Role of Hypoxic Stress in Regulating Tumor Immunogenicity, Resistance and Plasticity

 , , ,
, , , 
{kind=link}
Abstract
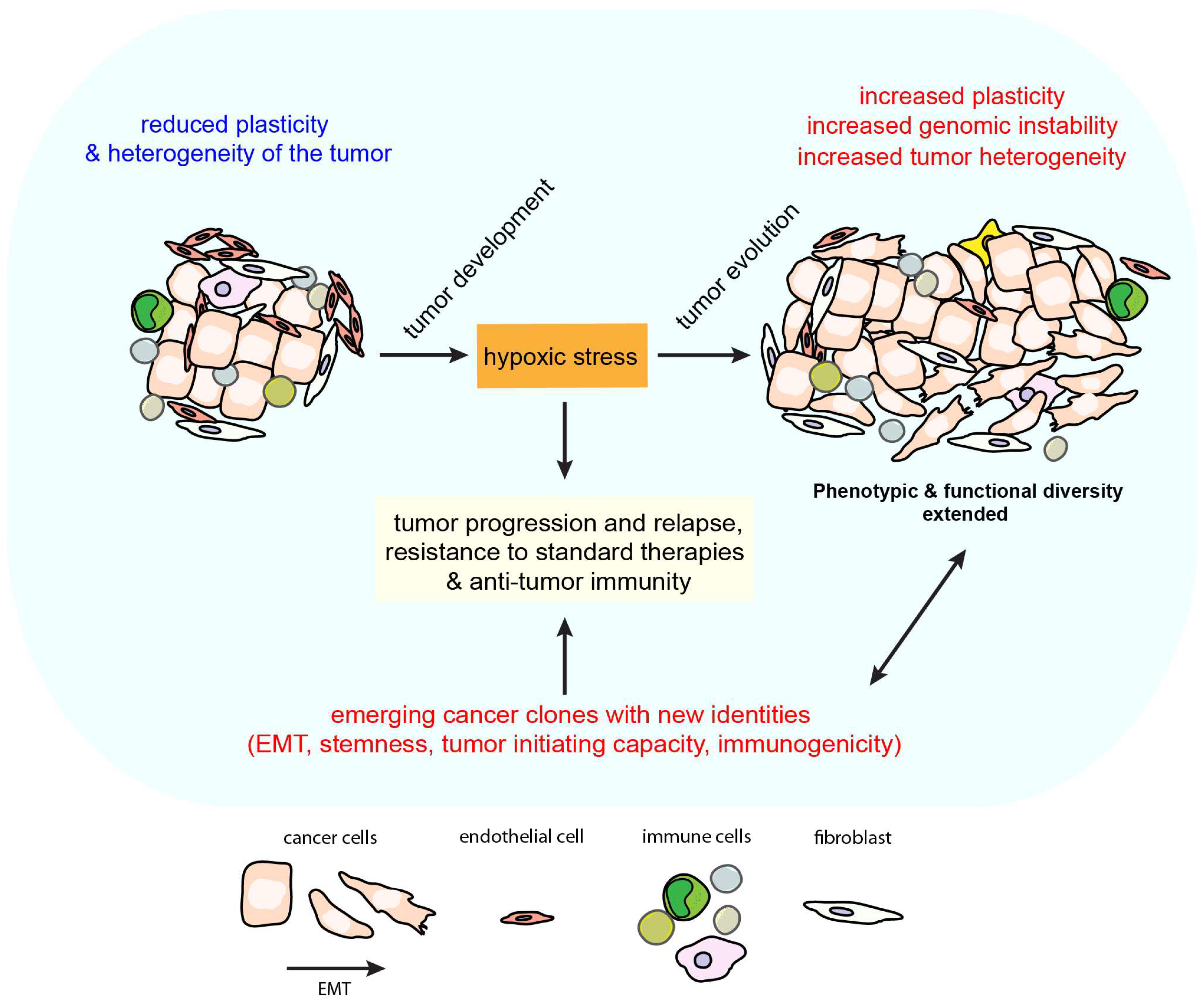
:1. Introduction
2. Hypoxia Induced Tumor Plasticity and Heterogeneity
3. Impact of Plasticity and Heterogeneity on Tumor Immune Escape
4. Mechanisms of Hypoxia-Induced Cancer Stem Cells
5. EMT at the Crossroad of Stemness
6. Hypoxia, DNA Repair and Genomic Instability
7. Therapeutic Targeting of Hypoxia in Cancer
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell. Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A review in the theme: cellular responses to hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Buart, S.; Chouaib, S. Hypoxic stress-induced tumor and immune plasticity, suppression, and impact on tumor heterogeneity. Front. Immunol. 2017, 8, 1625. [Google Scholar] [CrossRef] [PubMed]
- Taddei, M.L.; Giannoni, E.; Comito, G.; Chiarugi, P. Microenvironment and tumor cell plasticity: An easy way out. Cancer Lett. 2013, 341, 80–96. [Google Scholar] [CrossRef] [PubMed]
- Mouriaux, F.; Sanschagrin, F.; Diorio, C.; Landreville, S.; Comoz, F.; Petit, E.; Bernaudin, M.; Rousseau, A.P.; Bergeron, D.; Morcos, M. Increased HIF-1α expression correlates with cell proliferation and vascular markers CD31 and VEGF-A in uveal melanoma. Invest. Ophthalmol. Vis. Sci. 2014, 55, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Huang, S.; Wang, L.; Yuan, X.; Dong, Q.; Zhang, D.; Wang, X. Clinical and prognostic significance of HIF-1α overexpression in oral squamous cell carcinoma: A meta-analysis. World J. Surg. Oncol. 2017, 15, 104. [Google Scholar] [CrossRef] [PubMed]
- Buart, S.; Terry, S.; Noman, M.Z.; Lanoy, E.; Boutros, C.; Fogel, P.; Dessen, P.; Meurice, G.; Gaston-Mathe, Y.; Vielh, P.; et al. Transcriptional response to hypoxic stress in melanoma and prognostic potential of GBE1 and BNIP3. Oncotarget 2017, 8, 108786–108801. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.P.; Wu, K.J. Hypoxia-regulated target genes implicated in tumor metastasis. J. Biomed. Sci. 2012, 19, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.P.; Wu, K.J. Epigenetic regulation of hypoxia-responsive gene expression: Focusing on chromatin and DNA modifications. Int. J. Cancer 2014, 134, 249–256. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Holzel, M.; Bovier, A.; Tuting, T. Plasticity of tumour and immune cells: A source of heterogeneity and a cause for therapy resistance? Nat. Rev. Cancer 2013, 13, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Zitvogel, L.; Sautes-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; Fallahi-Sichani, M.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 2018, 33, 463–479. [Google Scholar] [CrossRef] [PubMed]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 2018, 174, 1293–1308. [Google Scholar] [CrossRef] [PubMed]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell Analyses. Cell 2017, 169, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Velica, P.; Rundqvist, H.; Fitzpatrick, S.; Vojnovic, N.; Phan, A.T.; Loman, N.; Hedenfalk, I.; et al. An HIF-1α/VEGF-A Axis in cytotoxic t cells regulates tumor progression. Cancer Cell 2017, 32, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Paolicchi, E.; Gemignani, F.; Krstic-Demonacos, M.; Dedhar, S.; Mutti, L.; Landi, S. Targeting hypoxic response for cancer therapy. Oncotarget 2016, 7, 13464–13478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, J.; Nakayama, K.; Cardiff, R.D.; Borowsky, A.D.; Kaul, K.; Williams, R.; Krajewski, S.; Mercola, D.; Carpenter, P.M.; Bowtell, D.; et al. Siah2-dependent concerted activity of HIF and FoxA2 regulates formation of neuroendocrine phenotype and neuroendocrine prostate tumors. Cancer Cell 2010, 18, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Danza, G.; Di Serio, C.; Rosati, F.; Lonetto, G.; Sturli, N.; Kacer, D.; Pennella, A.; Ventimiglia, G.; Barucci, R.; Piscazzi, A.; et al. Notch signaling modulates hypoxia-induced neuroendocrine differentiation of human prostate cancer cells. Mol. Cancer Res. 2012, 10, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.P.; Chang, Y.T.; Lee, S.Y.; Campbell, M.; Wang, T.C.; Shen, S.H.; Chung, H.J.; Chang, Y.H.; Chiu, A.W.; Pan, C.C.; et al. REST reduction is essential for hypoxia-induced neuroendocrine differentiation of prostate cancer cells by activating autophagy signaling. Oncotarget 2016, 7, 26137–26151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.-H.; Wu, M.-Z.; Chiou, S.-H.; Chen, P.-M.; Chang, S.-Y.; Liu, C.-J.; Teng, S.-C.; Wu, K.-J. Direct regulation of TWIST by HIF-1α promotes metastasis. Nature Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Scortegagna, M.; Martin, R.J.; Kladney, R.D.; Neumann, R.G.; Arbeit, J.M. Hypoxia-inducible factor-1α suppresses squamous carcinogenic progression and epithelial-mesenchymal transition. Cancer Res. 2009, 69, 2638–2646. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Wang, J.; Li, J.; Post, M. Mouse Snail Is a Target Gene for HIF. Mol. Cancer Res. 2011, 9, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schietke, R.; Warnecke, C.; Wacker, I.; Schodel, J.; Mole, D.R.; Campean, V.; Amann, K.; Goppelt-Struebe, M.; Behrens, J.; Eckardt, K.U.; et al. The lysyl oxidases LOX and LOXL2 are necessary and sufficient to repress E-cadherin in hypoxia: Insights into cellular transformation processes mediated by HIF-1. J. Biol. Chem. 2010, 285, 6658–6669. [Google Scholar] [CrossRef] [PubMed]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017, 171, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Buart, S.; Tan, T.Z.; Gros, G.; Noman, M.Z.; Lorens, J.B.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. Acquisition of tumor cell phenotypic diversity along the EMT spectrum under hypoxic pressure: Consequences on susceptibility to cell-mediated cytotoxicity. OncoImmunology 2017, 6, e1271858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, L.; Blackwell, K. Hypoxia and anemia: Factors in decreased sensitivity to radiation therapy and chemotherapy? Oncologist 2004, 9, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Te Boekhorst, V.; Odenthal, J.; Bianchi, R.; van Helvert, S.; Ikenberg, K.; Ilina, O.; Stoma, S.; Xandry, J.; Jiang, L.; et al. Hypoxia induces a HIF-1-dependent transition from collective-to-amoeboid dissemination in epithelial cancer cells. Curr. Biol. 2017, 27, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Keener, A.B. Shapeshifters in cancer: How some tumor cells change phenotype to evade therapy. Nat. Med. 2016, 22, 1194–1196. [Google Scholar] [CrossRef] [PubMed]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wolfel, T.; Holzel, M.; et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; Andre, F.; De Cremoux, P.; Bertheau, P.; Badoual, C.; Vielh, P.; Larsen, A.K.; et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res. 2013, 73, 2418–2427. [Google Scholar] [CrossRef] [PubMed]
- Akalay, I.; Tan, T.Z.; Kumar, P.; Janji, B.; Mami-Chouaib, F.; Charpy, C.; Vielh, P.; Larsen, A.K.; Thiery, J.P.; Sabbah, M.; et al. Targeting WNT1-inducible signaling pathway protein 2 alters human breast cancer cell susceptibility to specific lysis through regulation of KLF-4 and miR-7 expression. Oncogene 2015, 34, 2261–2271. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.C.; Peters, H.L.; Taguchi, A.; Katayama, H.; Wang, H.; Momin, A.; Jolly, M.K.; Celiktas, M.; Rodriguez-Canales, J.; Liu, H.; et al. Immunoproteasome deficiency is a feature of non-small cell lung cancer with a mesenchymal phenotype and is associated with a poor outcome. Proc. Natl. Acad. Sci. USA 2016, 113, E1555–E1564. [Google Scholar] [CrossRef] [PubMed]
- Le Floc’h, A.; Jalil, A.; Vergnon, I.; Le Maux Chansac, B.; Lazar, V.; Bismuth, G.; Chouaib, S.; Mami-Chouaib, F. A E β 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J. Exp. Med. 2007, 204, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Djenidi, F.; Adam, J.; Goubar, A.; Durgeau, A.; Meurice, G.; de Montpreville, V.; Validire, P.; Besse, B.; Mami-Chouaib, F. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J. Immunol. 2015, 194, 3475–3486. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noman, M.Z.; Janji, B.; Abdou, A.; Hasmim, M.; Terry, S.; Tan, T.Z.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. OncoImmunology 2017, 6, e1263412. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, D.H.; Huang, B.; Fernando, R.I.; Tsang, K.Y.; Palena, C. WEE1 inhibition alleviates resistance to immune attack of tumor cells undergoing epithelial-mesenchymal transition. Cancer Res. 2014, 74, 2510–2519. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, D.H.; Griner, L.M.; Keller, J.M.; Hu, X.; Southall, N.; Marugan, J.; David, J.M.; Ferrer, M.; Palena, C. Targeting estrogen receptor signaling with fulvestrant enhances immune and chemotherapy-mediated cytotoxicity of human lung cancer. Clin. Cancer Res. 2016, 22, 6204–6216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noman, M.Z.; Van Moer, K.; Marani, V.; Gemmill, R.M.; Tranchevent, L.C.; Azuaje, F.; Muller, A.; Chouaib, S.; Thiery, J.P.; Berchem, G.; et al. CD47 is a direct target of SNAI1 and ZEB1 and its blockade activates the phagocytosis of breast cancer cells undergoing EMT. OncoImmunology 2018, 7, e1345415. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, M.; Zanotto, M.; Malpeli, G.; Bassi, G.; Perbellini, O.; Chilosi, M.; Bifari, F.; Krampera, M. Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br. J. Cancer 2015, 112, 1067–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Huergo-Zapico, L.; Parodi, M.; Cantoni, C.; Lavarello, C.; Fernandez-Martinez, J.L.; Petretto, A.; DeAndres-Galiana, E.J.; Balsamo, M.; Lopez-Soto, A.; Pietra, G.; et al. NK-cell editing mediates epithelial-to-mesenchymal transition via phenotypic and proteomic changes in melanoma cell lines. Cancer Res. 2018, 78, 3913–3925. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Soto, A.; Huergo-Zapico, L.; Galvan, J.A.; Rodrigo, L.; de Herreros, A.G.; Astudillo, A.; Gonzalez, S. Epithelial-mesenchymal transition induces an antitumor immune response mediated by NKG2D receptor. J. Immunol. 2013, 190, 4408–4419. [Google Scholar] [CrossRef] [PubMed]
- Chockley, P.J.; Chen, J.; Chen, G.; Beer, D.G.; Standiford, T.J.; Keshamouni, V.G. Epithelial-mesenchymal transition leads to NK cell-mediated metastasis-specific immunosurveillance in lung cancer. J. Clin. Investig. 2018, 128, 1384–1396. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.Y.; Chen, W.; Bai, X.L.; Xu, X.Y.; Zhang, Q.; Xia, X.F.; Sun, X.; Li, G.G.; Hu, Q.D.; Fu, Q.H.; et al. Hypoxia-induced epithelial-to-mesenchymal transition in hepatocellular carcinoma induces an immunosuppressive tumor microenvironment to promote metastasis. Cancer Res. 2016, 76, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lu, H.; Xiang, L.; Bullen, J.W.; Zhang, C.; Samanta, D.; Gilkes, D.M.; He, J.; Semenza, G.L. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, E6215–E6223. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Park, Y.; Ni, X.; Li, H.; Zahnow, C.A.; Gabrielson, E.; Pan, F.; Semenza, G.L. Chemotherapy induces enrichment of CD47(+)/CD73(+)/PDL1(+) immune evasive triple-negative breast cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1239–E1248. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massague, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Hasmim, M.; Noman, M.Z.; Messai, Y.; Bordereaux, D.; Gros, G.; Baud, V.; Chouaib, S. Cutting edge: Hypoxia-induced Nanog favors the intratumoral infiltration of regulatory T cells and macrophages via direct regulation of TGF-β1. J. Immunol. 2013, 191, 5802–5806. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; Jhunjhunwala, S.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Aragones, J.; Morales-Kastresana, A.; de Landazuri, M.O.; Melero, I. Molecular pathways: Hypoxia response in immune cells fighting or promoting cancer. Clin. Cancer Res. 2012, 18, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7, 277. [Google Scholar] [CrossRef] [PubMed]
- Mancino, A.; Schioppa, T.; Larghi, P.; Pasqualini, F.; Nebuloni, M.; Chen, I.H.; Sozzani, S.; Austyn, J.M.; Mantovani, A.; Sica, A. Divergent effects of hypoxia on dendritic cell functions. Blood 2008, 112, 3723–3734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Elly, C.; Park, Y.; Liu, Y.C. E3 Ubiquitin Ligase VHL Regulates Hypoxia-Inducible Factor-1α to Maintain Regulatory T Cell Stability and Suppressive Capacity. Immunity 2015, 42, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Clambey, E.T.; McNamee, E.N.; Westrich, J.A.; Glover, L.E.; Campbell, E.L.; Jedlicka, P.; de Zoeten, E.F.; Cambier, J.C.; Stenmark, K.R.; Colgan, S.P.; et al. Hypoxia-inducible factor-1 α-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. USA 2012, 109, E2784–E2793. [Google Scholar] [CrossRef] [PubMed]
- Antonios, J.P.; Soto, H.; Everson, R.G.; Moughon, D.; Orpilla, J.R.; Shin, N.P.; Sedighim, S.; Treger, J.; Odesa, S.; Tucker, A.; et al. Immunosuppressive tumor-infiltrating myeloid cells mediate adaptive immune resistance via a PD-1/PD-L1 mechanism in glioblastoma. Neuro Oncol. 2017, 19, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Eubank, T.D.; Roda, J.M.; Liu, H.; O’Neil, T.; Marsh, C.B. Opposing roles for HIF-1α and HIF-2α in the regulation of angiogenesis by mononuclear phagocytes. Blood 2011, 117, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A.; Lleonart, M. The hypoxic microenvironment: A determinant of cancer stem cell evolution. Bioessays 2016, 38, S65–S74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Lin, Y.C.; Tsai, M.H.; Lin, C.S.; Murayama, Y.; Sato, R.; Yokoyama, K.K. Emerging roles of hypoxia-inducible factors and reactive oxygen species in cancer and pluripotent stem cells. Kaohsiung J. Med. Sci. 2015, 31, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Liu, Y. Hypoxia-inducible factors in cancer stem cells and inflammation. Trends Pharmacol. Sci. 2015, 36, 374–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2α regulates Oct-4: Effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006, 20, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, J.; Dondeti, V.; Simon, M.C. Hypoxia-inducible factors in stem cells and cancer. J. Cell. Mol. Med. 2009, 13, 4319–4328. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.; Cotten, C.; Lonardo, E.; Strizzi, L.; Baraty, C.; Mancino, M.; Gonzales, M.; Watanabe, K.; Nagaoka, T.; Berry, C.; et al. Cripto-1 is required for hypoxia to induce cardiac differentiation of mouse embryonic stem cells. Am. J. Pathol. 2009, 175, 2146–2158. [Google Scholar] [CrossRef] [PubMed]
- Westfall, S.D.; Sachdev, S.; Das, P.; Hearne, L.B.; Hannink, M.; Roberts, R.M.; Ezashi, T. Identification of oxygen-sensitive transcriptional programs in human embryonic stem cells. Stem Cells Dev. 2008, 17, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Wahab, S.M.R.; Islam, F.; Gopalan, V.; Lam, A.K. The Identifications and Clinical Implications of Cancer Stem Cells in Colorectal Cancer. Clin. Colorectal Cancer 2017, 16, 93–102. [Google Scholar] [CrossRef] [PubMed]
- van Schaijik, B.; Davis, P.F.; Wickremesekera, A.C.; Tan, S.T.; Itinteang, T. Subcellular localisation of the stem cell markers OCT4, SOX2, NANOG, KLF4 and c-MYC in cancer: A review. J. Clin. Pathol. 2018, 71, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.M.; Gandhi, S.C.; Bodmer, W.F. Hypoxia and lineage specification of cell line-derived colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4382–4387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Zayed, H.; Petersen, I. Stem cell transcription factor SOX2 in synovial sarcoma and other soft tissue tumors. Pathol. Res. Pract. 2018, 214, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; El-Sayed, I.Y.; Destouches, D.; Maille, P.; Nicolaiew, N.; Ploussard, G.; Semprez, F.; Pimpie, C.; Beltran, H.; Londono-Vallejo, A.; et al. CRIPTO overexpression promotes mesenchymal differentiation in prostate carcinoma cells through parallel regulation of AKT and FGFR activities. Oncotarget 2015, 6, 11994–12008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, M.Y.; Lemos, R., Jr.; Liu, X.; Powis, G. The hypoxia-associated factor switches cells from HIF-1α- to HIF-2α-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011, 71, 4015–4027. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, M.; Palanichamy, J.K.; Ramalingam, P.; Mudassir, M.; Irshad, K.; Chosdol, K.; Sarkar, C.; Seth, P.; Goswami, S.; Sinha, S.; et al. HIF-2α mediates a marked increase in migration and stemness characteristics in a subset of glioma cells under hypoxia by activating an Oct-4/Sox-2-Mena (INV) axis. Int. J. Biochem. Cell Biol. 2016, 74, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.A.; Chen, Y.F.; Bao, Y.; Mahara, S.; Yatim, S.; Oguz, G.; Lee, P.L.; Feng, M.; Cai, Y.; Tan, E.Y.; et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E5990–E5999. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.M.; Bandopadhyay, G.; Coyle, B.; Grabowska, A. A HIF-independent, CD133-mediated mechanism of cisplatin resistance in glioblastoma cells. Cell Oncol. 2018, 41, 319–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Samanta, D.; Lu, H.; Bullen, J.W.; Zhang, H.; Chen, I.; He, X.; Semenza, G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA 2016, 113, E2047–E2056. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Tran, L.; Park, Y.; Chen, I.; Lan, J.; Xie, Y.; Semenza, G.L. Reciprocal regulation of DUSP9 and DUSP16 expression by HIF1 controls ERK and p38 MAP kinase activity and mediates chemotherapy-induced breast cancer stem cell enrichment. Cancer Res. 2018, 78, 4191–4202. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Weinberg, R.A. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shi, X.; Peng, Y.; Wu, M.; Zhang, P.; Xie, R.; Wu, Y.; Yan, Q.; Liu, S.; Wang, J. HIF-1α Promotes Epithelial-Mesenchymal Transition and Metastasis through Direct Regulation of ZEB1 in Colorectal Cancer. PLoS ONE 2015, 10, e0129603. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhofer, N.; Kong, C.; Le, Q.T.; Chi, J.T.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Sharma, P.; Dogra, N.; Singh, S. Eradicating cancer stem cells: Concepts, issues, and challenges. Curr. Treat. Opt. Oncol. 2018, 19, 20. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Cao, Y.; Luo, Y.; Hu, H.; Ling, H. Signalling mechanism(s) of epithelial-mesenchymal transition and cancer stem cells in tumour therapeutic resistance. Clin. Chim. Acta 2018, 483, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Zavadil, J.; Cermak, L.; Soto-Nieves, N.; Bottinger, E.P. Integration of TGF-β/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004, 23, 1155–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, S.; Charbonneau, M.; Grandmont, S.; Richard, D.E.; Dubois, C.M. Transforming growth factor β1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J. Biol. Chem. 2006, 281, 24171–24181. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, C.; Caja, L.; Moustakas, A. Transforming growth factor β as regulator of cancer stemness and metastasis. Br. J. Cancer 2016, 115, 761–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Gifford, C.C.; Samarakoon, R.; Higgins, P.J. Deregulation of negative controls on TGF-β1 signaling in tumor progression. Cancers 2018, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Konge, J.; Leteurtre, F.; Goislard, M.; Biard, D.; Morel-Altmeyer, S.; Vaurijoux, A.; Gruel, G.; Chevillard, S.; Lebeau, J. Breast cancer stem cell-like cells generated during TGFβ-induced EMT are radioresistant. Oncotarget 2018, 9, 23519–23531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singla, M.; Kumar, A.; Bal, A.; Sarkar, S.; Bhattacharyya, S. Epithelial to mesenchymal transition induces stem cell like phenotype in renal cell carcinoma cells. Cancer Cell Int. 2018, 18, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Gu, X.; Xia, L.; Zhou, Y.; Bouamar, H.; Yang, J.; Ding, X.; Zwieb, C.; Zhang, J.; Hinck, A.P.; et al. A Novel TGFβ Trap Blocks Chemotherapeutics-Induced TGFβ1 Signaling and Enhances Their Anticancer Activity in Gynecologic Cancers. Clin. Cancer Res. 2018, 24, 2780–2793. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Li, T.; Han, L.; Qin, P.; Wu, Z.; Xu, B.; Gao, Q.; Song, Y. TGFβ1-induced down-regulation of microRNA-138 contributes to epithelial-mesenchymal transition in primary lung cancer cells. Biochem. Biophys. Res. Commun. 2018, 496, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Chruscik, A.; Gopalan, V.; Lam, A.K. The clinical and biological roles of transforming growth factor β in colon cancer stem cells: A systematic review. Eur. J. Cell Biol. 2018, 97, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Lee, S.; Kim, H.R.; Park, S.Y.; Lee, M.; Yoon, J.H.; Kim, Y.J. Transforming growth factor-β decreases side population cells in hepatocellular carcinoma in vitro. Oncol Lett. 2018, 15, 8723–8728. [Google Scholar] [CrossRef] [PubMed]
- Galoczova, M.; Coates, P.; Vojtesek, B. STAT3, stem cells, cancer stem cells and p63. Cell. Mol. Biol. Lett. 2018, 23, 12. [Google Scholar] [CrossRef] [PubMed]
- Groner, B.; von Manstein, V. Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Mol. Cell. Endocrinol. 2017, 451, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.E.; Lee, H.G.; Cho, I.H.; Chung, D.H.; Yoon, S.H.; Yang, Y.M.; Lee, J.W.; Choi, S.; Park, J.W.; Ye, S.K.; et al. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J. 2005, 19, 1296–1298. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Briggs, J.; Park, S.; Niu, G.; Kortylewski, M.; Zhang, S.; Gritsko, T.; Turkson, J.; Kay, H.; Semenza, G.L.; et al. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 2005, 24, 5552–5560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.E.; Kim, H.S.; Lee, C.S.; Shin, Y.J.; Kim, Y.N.; Kang, G.H.; Kim, T.Y.; Juhnn, Y.S.; Kim, S.J.; Park, J.W.; et al. STAT3 inhibits the degradation of HIF-1α by pVHL-mediated ubiquitination. Exp. Mol. Med. 2008, 40, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Pawlus, M.R.; Wang, L.; Hu, C.J. STAT3 and HIF1α cooperatively activate HIF1 target genes in MDA-MB-231 and RCC4 cells. Oncogene 2014, 33, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.L.A.; Hirpara, J.L.; Pervaiz, S.; Eu, J.Q.; Sethi, G.; Goh, B.C. Do STAT3 inhibitors have potential in the future for cancer therapy? Exp. Opin. Investig. Drugs 2017, 26, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Wu, S.Y.; Pradeep, S.; Ivan, C.; Pecot, C.V.; Gharpure, K.M.; Nagaraja, A.S.; Armaiz-Pena, G.N.; McGuire, M.; Zand, B.; et al. Hypoxia-mediated downregulation of miRNA biogenesis promotes tumour progression. Nat. Commun. 2014, 5, 5202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Beucken, T.; Koch, E.; Chu, K.; Rupaimoole, R.; Prickaerts, P.; Adriaens, M.; Voncken, J.W.; Harris, A.L.; Buffa, F.M.; Haider, S.; et al. Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER. Nat. Commun. 2014, 5, 5203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, T.J.; Souza, A.L.; Clish, C.B.; Puigserver, P. A hypoxia-induced positive feedback loop promotes hypoxia-inducible factor 1α stability through miR-210 suppression of glycerol-3-phosphate dehydrogenase 1-like. Mol. Cell. Biol 2011, 31, 2696–2706. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Buart, S.; Romero, P.; Ketari, S.; Janji, B.; Mari, B.; Mami-Chouaib, F.; Chouaib, S. Hypoxia-inducible miR-210 regulates the susceptibility of tumor cells to lysis by cytotoxic T cells. Cancer Res. 2012, 72, 4629–4641. [Google Scholar] [CrossRef] [PubMed]
- Bavelloni, A.; Ramazzotti, G.; Poli, A.; Piazzi, M.; Focaccia, E.; Blalock, W.; Faenza, I. MiRNA-210: A Current Overview. Anticancer Res. 2017, 37, 6511–6521. [Google Scholar] [PubMed]
- Costales, M.G.; Haga, C.L.; Velagapudi, S.P.; Childs-Disney, J.L.; Phinney, D.G.; Disney, M.D. Small molecule inhibition of microRNA-210 reprograms an oncogenic hypoxic circuit. J. Am. Chem. Soc. 2017, 139, 3446–3455. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Zhao, L.; Chen, W.; Liu, T.; Li, Z.; Li, X. miR-210, a modulator of hypoxia-induced epithelial-mesenchymal transition in ovarian cancer cell. Int. J. Clin. Exp. Med. 2015, 8, 2299–2307. [Google Scholar] [PubMed]
- Bindra, R.S.; Glazer, P.M. Genetic instability and the tumor microenvironment: Towards the concept of microenvironment-induced mutagenesis. Mutat. Res. 2005, 569, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Scanlon, S.E.; Glazer, P.M. Multifaceted control of DNA repair pathways by the hypoxic tumor microenvironment. DNA Repair 2015, 32, 180–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luoto, K.R.; Kumareswaran, R.; Bristow, R.G. Tumor hypoxia as a driving force in genetic instability. Genome Integr. 2013, 4, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, T.Y.; Rockwell, S.; Glazer, P.M. Genetic instability induced by the tumor microenvironment. Cancer Res. 1996, 56, 5754–5757. [Google Scholar] [PubMed]
- Li, C.Y.; Little, J.B.; Hu, K.; Zhang, W.; Zhang, L.; Dewhirst, M.W.; Huang, Q. Persistent genetic instability in cancer cells induced by non-DNA-damaging stress exposures. Cancer Res. 2001, 61, 428–432. [Google Scholar] [PubMed]
- Keysar, S.B.; Trncic, N.; Larue, S.M.; Fox, M.H. Hypoxia/reoxygenation-induced mutations in mammalian cells detected by the flow cytometry mutation assay and characterized by mutant spectrum. Radiat. Res. 2010, 173, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, M.; Ohga, N.; Akiyama, K.; Hida, Y.; Maishi, N.; Towfik, A.M.; Inoue, N.; Shindoh, M.; Hida, K. Hypoxia-induced reactive oxygen species cause chromosomal abnormalities in endothelial cells in the tumor microenvironment. PLoS ONE 2013, 8, e80349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cao, S.; Toole, B.P.; Xu, Y. Cancer may be a pathway to cell survival under persistent hypoxia and elevated ROS: A model for solid-cancer initiation and early development. Int. J. Cancer 2015, 136, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Pires, I.M.; Bencokova, Z.; Milani, M.; Folkes, L.K.; Li, J.L.; Stratford, M.R.; Harris, A.L.; Hammond, E.M. Effects of acute versus chronic hypoxia on DNA damage responses and genomic instability. Cancer Res. 2010, 70, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Graeber, T.G.; Osmanian, C.; Jacks, T.; Housman, D.E.; Koch, C.J.; Lowe, S.W.; Giaccia, A.J. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 1996, 379, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Bindra, R.S.; Schaffer, P.J.; Meng, A.; Woo, J.; Maseide, K.; Roth, M.E.; Lizardi, P.; Hedley, D.W.; Bristow, R.G.; Glazer, P.M. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol. Cell. Biol. 2004, 24, 8504–8518. [Google Scholar] [CrossRef] [PubMed]
- Tsuchimoto, T.; Sakata, K.; Someya, M.; Yamamoto, H.; Hirayama, R.; Matsumoto, Y.; Furusawa, Y.; Hareyama, M. Gene expression associated with DNA-dependent protein kinase activity under normoxia, hypoxia, and reoxygenation. J. Radiat. Res. 2011, 52, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, V.T.; Bindra, R.S.; Yuan, J.; Campisi, D.; Narayanan, L.; Jensen, R.; Giordano, F.; Johnson, R.S.; Rockwell, S.; Glazer, P.M. Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol.Cell. Biol 2003, 23, 3265–3273. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Robbins, P.F. Cancer immunotherapy targeting neoantigens. Semin. Immunol. 2016, 28, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magri, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Anker, J.F.; Bais, P.; Namburi, S.; Giles, F.J.; Chuang, J.H. Mutations in DNA repair genes are associated with increased neo-antigen load and activated T cell infiltration in lung adenocarcinoma. Oncotarget 2018, 9, 7949–7960. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—What challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Jahanban-Esfahlan, R.; de la Guardia, M.; Ahmadi, D.; Yousefi, B. Modulating tumor hypoxia by nanomedicine for effective cancer therapy. J. Cell. Physiol. 2018, 233, 2019–2031. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, W.; Li, H.; Yan, W.; Yang, X.; Liu, D.; Wang, S.; Zhang, J. Two-photon fluorescent probe for detection of nitroreductase and hypoxia-specific microenvironment of cancer stem cell. Anal. Chim. Acta 2018, 1024, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Wigerup, C.; Pahlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Baran, N.; Konopleva, M. Molecular pathways: Hypoxia-activated prodrugs in cancer therapy. Clin. Cancer Res. 2017, 23, 2382–2390. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Cripps, A.; Wei, M.Q. New strategies for cancer gene therapy: Progress and opportunities. Clin. Exp. Pharmacol. Physiol. 2010, 37, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Zhang, H.; Qian, D.Z.; Rey, S.; Liu, J.O.; Semenza, G.L. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc. Natl. Acad. Sci. USA 2009, 106, 17910–17915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Saez, O.; Gajate Borau, P.; Alonso-Gordoa, T.; Molina-Cerrillo, J.; Grande, E. Targeting HIF-2 α in clear cell renal cell carcinoma: A promising therapeutic strategy. Crit. Rev. Oncol. Hematol. 2017, 111, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Kopecka, J.; Porto, S.; Lusa, S.; Gazzano, E.; Salzano, G.; Giordano, A.; Desiderio, V.; Ghigo, D.; Caraglia, M.; De Rosa, G.; et al. Self-assembling nanoparticles encapsulating zoledronic acid revert multidrug resistance in cancer cells. Oncotarget 2015, 6, 31461–31478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopecka, J.; Porto, S.; Lusa, S.; Gazzano, E.; Salzano, G.; Pinzon-Daza, M.L.; Giordano, A.; Desiderio, V.; Ghigo, D.; De Rosa, G.; et al. Zoledronic acid-encapsulating self-assembling nanoparticles and doxorubicin: A combinatorial approach to overcome simultaneously chemoresistance and immunoresistance in breast tumors. Oncotarget 2016, 7, 20753–20772. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qian, D.Z.; Tan, Y.S.; Lee, K.; Gao, P.; Ren, Y.R.; Rey, S.; Hammers, H.; Chang, D.; Pili, R.; et al. Digoxin and other cardiac glycosides inhibit HIF-1α synthesis and block tumor growth. Proc. Natl. Acad. Sci. USA 2008, 105, 19579–19586. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Rey, S.; Tafani, M.; Zhang, H.; Wong, C.C.; Russo, A.; Russo, M.A.; Semenza, G.L. Hypoxia-inducible factor 1-dependent expression of platelet-derived growth factor B promotes lymphatic metastasis of hypoxic breast cancer cells. Proc. Natl. Acad. Sci. USA 2012, 109, E2707–E2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beppu, K.; Nakamura, K.; Linehan, W.M.; Rapisarda, A.; Thiele, C.J. Topotecan blocks hypoxia-inducible factor-1α and vascular endothelial growth factor expression induced by insulin-like growth factor-I in neuroblastoma cells. Cancer Res. 2005, 65, 4775–4781. [Google Scholar] [CrossRef] [PubMed]
- Rapisarda, A.; Uranchimeg, B.; Sordet, O.; Pommier, Y.; Shoemaker, R.H.; Melillo, G. Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: Mechanism and therapeutic implications. Cancer Res. 2004, 64, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Rapisarda, A.; Uranchimeg, B.; Scudiero, D.A.; Selby, M.; Sausville, E.A.; Shoemaker, R.H.; Melillo, G. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res. 2002, 62, 4316–4324. [Google Scholar] [PubMed]
- Koh, M.Y.; Spivak-Kroizman, T.; Venturini, S.; Welsh, S.; Williams, R.R.; Kirkpatrick, D.L.; Powis, G. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1α. Mol. Cancer Ther. 2008, 7, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.; Williams, R.; Kirkpatrick, L.; Paine-Murrieta, G.; Powis, G. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1α. Mol. Cancer Ther. 2004, 3, 233–244. [Google Scholar] [PubMed]
- Liang, S.; Medina, E.A.; Li, B.; Habib, S.L. Preclinical evidence of the enhanced effectiveness of combined rapamycin and AICAR in reducing kidney cancer. Mol. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Zhang, S.; Zhang, Z.; Xu, P.; Mao, D.; Huang, S.; Chen, B.; Zhang, C.; Zhang, S. Jianpi Jiedu decoction, a traditional Chinese medicine formula, inhibits tumorigenesis, metastasis, and angiogenesis through the mTOR/HIF-1α/VEGF pathway. J. Ethnopharmacol. 2018, 224, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Mi, C.; Wang, Z.; Zhang, Z.H.; Li, M.Y.; Zuo, H.X.; Wang, J.Y.; Jin, X.; Ma, J. Fraxinellone has anticancer activity in vivo by inhibiting programmed cell death-ligand 1 expression by reducing hypoxia-inducible factor-1α and STAT3. Pharmacol. Res. 2018, 135, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Feliz, L.R.; Tsimberidou, A.M. Anti-vascular endothelial growth factor therapy in the era of personalized medicine. Cancer Chemother. Pharmacol. 2013, 72, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuvillier, O. The therapeutic potential of HIF-2 antagonism in renal cell carcinoma. Transl. Androl. Urol. 2017, 6, 131–133. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terry, S.; Faouzi Zaarour, R.; Hassan Venkatesh, G.; Francis, A.; El-Sayed, W.; Buart, S.; Bravo, P.; Thiery, J.; Chouaib, S. Role of Hypoxic Stress in Regulating Tumor Immunogenicity, Resistance and Plasticity. Int. J. Mol. Sci. 2018, 19, 3044. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103044
Terry S, Faouzi Zaarour R, Hassan Venkatesh G, Francis A, El-Sayed W, Buart S, Bravo P, Thiery J, Chouaib S. Role of Hypoxic Stress in Regulating Tumor Immunogenicity, Resistance and Plasticity. International Journal of Molecular Sciences. 2018; 19(10):3044. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103044
Chicago/Turabian StyleTerry, Stéphane, Rania Faouzi Zaarour, Goutham Hassan Venkatesh, Amirtharaj Francis, Walid El-Sayed, Stéphanie Buart, Pamela Bravo, Jérome Thiery, and Salem Chouaib. 2018. "Role of Hypoxic Stress in Regulating Tumor Immunogenicity, Resistance and Plasticity" International Journal of Molecular Sciences 19, no. 10: 3044. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103044