The choice of appropriate endogenous controls to normalize expression levels is one of the key factors greatly influencing the results of RT-qPCR expression profiling [
53]. Optimal endogenous normalizers are crucial for the comparison of miRNA expression levels from different RT-qPCR experiments. Such interexperimental reproducibility of expression data is particularly important when miRNAs are considered as candidate biomarkers to be implemented in standardized diagnostic or treatment stratification procedures [
54].
3.1. Strategy for the Identification of Optimal Endogenous Normalizer miRNAs for RT-qPCR
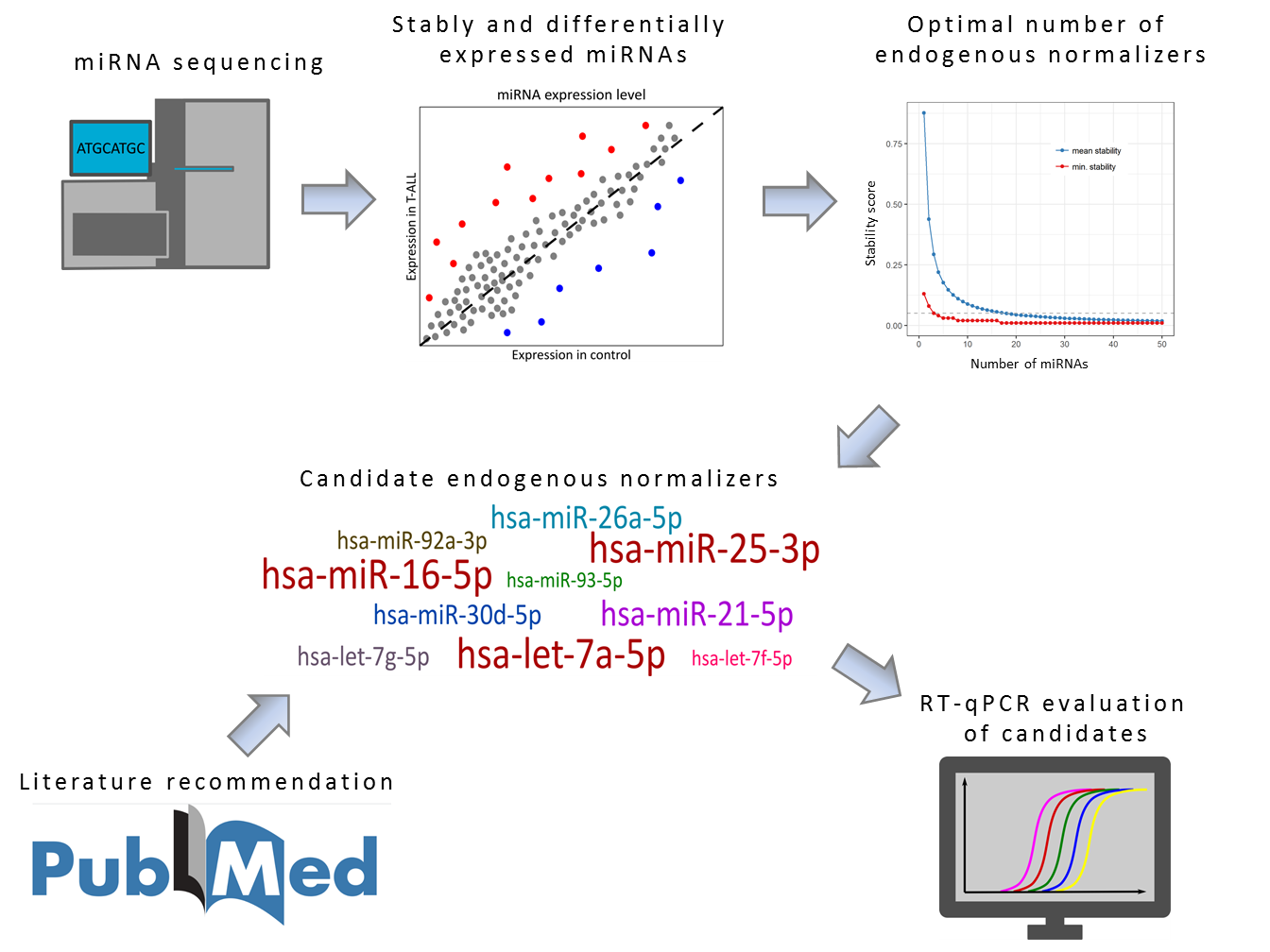
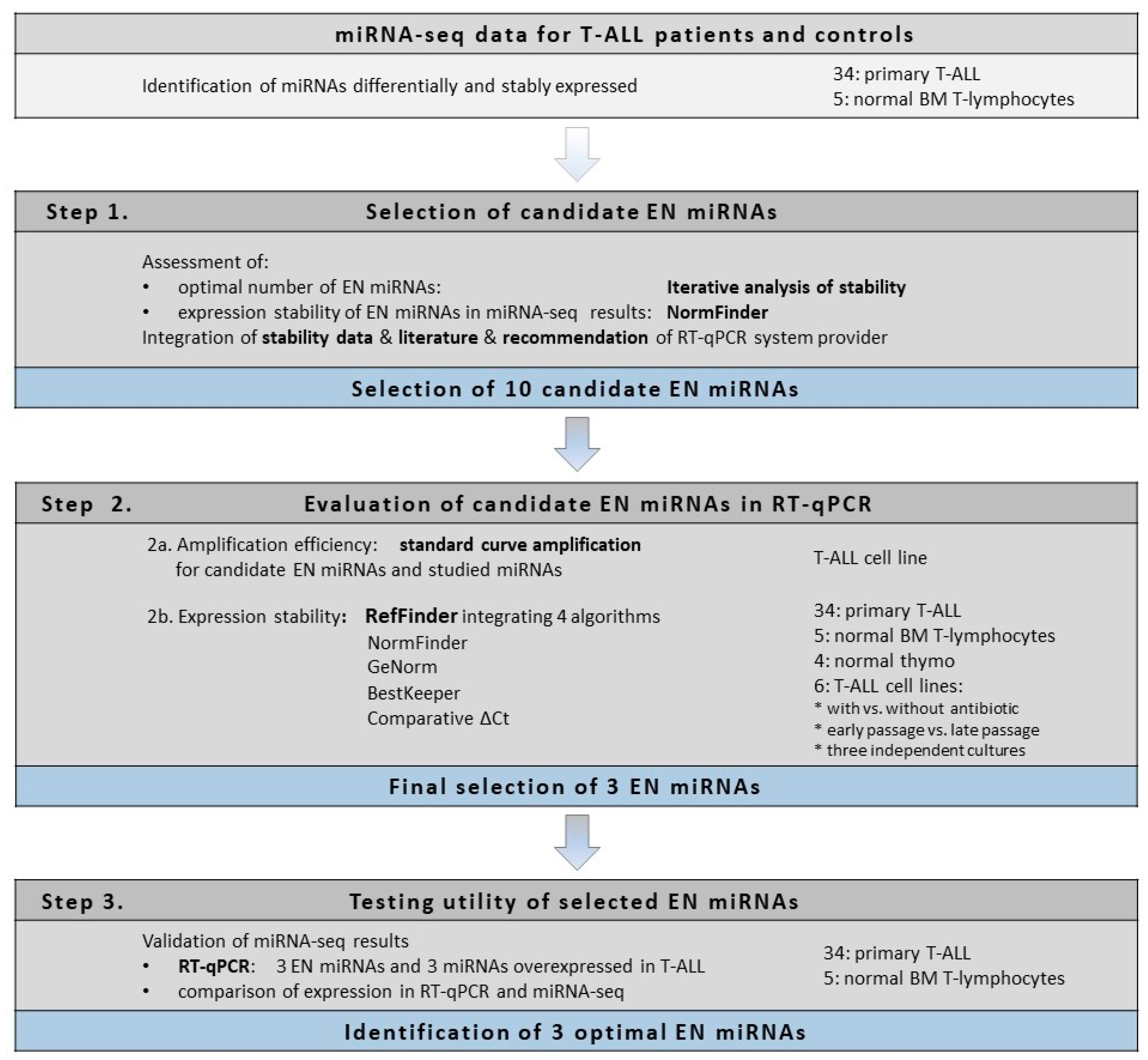
Here we present the strategy used for the identification of optimal endogenous normalizer (EN) miRNAs for RT-qPCR miRNA profiling in cells of T-lineage. The study design and experimental workflow are presented in
Figure 1. The starting point of our study was the results of miRNA transcriptome profiling with use of next-generation sequencing (miRNA-seq) performed in pediatric T-ALL samples and normal controls [
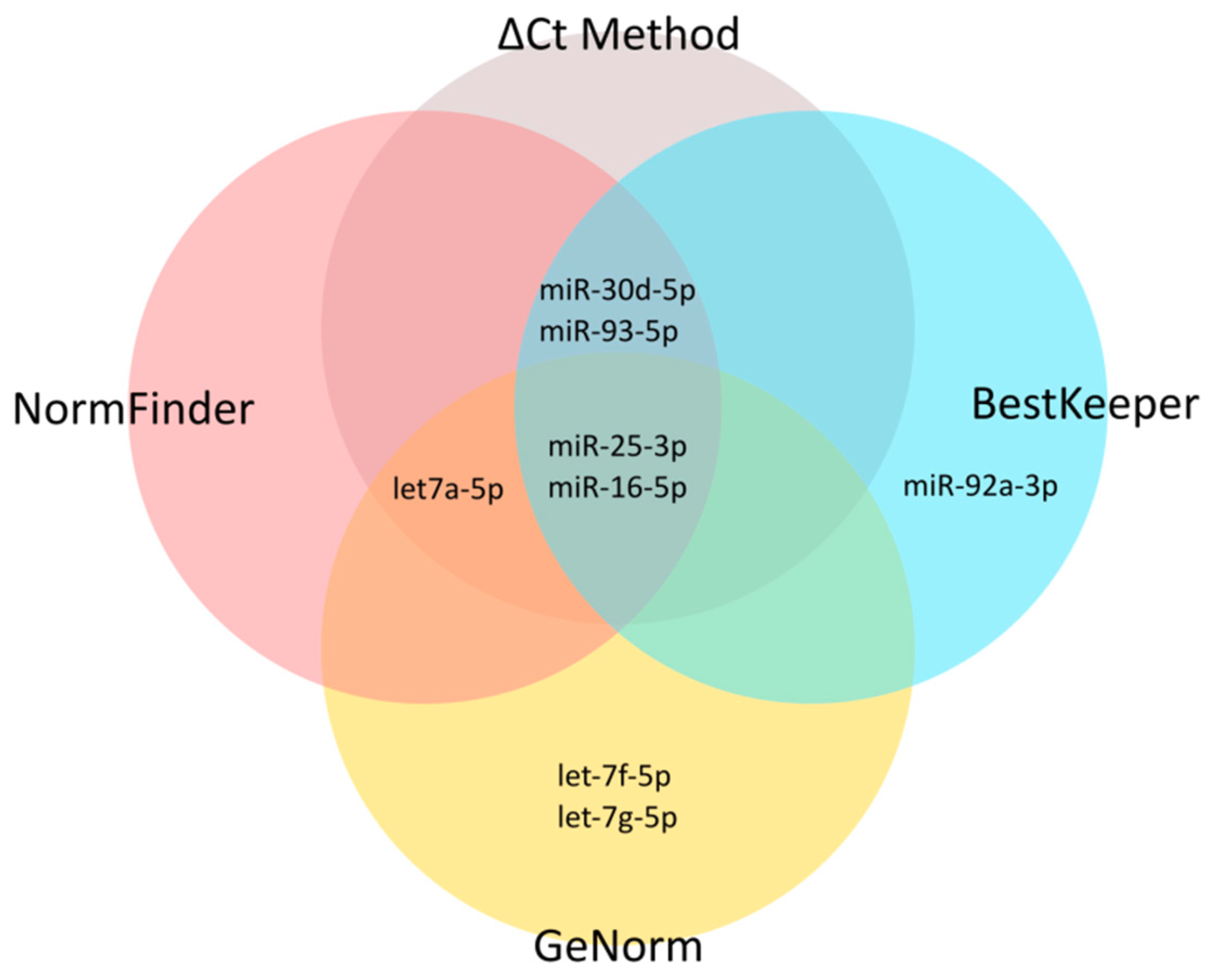
43]. First, using an iterative analysis of the stability of candidate ENs, we established three as an applicable number of miRNAs to be combined as ENs. Based on the expression stability in our miRNA-seq data, we selected 10 candidate EN miRNAs. Out of these, seven were recommended by Thermo Fisher Scientific as endogenous controls for TaqMan Advanced miRNA Assays (
www.thermofisher.com/advancedmirna). Four miRNAs were also reported in the literature as suitable endogenous controls for different cancer samples [
44,
45,
46,
47,
48]. To get insight into the potential utility of these candidate EN miRNAs in other experimental settings relative to cells of T-lineage, we included in our study four types of material commonly used in this research: primary T-ALL samples, normal mature T-lymphocytes from bone marrow, immature thymocyte samples, and six T-ALL cell lines. We also included varying cell line culture conditions for a more in-depth analysis of miRNA stability. Next, we analyzed the expression of these candidate EN miRNAs by RT-qPCR, and with the use of four algorithms we assessed the expression stability across all samples with respect to different material types. We selected three EN miRNAs (hsa-miR-16-5p, hsa-miR-25-3p, and hsa-let-7a-5p) exhibiting stable and abundant expression across all samples under study and successfully used them in the RT-qPCR validation of our miRNA-seq results.
Note that two of the three miRNAs we identified as the most optimal ENs for T-lineage cells, hsa-miR-25-3p and hsa-miR-16-5p, were among those proposed as universal ENs for TaqMan Advanced miRNA Assays (
www.thermofisher.com/advancedmirna). These two miRNAs were also reported in the literature as optimal ENs used for miRNA expression profiling in cancer cells. Specifically, hsa-miR-25-3p was shown to be stably expressed in several human cancer cell lines, including cervical, breast, and colorectal cancer, acute lymphoblastic leukemia, and testicular embryonal carcinoma [
45]. hsa-miR-16-5p was reported to be a suitable EN for miRNA expression profiling in malignant, benign, and normal breast tissues [
44], and was also used as an EN for the profiling of exosomal miRNA expression in blood serum of patients with breast and gastric cancer [
46,
47,
48]. The third miRNA we identified as an optimal EN, hsa-let-7a-5p, was reported to be suitable for miRNA expression analysis in human breast cancer [
44]. Here we show that these three miRNAs are also optimal ENs for miRNA profiling in T-ALL.
Below, we discuss several important aspects of the selection of optimal normalizers for miRNA expression in light of the proposed strategy.
3.2. Number of miRNAs to Be Used as Endogenous Normalizers in RT-qPCR
There are several approaches to the normalization of expression data in RT-qPCR, such as the use of a single endogenous control gene, the geometric mean of expression of several endogenous controls, and the global mean of expression of all transcripts under study. Below, we discuss the issue of the number of endogenous normalizers to be used in RT-qPCR.
One of the approaches to normalization in RT-qPCR is the use of a single endogenous control gene. However, this approach may generate a large normalization error [
16]. Therefore, normalization against a geometric mean of expression of several validated endogenous control genes is a more appropriate approach and greatly reduces the error rate [
53,
55].
Another possible strategy for RT-qPCR normalization is the use of a global mean expression value of all miRNAs under study (or genes, in the case of mRNA profiling) [
56,
57,
58]. This approach was demonstrated by Mestdagh et al. to be accurate and reliable for miRNA profiling in RT-qPCR experiments [
56]. In this study, the stability of a global mean was higher than the stability of commonly used endogenous control small RNAs. However, this approach is not suitable for every dataset, especially those in which a significant proportion of miRNAs show a positive correlation of signal intensities, which will limit the effectiveness of the approach. Additionally, a limitation of this method is the need to analyze a large and unbiased group of examined miRNAs or genes [
56]. Another option for normalization of miRNA expression is a method based on the use of weight mean of miRNA expression to generate an artificial endogenous control used to calculate ΔCq values. The standard deviation of miRNA expression across all samples is used as a measure of stability, and the expression of each miRNA is weighted by its stability [
59]. Yet, the utility of these approaches is limited to high- and medium-throughput expression profiling experiments like RT-qPCR-based arrays, and they are not suitable for small numbers of studied miRNAs or genes. Thus, the valid question is, what number of miRNAs should be used as ENs in a particular experiment?
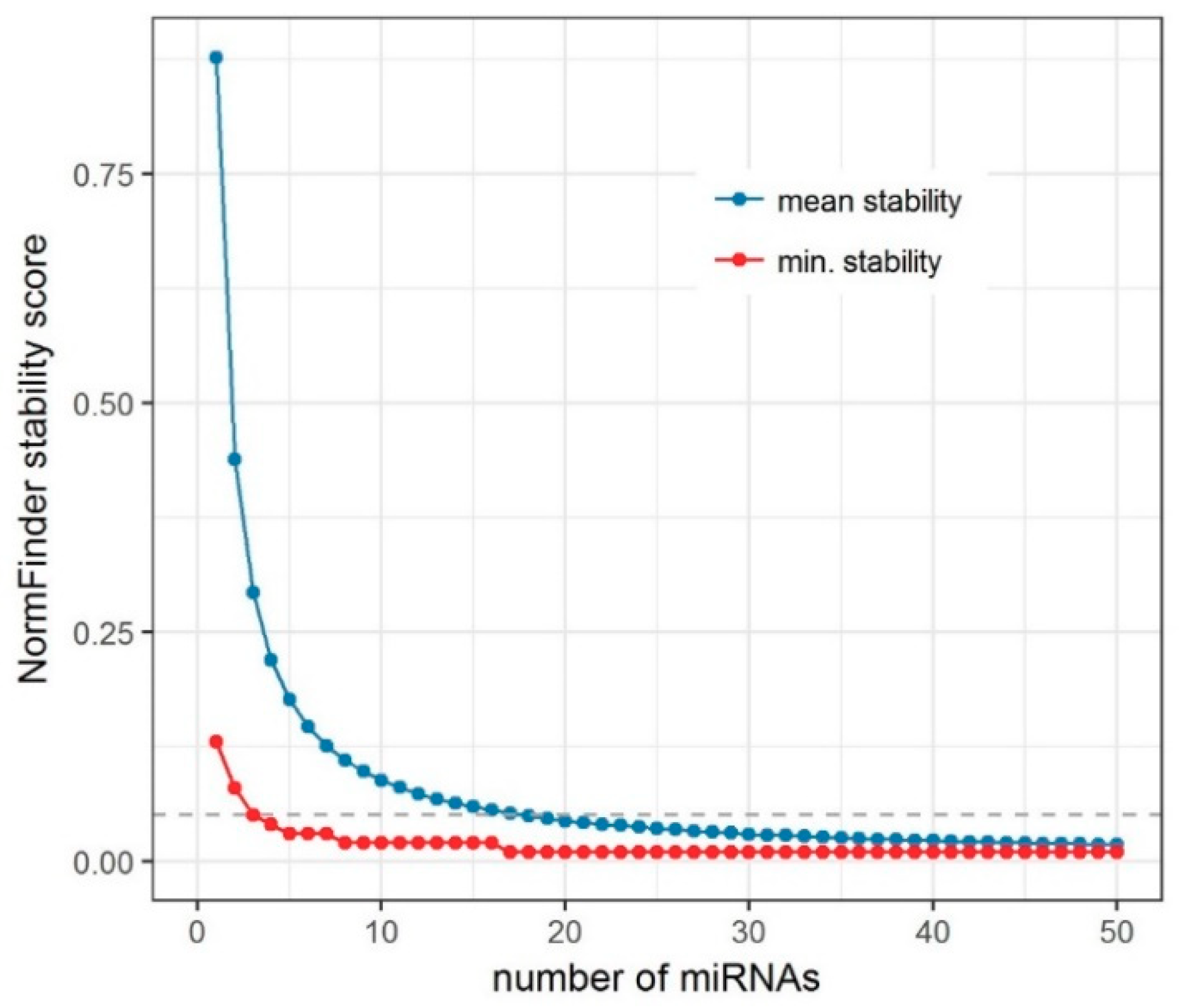
To address this question, we created an algorithm for an iterative analysis of miRNA expression stability (
Supplementary Material, IterativeStability_v1.0.R) and applied it to our miRNA-seq data. Several aspects should be considered when translating findings from miRNA-seq to an RT-qPCR experimental setting. The stability of miRNAs as measured by miRNA-seq depends not only on the intersample variability in miRNA concentrations, but also on the accuracy of the measurement method. The accuracy depends on the RNA isolation, the library preparation, the process of sequencing, and the data processing algorithms, including the data standardization strategy and its assumptions [
17]. Furthermore, the expression of low-level miRNAs might be significantly affected by the sampling process, caused by the limited capacity of the flow-cell used in the sequencing process, as in RNA-seq [
60]. For these reasons, even the most stable miRNAs will show significant variance between samples, caused by noise of technical origin. In order to reduce this effect, it is often beneficial to combine several endogenous reference RNAs for the normalization of expression data in RT-qPCR. The goal is to establish the number and optimal set of miRNAs to be combined. Using our miRNA-seq data, we selected the best
k-number of miRNAs. However, in other experiments, a
k + 1 number of combined miRNAs might provide a more stable signal. In general, in this iterative approach the stability is expected to increase as
k increases, until we reach the point at which we start to incorporate differentially expressed miRNAs. However, in most cases, this will happen long after we exceed the practical limits of the number of miRNAs that can be used as reference in RT-qPCR experiments.
In our iterative analysis of miRNA expression stability, we demonstrated that the use of more than three EN miRNAs could provide only a limited increase of stability. The advantage of combining more miRNAs is likely disproportionate in view of additional cost. The use of three miRNAs seems to give a reasonable balance between good stability for endogenous normalization and cost-effectiveness of the study.
3.3. Expression Stability Relative to Sample Type and Culture Conditions
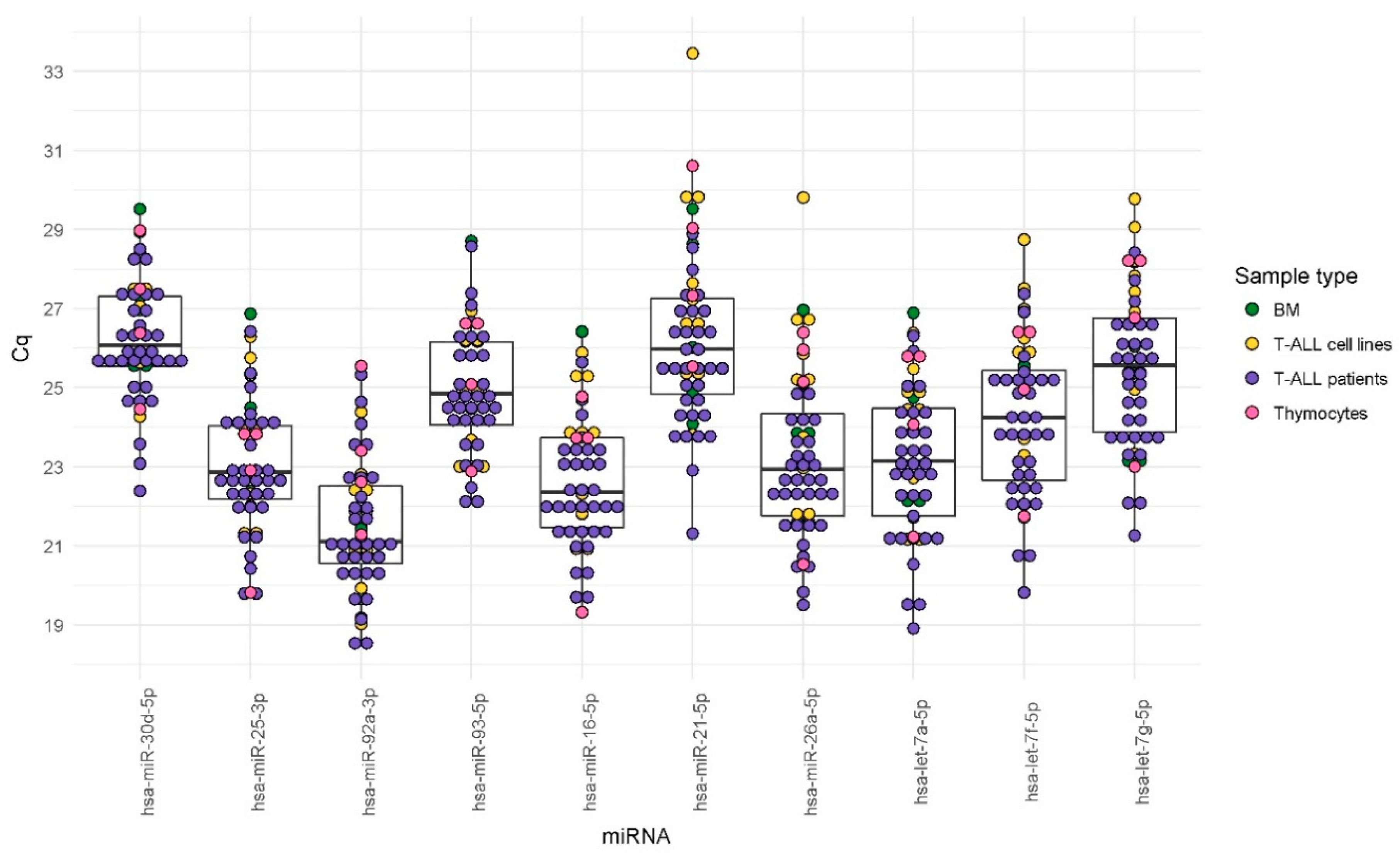
In our study, we analyzed the expression stability of 10 candidate EN miRNAs in several types of material commonly used in research on T-lineage cells, both normal and malignant. We included T-ALL samples, normal thymocytes, normal mature T-lymphocytes, and six T-ALL cell lines, cultured in varying conditions (as presented in
Figure 1 and described in detail in the Materials section). We did not observe significant differences in the expression levels of candidate EN miRNAs between different types of biological material tested, as presented in
Table 3. This observation also applies to T-ALL cell line samples cultured in different conditions, as presented in
Supplementary Table S2. The variability of expression of these miRNAs is rather sample-dependent, as illustrated in
Supplementary Figure S1.
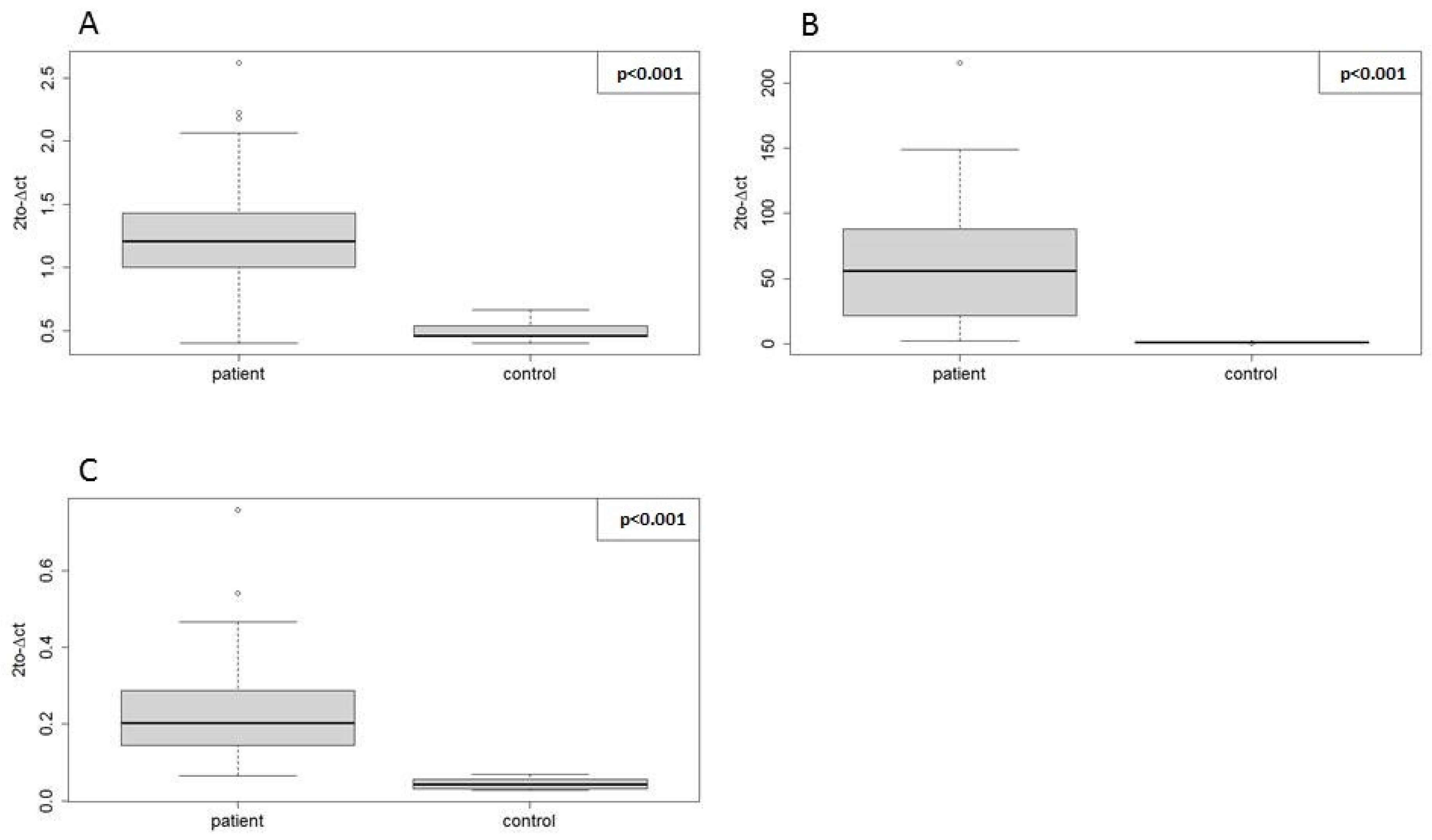
We also demonstrated that our three selected EN miRNAs were suitable for miRNA profiling across different T-ALL cell lines and varying culture conditions by evaluating the relative expression of hsa-miR-128-3p in these samples in reference to normal bone marrow T-lymphocytes. As shown in
Supplementary Figure S2, the hsa-miR-128-3p expression levels were very close in the MOLT-4 cells cultured with and without antibiotic. Similarly, the time of culture (early vs. late passage analyzed in the case of the CCRF-CEM cell line) did not affect the expression level of this miRNA. We observed the difference in fold change in one of the three independent cultures of Jurkat cells and, naturally, differences of fold change values across different cell lines. Nevertheless, using our three EN miRNAs, we showed that hsa-miR-128-3p was overexpressed in all six T-ALL cell lines, which is in line with literature data [
35,
61].
3.4. RT-qPCR Validation of miRNA-Seq Results
One of the discrepancies between miRNA-seq and RT-qPCR results when used for validation purposes is the difference of fold change values observed for differentially expressed miRNAs [
62]. This can be explained by the fact that the operating principles for the two methods of quantification are different. miRNA-seq is based on the read count number of a particular transcript, whereas RT-qPCR is based on hybridization with primers and hydrolysis probe and amplification. The design of amplification primers and probes is particularly challenging in the case of miRNAs due to the fact that mature miRNA sequences are short. In combination with the high similarity between sequences of isomiRs, this limits the options for the optimal design of a miRNA assay to ensure the highest sensitivity and specificity [
17]. These factors can potentially lead to nonspecific hybridization and eventually to biased RT-qPCR results as compared to miRNA-seq [
62]. In our experimental setting, the difference in the fold change values was most striking in hsa-miR-181a-5p, as shown in
Table 5. Based on the analysis of similarity between mature miRNA sequences, their seed sequences, and the Pearson’s correlation of expression in our miRNA-seq data, we concluded that this discrepancy might be attributed to high similarity of hsa-miR-181a-5p and its isomiRs, as presented in
Supplementary Table S7.
Nevertheless, the direction of expression change and statistical significance were concordant between the two methods for all three miRNAs, which proves the positive validation of miRNA-seq findings with RT-qPCR using our EN miRNAs.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}