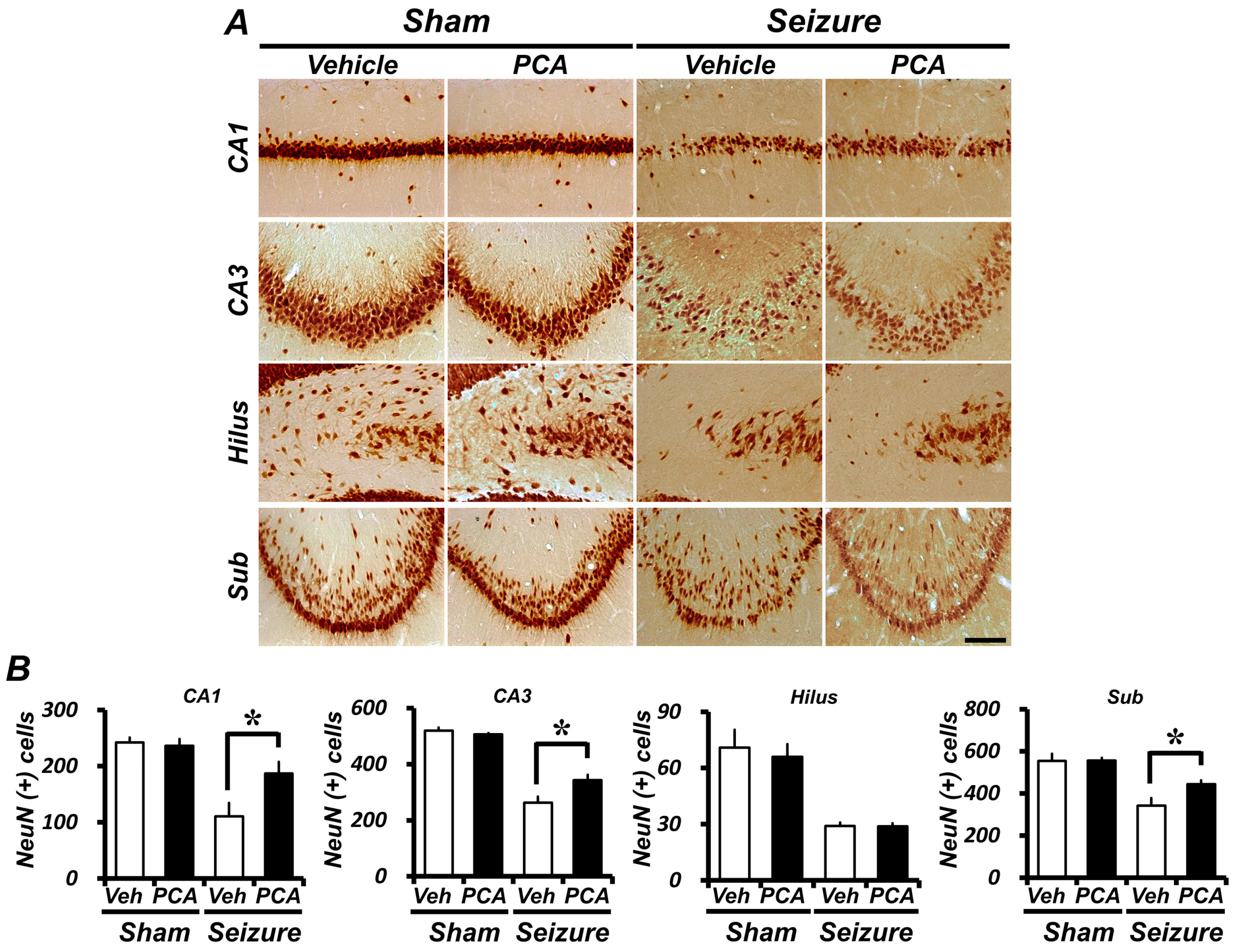
2.4. Seizure-Induced Hippocampal Neuronal Loss Is Reduced by PCA Treatment
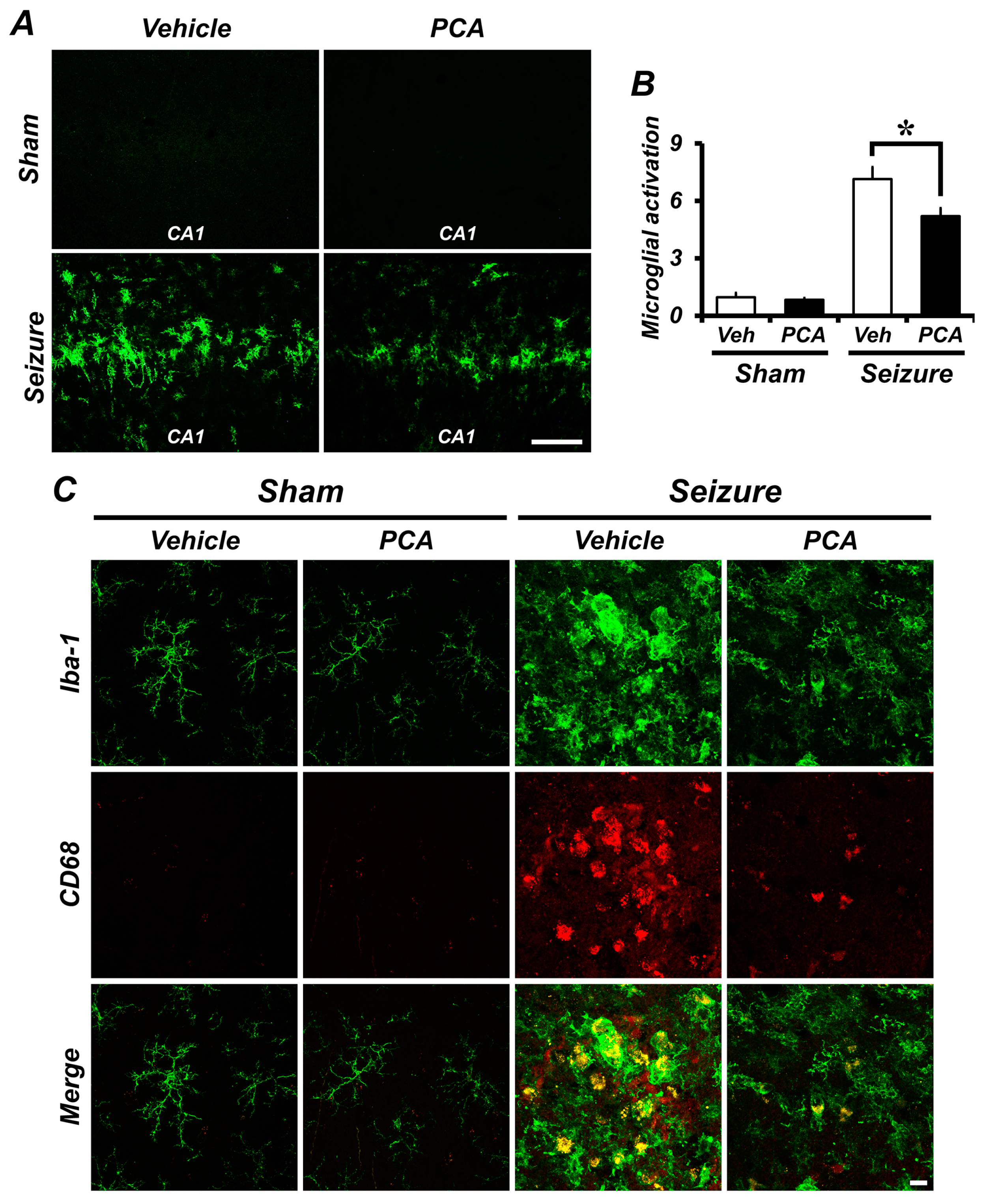
2.5. Seizure-Induced Microglia Activation Was Reduced by PCA
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Experimental Animals
4.3. Seizure Induction
4.4. Protocatechuic Acid (PCA) Injection
4.5. Brain Sample Preparation
4.6. Detection of Live Neurons
4.7. Detection of Degenerating Neurons
4.8. Detection of Microglia Activation Macrohage Activation
4.9. Detection of GSH levels
4.10. Detection of Oxidative Injury
4.11. Data Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chang, Y.C.; Huang, A.M.; Kuo, Y.M.; Wang, S.T.; Chang, Y.Y.; Huang, C.C. Febrile seizures impair memory and cAMP response-element binding protein activation. Ann. Neurol. 2003, 54, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J., Jr.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Thurman, D.J.; Beghi, E.; Begley, C.E.; Berg, A.T.; Buchhalter, J.R.; Ding, D.; Hesdorffer, D.C.; Hauser, W.A.; Kazis, L.; Kobau, R.; et al. Standards for epidemiologic studies and surveillance of epilepsy. Epilepsia 2011, 52 (Suppl. S7), S2–S26. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, E.M.; Coulter, D.A. Mechanisms of epileptogenesis: A convergence on neural circuit dysfunction. Nat. Rev. Neurosci. 2013, 14, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Rogawski, M.A.; Loscher, W. The neurobiology of antiepileptic drugs. Nat. Rev. Neurosci. 2004, 5, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Patel, M. Mitochondrial dysfunction and oxidative stress: Cause and consequence of epileptic seizures. Free Radic. Biol. Med. 2004, 37, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, M.R.; Shams, S.; Nouri, M.; Mohseni, M.; Shabanian, R.; Yekaninejad, M.S.; Chegini, N.; Khodadad, A.; Safaralizadeh, R. A probable causative factor for an old problem: Selenium and glutathione peroxidase appear to play important roles in epilepsy pathogenesis. Epilepsia 2007, 48, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef]
- Hayes, J.D.; Ellis, E.M.; Neal, G.E.; Harrison, D.J.; Manson, M.M. Cellular response to cancer chemopreventive agents: Contribution of the antioxidant responsive element to the adaptive response to oxidative and chemical stress. Biochem. Soc. Symp. 1999, 64, 141–168. [Google Scholar] [PubMed]
- Mueller, S.G.; Trabesinger, A.H.; Boesiger, P.; Wieser, H.G. Brain glutathione levels in patients with epilepsy measured by in vivo 1H-MRS. Neurology 2001, 57, 1422–1427. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [PubMed]
- Lovell, M.A.; Xie, C.; Markesbery, W.R. Decreased glutathione transferase activity in brain and ventricular fluid in Alzheimer’s disease. Neurology 1998, 51, 1562–1566. [Google Scholar] [CrossRef] [PubMed]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.Y.; Yin, M.C. Antibacterial effects of roselle calyx extracts and protocatechuic acid in ground beef and apple juice. Foodborne Pathog. Dis. 2009, 6, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Ciftci, O.; Disli, O.M.; Timurkaan, N. Protective effects of protocatechuic acid on TCDD-induced oxidative and histopathological damage in the heart tissue of rats. Toxicol. Ind. Health 2013, 29, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Lende, A.B.; Kshirsagar, A.D.; Deshpande, A.D.; Muley, M.M.; Patil, R.R.; Bafna, P.A.; Naik, S.R. Anti-inflammatory and analgesic activity of protocatechuic acid in rats and mice. Inflammopharmacology 2011, 19, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jiang, W.; Zhu, H.; Hou, J. Antifibrotic effects of protocatechuic aldehyde on experimental liver fibrosis. Pharm. Biol. 2012, 50, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lin, J.; Han, W.; Mai, W.; Wang, L.; Li, Q.; Lin, M.; Bai, M.; Zhang, L.; Chen, D. Antioxidant ability and mechanism of rhizoma Atractylodes macrocephala. Molecules 2012, 17, 13457–13472. [Google Scholar] [CrossRef] [PubMed]
- Scazzocchio, B.; Vari, R.; Filesi, C.; D’Archivio, M.; Santangelo, C.; Giovannini, C.; Iacovelli, A.; Silecchia, G.; Li Volti, G.; Galvano, F.; et al. Cyanidin-3-O-β-glucoside and protocatechuic acid exert insulin-like effects by upregulating PPAR-γ activity in human omental adipocytes. Diabetes 2011, 60, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.F.; An, L.J.; Jiang, B.; Guan, S.; Bao, Y.M. Alpinia protocatechuic acid protects against oxidative damage in vitro and reduces oxidative stress in vivo. Neurosci. Lett. 2006, 403, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Choi, B.Y.; Kho, A.R.; Jeong, J.H.; Hong, D.K.; Suh, S.W. Administration of Protocatechuic Acid Reduces Traumatic Brain Injury-Induced Neuronal Death. Int. J. Mol. Sci. 2017, 18, 2510. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, T.M.; Swanson, R.A. Poly(ADP-ribose) polymerase-1 promotes microglial activation, proliferation, and matrix metalloproteinase-9-mediated neuron death. J. Immunol. 2005, 174, 2288–2296. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, T.M.; Higashi, Y.; Suh, S.W.; Escartin, C.; Nagasawa, K.; Swanson, R.A. Zinc triggers microglial activation. J. Neurosci. 2008, 28, 5827–5835. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Muley, M.M.; Thakare, V.N.; Patil, R.R.; Bafna, P.A.; Naik, S.R. Amelioration of cognitive, motor and endogenous defense functions with silymarin, piracetam and protocatechuic acid in the cerebral global ischemic rat model. Life Sci. 2013, 93, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, G.; Szeto, S.S.; Chong, C.M.; Quan, Q.; Huang, C.; Cui, W.; Guo, B.; Wang, Y.; Han, Y.; et al. Examining the neuroprotective effects of protocatechuic acid and chrysin on in vitro and in vivo models of Parkinson disease. Free Radic. Biol. Med. 2015, 84, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Bains, J.S.; Shaw, C.A. Neurodegenerative disorders in humans: The role of glutathione in oxidative stress-mediated neuronal death. Brain Res. Brain Res. Rev. 1997, 25, 335–358. [Google Scholar] [CrossRef]
- Klein, J.A.; Ackerman, S.L. Oxidative stress, cell cycle, and neurodegeneration. J. Clin. Investig. 2003, 111, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, A.; Li, L.; Huang, Y.; Xue, P.; Hao, A. Oxidative stress mediates hippocampal neuron death in rats after lithium-pilocarpine-induced status epilepticus. Seizure 2010, 19, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Won, S.J.; Kim, J.E.; Cittolin-Santos, G.F.; Swanson, R.A. Assessment at the single-cell level identifies neuronal glutathione depletion as both a cause and effect of ischemia-reperfusion oxidative stress. J. Neurosci. 2015, 35, 7143–7152. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.J.; Jeong, J.H.; Chung, Y.H.; Kim, W.K.; Ko, K.H.; Bach, J.H.; Hong, J.S.; Yoneda, Y.; Kim, H.C. Role of oxidative stress in epileptic seizures. Neurochem. Int. 2011, 59, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Gras, G.; Samah, B.; Hubert, A.; Leone, C.; Porcheray, F.; Rimaniol, A.C. EAAT expression by macrophages and microglia: Still more questions than answers. Amino Acids 2012, 42, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvathenani, L.K.; Tertyshnikova, S.; Greco, C.R.; Roberts, S.B.; Robertson, B.; Posmantur, R. P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer’s disease. J. Biol. Chem. 2003, 278, 13309–13317. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, L.A.; Wang, L.; Ribak, C.E. Rapid astrocyte and microglial activation following pilocarpine-induced seizures in rats. Epilepsia 2008, 49 (Suppl. S2), S33–S41. [Google Scholar] [CrossRef] [PubMed]
- Frantseva, M.V.; Perez Velazquez, J.L.; Tsoraklidis, G.; Mendonca, A.J.; Adamchik, Y.; Mills, L.R.; Carlen, P.L.; Burnham, M.W. Oxidative stress is involved in seizure-induced neurodegeneration in the kindling model of epilepsy. Neuroscience 2000, 97, 431–435. [Google Scholar] [CrossRef]
- Barros, D.O.; Xavier, S.M.; Barbosa, C.O.; Silva, R.F.; Freitas, R.L.; Maia, F.D.; Oliveira, A.A.; Freitas, R.M.; Takahashi, R.N. Effects of the vitamin E in catalase activities in hippocampus after status epilepticus induced by pilocarpine in Wistar rats. Neurosci. lett. 2007, 416, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Pence, S.; Erkutlu, I.; Kurtul, N.; Bosnak, M.; Tan, U. Total brain tissue sialic acid levels due to glutathione effect in experimental epilepsy. Int. J. Neurosci. 2007, 117, 1523–1535. [Google Scholar] [CrossRef] [PubMed]
- Santos, I.M.; Tome Ada, R.; Saldanha, G.B.; Ferreira, P.M.; Militao, G.C.; Freitas, R.M. Oxidative stress in the hippocampus during experimental seizures can be ameliorated with the antioxidant ascorbic acid. Oxid. Med. Cell. Longev. 2009, 2, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Militao, G.C.; Ferreira, P.M.; de Freitas, R.M. Effects of lipoic acid on oxidative stress in rat striatum after pilocarpine-induced seizures. Neurochem. Int. 2010, 56, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Tome Ada, R.; Ferreira, P.M.; Freitas, R.M. Inhibitory action of antioxidants (ascorbic acid or α-tocopherol) on seizures and brain damage induced by pilocarpine in rats. Arquivos de Neuro-Psiquiatria 2010, 68, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Nehru, B.; Munshi, A.; Jyothy, A. Antioxidant potential of curcumin against oxidative insult induced by pentylenetetrazol in epileptic rats. Methods Find. Exp. Clin. Pharmacol. 2010, 32, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Van Rijckevorsel, K. Cognitive problems related to epilepsy syndromes, especially malignant epilepsies. Seizure 2006, 15, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Persinger, M.A.; Makarec, K.; Bradley, J.C. Characteristics of limbic seizures evoked by peripheral injections of lithium and pilocarpine. Physiol. Behav. 1988, 44, 27–37. [Google Scholar] [CrossRef]
- Racine, R.J.; Gartner, J.G.; Burnham, W.M. Epileptiform activity and neural plasticity in limbic structures. Brain Res. 1972, 47, 262–268. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, Y.J.; Kim, J.Y.; Kang, T.C. PARP1 activation/expression modulates regional-specific neuronal and glial responses to seizure in a hemodynamic-independent manner. Cell Death Dis. 2014, 5, e1362. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jang, B.G.; Choi, B.Y.; Kim, H.S.; Sohn, M.; Chung, T.N.; Choi, H.C.; Song, H.K.; Suh, S.W. Post-treatment of an NADPH oxidase inhibitor prevents seizure-induced neuronal death. Brain Res. 2013, 1499, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Kim, J.H.; Kim, H.J.; Lee, B.E.; Kim, I.Y.; Sohn, M.; Suh, S.W. Zinc chelation reduces traumatic brain injury-induced neurogenesis in the subgranular zone of the hippocampal dentate gyrus. J. Trace Elem. Med. Biol. 2014, 28, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Schmued, L.C.; Hopkins, K.J. Fluoro-Jade: Novel fluorochromes for detecting toxicant-induced neuronal degeneration. Toxicol. Pathol. 2000, 28, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Aoyama, K.; Chen, Y.; Garnier, P.; Matsumori, Y.; Gum, E.; Liu, J.; Swanson, R.A. Hypoglycemic neuronal death and cognitive impairment are prevented by poly(ADP-ribose) polymerase inhibitors administered after hypoglycemia. J. Neurosci. 2003, 23, 10681–10690. [Google Scholar] [PubMed]
- Suh, S.W.; Gum, E.T.; Hamby, A.M.; Chan, P.H.; Swanson, R.A. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J. Clin. Investig. 2007, 117, 910–918. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.H.; Choi, B.Y.; Kho, A.R.; Jeong, J.H.; Hong, D.K.; Lee, S.H.; Lee, S.Y.; Lee, M.W.; Song, H.K.; Choi, H.C.; et al. Protective Effects of Protocatechuic Acid on Seizure-Induced Neuronal Death. Int. J. Mol. Sci. 2018, 19, 187. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19010187
Lee SH, Choi BY, Kho AR, Jeong JH, Hong DK, Lee SH, Lee SY, Lee MW, Song HK, Choi HC, et al. Protective Effects of Protocatechuic Acid on Seizure-Induced Neuronal Death. International Journal of Molecular Sciences. 2018; 19(1):187. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19010187
Chicago/Turabian StyleLee, Song Hee, Bo Young Choi, A Ra Kho, Jeong Hyun Jeong, Dae Ki Hong, Sang Hwon Lee, Sang Yup Lee, Min Woo Lee, Hong Ki Song, Hui Chul Choi, and et al. 2018. "Protective Effects of Protocatechuic Acid on Seizure-Induced Neuronal Death" International Journal of Molecular Sciences 19, no. 1: 187. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19010187