The Importance of the Right Framework: Mitogen-Activated Protein Kinase Pathway and the Scaffolding Protein PTPIP51

Abstract
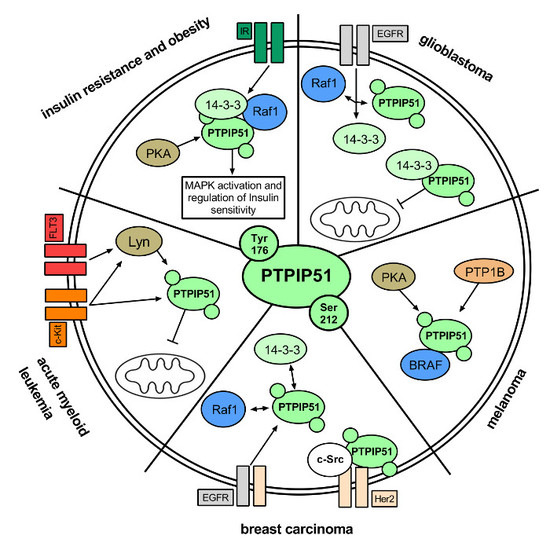
:
1. Background
2. Regulation of PTPIP51 in Metabolic Signaling
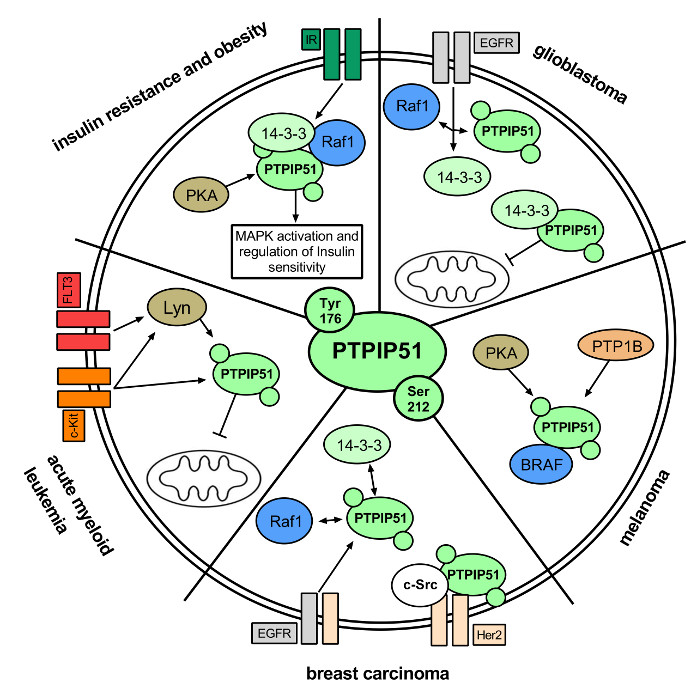
2.1. Insulin Resistance and Obesity
PTPIP51 in Insulin Resistance and Obesity
3. Regulation of PTPIP51 in Cancer
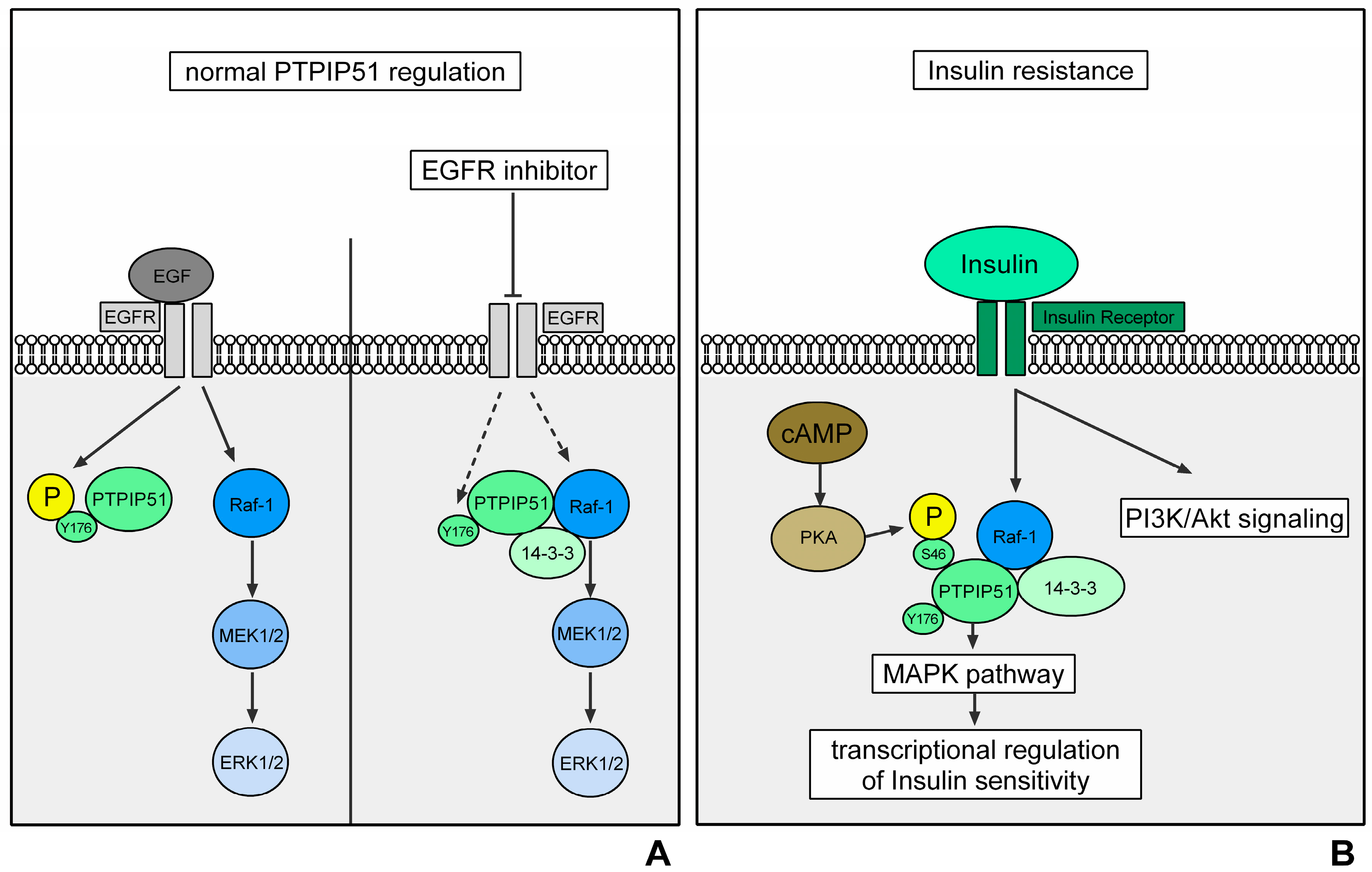
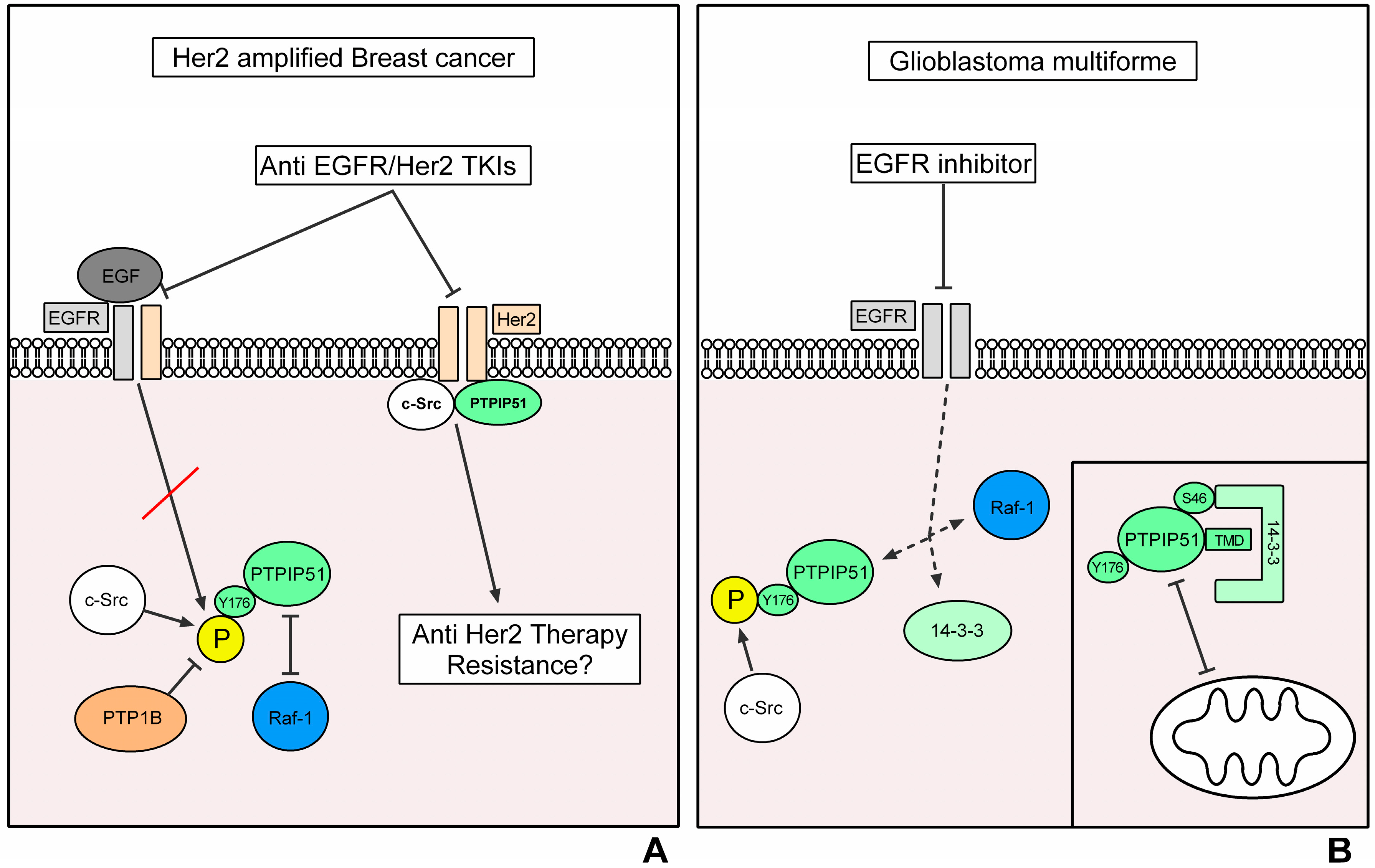
3.1. Breast Cancer
PTPIP51 in Breast Carcinoma
3.2. Glioblastoma Multiforme
PTPIP51 in Glioblastoma
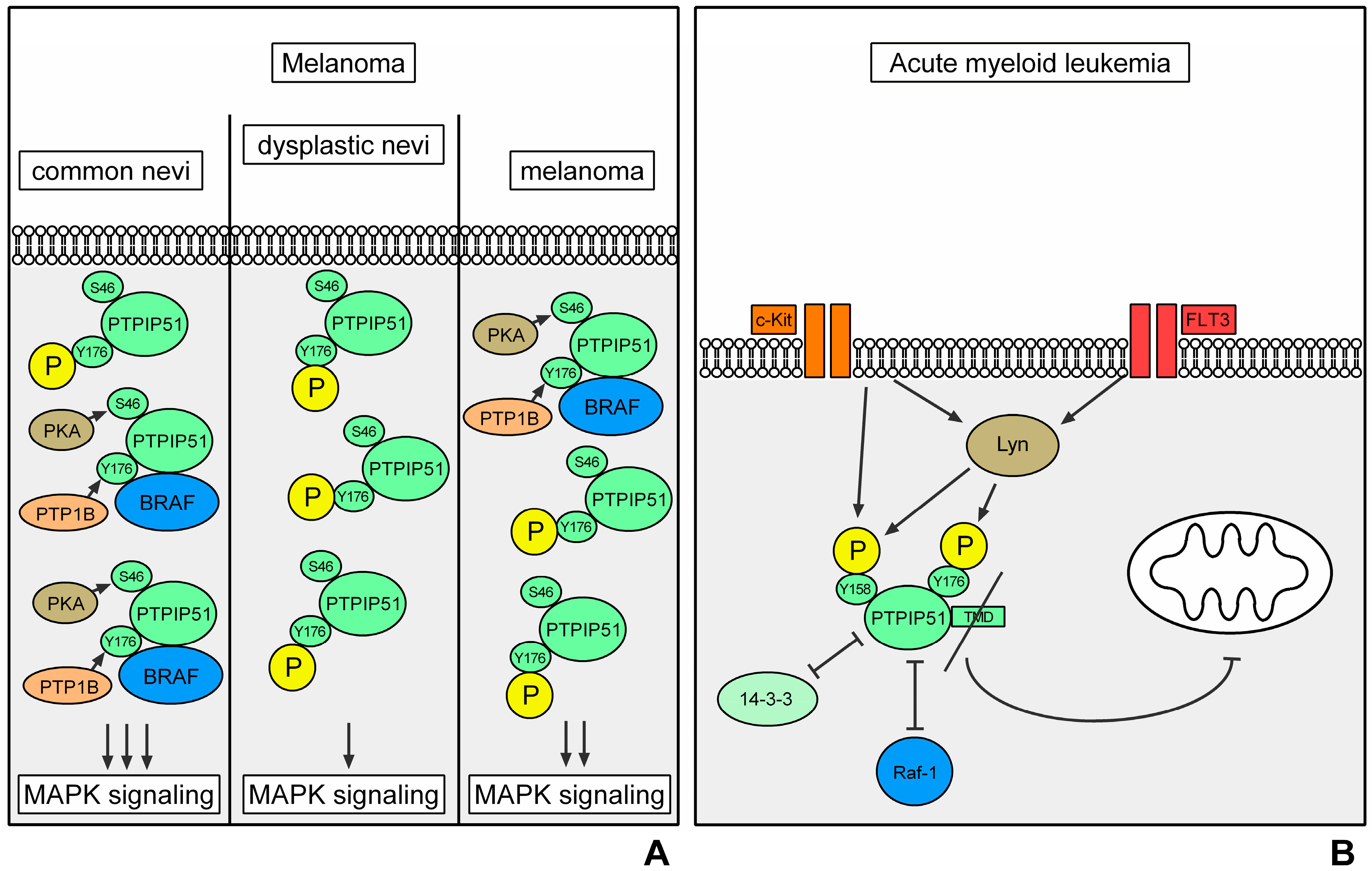
3.3. Melanoma
PTPIP51 in Melanoma
3.4. Acute Myeloid Leukemia
PTPIP51 in AML
4. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meister, M.; Tomasovic, A.; Banning, A.; Tikkanen, R. Mitogen-Activated Protein (MAP) Kinase Scaffolding Proteins: A Recount. Int. J. Mol. Sci. 2013, 14, 4854–4884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Tosuner, Z.; Geçer, M.Ö.; Hatiboğlu, M.A.; Abdallah, A.; Turna, S. BRAF V600E mutation and BRAF VE1 immunoexpression profiles in different types of glioblastoma. Oncol. Lett. 2018, 16, 2402–2408. [Google Scholar] [CrossRef] [PubMed]
- Thorson, J.A.; Yu, L.W.; Hsu, A.L.; Shih, N.Y.; Graves, P.R.; Tanner, J.W.; Allen, P.M.; Piwnica-Worms, H.; Shaw, A.S. 14-3-3 proteins are required for maintenance of Raf-1 phosphorylation and kinase activity. Mol. Cell. Biol. 1998, 18, 5229–5238. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Han, W.; Shi, T.; Lv, B.; He, Q.; Zhang, Y.; Li, T.; Zhang, Y.; Song, Q.; Wang, L.; et al. PTPIP51, a novel 14-3-3 binding protein, regulates cell morphology and motility via Raf-ERK pathway. Cell Signal 2008, 20, 2208–2220. [Google Scholar] [CrossRef] [PubMed]
- Brobeil, A.; Bobrich, M.; Tag, C.; Wimmer, M. PTPIP51 in protein interactions: Regulation and in situ interacting partners. Cell Biochem. Biophys. 2012, 63, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Brobeil, A.; Bobrich, M.; Wimmer, M. Protein tyrosine phosphatase interacting protein 51--a jack-of-all-trades protein. Cell Tissue Res. 2011, 344, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Stenzinger, A.; Schreiner, D.; Koch, P.; Hofer, H.-W.; Wimmer, M. Chapter 6 Cell and Molecular Biology of the Novel Protein Tyrosine-Phosphatase-Interacting Protein 51; Elsevier: Amsterdam, The Netherlands, 2009; pp. 183–246. [Google Scholar]
- Brobeil, A.; Koch, P.; Eiber, M.; Tag, C.; Wimmer, M. The known interactome of PTPIP51 in HaCaT cells—Inhibition of kinases and receptors. Int. J. Biochem. Cell Biol. 2014, 46, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Brobeil, A.; Dietel, E.; Gattenlöhner, S.; Wimmer, M. Orchestrating cellular signaling pathways-the cellular “conductor” protein tyrosine phosphatase interacting protein 51 (PTPIP51). Cell Tissue Res. 2017, 368, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Stoica, R.; de Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.-F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J.; et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoica, R.; Paillusson, S.; Gomez-Suaga, P.; Mitchell, J.C.; Lau, D.H.; Gray, E.H.; Sancho, R.M.; Vizcay-Barrena, G.; de Vos, K.J.; Shaw, C.E.; et al. ALS/FTD-associated FUS activates GSK-3β to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep. 2016, 17, 1326–1342. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Morotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.-F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.C.J. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C.J. The ER-Mitochondria Tethering Complex VAPB-PTPIP51 Regulates Autophagy. Curr. Biol. 2017, 27, 371–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Suaga, P.; Paillusson, S.; Miller, C.C.J. ER-mitochondria signaling regulates autophagy. Autophagy 2017, 13, 1250–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galmes, R.; Houcine, A.; van Vliet, A.R.; Agostinis, P.; Jackson, C.L.; Giordano, F. ORP5/ORP8 localize to endoplasmic reticulum-mitochondria contacts and are involved in mitochondrial function. EMBO Rep. 2016, 17, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Brobeil, A.; Kämmerer, F.; Tag, C.; Steger, K.; Gattenlöhner, S.; Wimmer, M. PTPIP51-A New RelA-tionship with the NFκB Signaling Pathway. Biomolecules 2015, 5, 485–504. [Google Scholar] [CrossRef] [PubMed]
- Brobeil, A.; Chehab, R.; Dietel, E.; Gattenlöhner, S.; Wimmer, M. Altered Protein Interactions of the Endogenous Interactome of PTPIP51 towards MAPK Signaling. Biomolecules 2017, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Bhupathiraju, S.N.; Hu, F.B. Epidemiology of Obesity and Diabetes and Their Cardiovascular Complications. Circ. Res. 2016, 118, 1723–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobrich, M.; Brobeil, A.; Mooren, F.C.; Krüger, K.; Steger, K.; Tag, C.; Wimmer, M. PTPIP51 interaction with PTP1B and 14-3-3β in adipose tissue of insulin-resistant mice. Int. J. Obes. 2011, 35, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Pessin, J.E.; Saltiel, A.R. Signaling pathways in insulin action: Molecular targets of insulin resistance. J. Clin. Investig. 2000, 106, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Thompson, B.J.; Hietakangas, V.; Cohen, S.M. MAPK/ERK signaling regulates insulin sensitivity to control glucose metabolism in Drosophila. PLoS Genet. 2011, 7, e1002429. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.-I.; Awazu, M.; Tamiya, M.; Iwasaki, Y.; Harada, A.; Kugisaki, S.; Tanimura, S.; Kohno, M. Targeting the ERK signaling pathway as a potential treatment for insulin resistance and type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E643–E651. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Fantus, I.G. Inhibition of the protein tyrosine phosphatase PTP1B: Potential therapy for obesity, insulin resistance and type-2 diabetes mellitus. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 621–640. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K.; Muthuswamy, S.K. A brake becomes an accelerator: PTP1B—A new therapeutic target for breast cancer. Cancer Cell 2007, 11, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. PTP1B: From the sidelines to the front lines! FEBS Lett. 2003, 546, 140–148. [Google Scholar] [CrossRef] [Green Version]
- Das, R.; Esposito, V.; Abu-Abed, M.; Anand, G.S.; Taylor, S.S.; Melacini, G. cAMP activation of PKA defines an ancient signaling mechanism. Proc. Natl. Acad. Sci. USA 2007, 104, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Shi, A.; Lu, C.; Song, T.; Zhang, Z.; Zhao, J. Breast Cancer: Epidemiology and Etiology. Cell Biochem. Biophys. 2015, 72, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.; Clark, G.; Wong, S.; Levin, W.; Ullrich, A.; McGuire, W. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.; Eiermann, W.; Robert, N.; Pienkowski, T.; Martin, M.; Press, M.; Mackey, J.; Glaspy, J.; Chan, A.; Pawlicki, M.; et al. Adjuvant Trastuzumab in HER2-Positive Breast Cancer. N. Engl. J. Med. 2011, 365, 1273–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moasser, M.M. The oncogene HER2: Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007, 26, 6469–6487. [Google Scholar] [CrossRef] [PubMed]
- Moasser, M.M. Targeting the function of the HER2 oncogene in human cancer therapeutics. Oncogene 2007, 26, 6577–6592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus Capecitabine for HER2-Positive Advanced Breast Cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef] [PubMed]
- Marty, M.; Cognetti, F.; Maraninchi, D.; Snyder, R.; Mauriac, L.; Tubiana-Hulin, M.; Chan, S.; Grimes, D.; Anton, A.; Lluch, A.; et al. Randomized phase II trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer administered as first-line treatment: The M77001 study group. J. Clin. Oncol. 2005, 23, 4265–4274. [Google Scholar] [CrossRef] [PubMed]
- Gajria, D.; Chandarlapaty, S. HER2-amplified breast cancer: Mechanisms of trastuzumab resistance and novel targeted therapies. Expert. Rev. Anticancer Ther. 2011, 11, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Canonici, A.; Gijsen, M.; Mullooly, M.; Bennett, R.; Bouguern, N.; Pedersen, K.; O’Brien, N.A.; Roxanis, I.; Li, J.-L.; Bridge, E.; et al. Neratinib overcomes trastuzumab resistance in HER2 amplified breast cancer. Oncotarget 2013, 4, 1592–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagliato, D.d.M.; Jardim, D.L.F.; Marchesi, M.S.P.; Hortobagyi, G.N. Mechanisms of resistance and sensitivity to anti-HER2 therapies in HER2+ breast cancer. Oncotarget 2016, 27, 64431–64446. [Google Scholar] [CrossRef] [PubMed]
- Peiró, G.; Ortiz-Martínez, F.; Gallardo, A.; Pérez-Balaguer, A.; Sánchez-Payá, J.; Ponce, J.J.; Tibau, A.; López-Vilaro, L.; Escuin, D.; Adrover, E.; et al. Src, a potential target for overcoming trastuzumab resistance in HER2-positive breast carcinoma. Br. J. Cancer 2014, 111, 689–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, K.L.; Hunter, L.A.; Ethier, S.P.; Boerner, J.L. Met and c-Src cooperate to compensate for loss of epidermal growth factor receptor kinase activity in breast cancer cells. Cancer Res. 2008, 68, 3314–3322. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.S. The hunting of the Src. Nat. Rev. Mol. Cell Biol. 2001, 2, 467–475. [Google Scholar]
- Zhang, S.; Huang, W.-C.; Li, P.; Guo, H.; Poh, S.-B.; Brady, S.W.; Xiong, Y.; Tseng, L.-M.; Li, S.-H.; Ding, Z.; et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med. 2011, 17, 461–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formisano, L.; Nappi, L.; Rosa, R.; Marciano, R.; D’Amato, C.; D’Amato, V.; Damiano, V.; Raimondo, L.; Iommelli, F.; Scorziello, A.; et al. Epidermal growth factor-receptor activation modulates Src-dependent resistance to lapatinib in breast cancer models. Breast Cancer Res. 2014, 16, R45. [Google Scholar] [CrossRef] [PubMed]
- Belsches-Jablonski, A.P.; Biscardi, J.S.; Peavy, D.R.; Tice, D.A.; Romney, D.A.; Parsons, S.J. Src family kinases and HER2 interactions in human breast cancer cell growth and survival. Oncogene 2001, 20, 1465–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldhammer, M.; Uetani, N.; Miranda-Saavedra, D.; Tremblay, M.L. PTP1B: A simple enzyme for a complex world. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 430–445. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.G.; Dubé, N.; Read, M.; Penney, J.; Paquet, M.; Han, Y.; Kennedy, B.P.; Muller, W.J.; Tremblay, M.L. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat. Genet. 2007, 39, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Hilmarsdottir, B.; Briem, E.; Halldorsson, S.; Kricker, J.; Ingthorsson, S.; Gustafsdottir, S.; Mælandsmo, G.M.; Magnusson, M.K.; Gudjonsson, T. Inhibition of PTP1B disrupts cell-cell adhesion and induces anoikis in breast epithelial cells. Cell Death Dis. 2017, 8, e2769. [Google Scholar] [CrossRef] [PubMed]
- Dietel, E.; Brobeil, A.; Tag, C.; Gattenloehner, S.; Wimmer, M. Effectiveness of EGFR/HER2-targeted drugs is influenced by the downstream interaction shifts of PTPIP51 in HER2-amplified breast cancer cells. Oncogenesis 2018, 7, 64. [Google Scholar] [CrossRef] [PubMed]
- McLendon, R.E.; Turner, K.; Perkinson, K.; Rich, J. Second messenger systems in human gliomas. Arch. Pathol. Lab. Med. 2007, 131, 1585–1590. [Google Scholar] [PubMed]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Jovčevska, I.; Kočevar, N.; Komel, R. Glioma and glioblastoma—How much do we (not) know? Mol. Clin. Oncol. 2013, 1, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.; Majewska, P.; Ioannidis, S.; Raza, M.H.; Williams, M. Estimating progression-free survival in patients with glioblastoma using routinely collected data. J. Neurooncol. 2017, 135, 621–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Vleeschouwer, S.D. Glioblastoma; Codon Publications: Brisbane, Australia, 2017. [Google Scholar]
- Yang, J.; Yan, J.; Liu, B. Targeting EGFRvIII for glioblastoma multiforme. Cancer Lett. 2017, 403, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Loew, S.; Schmidt, U.; Unterberg, A.; Halatsch, M.-E. The epidermal growth factor receptor as a therapeutic target in glioblastoma multiforme and other malignant neoplasms. Anticancer Agents Med. Chem. 2009, 9, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Clark, P.A.; Iida, M.; Treisman, D.M.; Kalluri, H.; Ezhilan, S.; Zorniak, M.; Wheeler, D.L.; Kuo, J.S. Activation of multiple ERBB family receptors mediates glioblastoma cancer stem-like cell resistance to EGFR-targeted inhibition. Neoplasia (N. Y.) 2012, 14, 420–428. [Google Scholar] [CrossRef]
- Golding, S.E.; Morgan, R.N.; Adams, B.R.; Hawkins, A.J.; Povirk, L.F.; Valerie, K. Pro-survival AKT and ERK signaling from EGFR and mutant EGFRvIII enhances DNA double-strand break repair in human glioma cells. Cancer Biol. Ther. 2009, 8, 730–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaffe, M.B. How do 14-3-3 proteins work?—Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett. 2002, 513, 53–57. [Google Scholar] [CrossRef]
- Yang, X.; Cao, W.; Lin, H.; Zhang, W.; Lin, W.; Cao, L.; Zhen, H.; Huo, J.; Zhang, X. Isoform-specific expression of 14-3-3 proteins in human astrocytoma. J. Neurol. Sci. 2009, 276, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Park, G.-Y.; Han, J.Y.; Han, Y.K.; Kim, S.D.; Kim, J.S.; Jo, W.S.; Chun, S.H.; Jeong, D.H.; Lee, C.-W.; Yang, K.; et al. 14-3-3 eta depletion sensitizes glioblastoma cells to irradiation due to enhanced mitotic cell death. Cancer Gene Ther. 2014, 21, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.K.; Koch, P.; Stenzinger, A.; Kuchelmeister, K.; Nestler, U.; Paradowska, A.; Steger, K.; Brobeil, A.; Viard, M.; Wimmer, M. PTPIP51, a positive modulator of the MAPK/Erk pathway, is upregulated in glioblastoma and interacts with 14-3-3β and PTP1B in situ. Histol. Histopathol. 2011, 26, 1531–1543. [Google Scholar] [PubMed]
- Lv, B.F.; Yu, C.F.; Chen, Y.Y.; Lu, Y.; Guo, J.H.; Song, Q.S.; Ma, D.L.; Shi, T.P.; Wang, L. Protein tyrosine phosphatase interacting protein 51 (PTPIP51) is a novel mitochondria protein with an N-terminal mitochondrial targeting sequence and induces apoptosis. Apoptosis 2006, 11, 1489–1501. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.K.; Brobeil, A.; Planz, J.; Bräuninger, A.; Gattenlöhner, S.; Nestler, U.; Stenzinger, A.; Paradowska, A.; Wimmer, M. PTPIP51 levels in glioblastoma cells depend on inhibition of the EGF-receptor. J. Neurooncol. 2015, 123, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Tuffin, L.J.; Feathers, R.; Hari, P.; Durand, N.; Li, Z.; Rodriguez, F.J.; Bakken, K.; Carlson, B.; Schroeder, M.; Sarkaria, J.N.; et al. Src family kinases differentially influence glioma growth and motility. Mol. Oncol. 2015, 9, 1783–1798. [Google Scholar] [CrossRef] [PubMed]
- Chan, X.Y.; Singh, A.; Osman, N.; Piva, T.J. Role Played by Signalling Pathways in Overcoming BRAF Inhibitor Resistance in Melanoma. Int. J. Mol. Sci. 2017, 18, E1527. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.J.; Johnson, D.B.; Sosman, J.A.; Chandra, S. Melanoma: What do all the mutations mean? Cancer 2018, 124, 3490–3499. [Google Scholar] [CrossRef] [PubMed]
- Suki, D. The Epidemiology of Brain Metastasis. In Intracranial Metastases: Current Management Strategies; Sawaya, R., Ed.; Blackwell Futura: Malden, MA, USA, 2004; pp. 20–34. [Google Scholar]
- Siu, T.L.; Huang, S. Cerebral metastases from malignant melanoma: Current treatment strategies, advances in novel therapeutics and future directions. Cancers 2010, 2, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Amaral, T.; Sinnberg, T.; Meier, F.; Krepler, C.; Levesque, M.; Niessner, H.; Garbe, C. The mitogen-activated protein kinase pathway in melanoma part I-Activation and primary resistance mechanisms to BRAF inhibition. Eur. J. Cancer 2017, 73, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Sperduto, P.W.; Jiang, W.; Brown, P.D.; Braunstein, S.; Sneed, P.; Wattson, D.A.; Shih, H.A.; Bangdiwala, A.; Shanley, R.; Lockney, N.A.; et al. The Prognostic Value of BRAF, C-KIT, and NRAS Mutations in Melanoma Patients with Brain Metastases. Int. J. Radiat. Oncol. Biol. Phys. 2017, 98, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.J. The role of mitogen-activated protein targeting in melanoma beyond BRAFV600. Curr. Opin. Oncol. 2016, 28, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Emuss, V.; Garnett, M.; Mason, C.; Marais, R. Mutations of C-RAF are rare in human cancer because C-RAF has a low basal kinase activity compared with B-RAF. Cancer Res. 2005, 65, 9719–9726. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Menzies, A.M.; Rizos, H. Mechanisms and strategies to overcome resistance to molecularly targeted therapy for melanoma. Cancer 2017, 123, 2118–2129. [Google Scholar] [CrossRef] [PubMed]
- Pilat, M.J.; Bumma, N.; Back, J.B.; Pandit, T.; Weise, A.M. BRAF: From Discovery to Drug Resistance. J. Sarcoma Res. 2018, 2, 1007. [Google Scholar]
- Si, L.; Zhang, X.; Xu, Z.; Jiang, Q.; Bu, L.; Wang, X.; Mao, L.; Zhang, W.; Richie, N.; Guo, J. Vemurafenib in Chinese patients with BRAFV600 mutation-positive unresectable or metastatic melanoma: An open-label, multicenter phase I study. BMC Cancer 2018, 18, 520. [Google Scholar] [CrossRef] [PubMed]
- Kirchberger, M.C.; Ugurel, S.; Mangana, J.; Heppt, M.V.; Eigentler, T.K.; Berking, C.; Schadendorf, D.; Schuler, G.; Dummer, R.; Heinzerling, L. MEK inhibition may increase survival of NRAS-mutated melanoma patients treated with checkpoint blockade: Results of a retrospective multicentre analysis of 364 patients. Eur. J. Cancer 2018, 98, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Choi, J.-W.; Kim, Y.-S. Frequencies of BRAF and NRAS mutations are different in histological types and sites of origin of cutaneous melanoma: A meta-analysis. Br. J. Dermatol. 2011, 164, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.E.; Liu, W.; Huynh, M.V.; Waqas, M.A.; Gillahan, J.E.; Clark, K.S.; Fu, K.; Martin, B.L.; Jeck, W.R.; Souroullas, G.P.; et al. Mutation-specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer Discov. 2014, 4, 1418–1429. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; García, J.S.; Perez-Garcia, J. NRAS-mutant melanoma: Current challenges and future prospect. Oncol. Targets Ther. 2017, 10, 3941–3947. [Google Scholar]
- Boespflug, A.; Caramel, J.; Dalle, S.; Thomas, L. Treatment of NRAS-mutated advanced or metastatic melanoma: Rationale, current trials and evidence to date. Ther. Adv. Med. Oncol. 2017, 9, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Lovly, C.M.; Sullivan, R.J.; Carvajal, R.D.; Sosman, J.A. Melanoma driver mutations and immune therapy. Oncoimmunology 2016, 5, e1051299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, N.E.; Edmiston, S.N.; Alexander, A.; Groben, P.A.; Parrish, E.; Kricker, A.; Armstrong, B.K.; Anton-Culver, H.; Gruber, S.B.; From, L.; et al. Association Between NRAS and BRAF Mutational Status and Melanoma-Specific Survival Among Patients with Higher-Risk Primary Melanoma. JAMA Oncol. 2015, 1, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Iorgulescu, J.B.; Harary, M.; Zogg, C.K.; Ligon, K.L.; Reardon, D.A.; Hodi, F.S.; Aizer, A.A.; Smith, T.R. Improved Risk-Adjusted Survival for Melanoma Brain Metastases in the Era of Checkpoint Blockade Immunotherapies: Results from a National Cohort. Cancer Immunol. Res. 2018, 6, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Echevarría-Vargas, I.M.; Reyes-Uribe, P.I.; Guterres, A.N.; Yin, X.; Kossenkov, A.V.; Liu, Q.; Zhang, G.; Krepler, C.; Cheng, C.; Wei, Z.; et al. Co-targeting BET and MEK as salvage therapy for MAPK and checkpoint inhibitor-resistant melanoma. EMBO Mol. Med. 2018, 10, e8446. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.M.; Tucker, M.A. Dysplastic nevi and melanoma. Cancer Epidemiol. Biomark. Prev. 2013, 22, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; O’Brien, S.; Jabbour, E.; Garcia-Manero, G.; Quintas-Cardama, A.; Shan, J.; Rios, M.B.; Ravandi, F.; Faderl, S.; Kadia, T.; et al. Improved survival in chronic myeloid leukemia since the introduction of imatinib therapy: A single-institution historical experience. Blood 2012, 119, 1981–1987. [Google Scholar] [CrossRef] [PubMed]
- Deschler, B.; Lübbert, M. Acute myeloid leukemia: Epidemiology and etiology. Cancer 2006, 107, 2099–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacher, U.; Schnittger, S.; Haferlach, C.; Haferlach, T. Molecular diagnostics in acute leukemias. Clin. Chem. Lab. Med. 2009, 47, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Mrózek, K.; Bloomfield, C.D. Chromosome aberrations, gene mutations and expression changes, and prognosis in adult acute myeloid leukemia. Hematol. Am. Soc. Hematol. Educ. Program. 2006, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Grafone, T.; Palmisano, M.; Nicci, C.; Storti, S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: Biology and treatment. Oncol. Rev. 2012, 6, e8. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, C.; Demur, C.; Bardet, V.; Prade-Houdellier, N.; Payrastre, B.; Récher, C. A critical role for Lyn in acute myeloid leukemia. Blood 2008, 111, 2269–2279. [Google Scholar] [CrossRef] [PubMed]
- Stirewalt, D.L.; Meshinchi, S.; Radich, J.P. Molecular targets in acute myelogenous leukemia. Blood Rev. 2003, 17, 15–23. [Google Scholar] [CrossRef]
- El Fakih, R.; Rasheed, W.; Hawsawi, Y.; Alsermani, M.; Hassanein, M. Targeting FLT3 Mutations in Acute Myeloid Leukemia. Cells 2018, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Levis, M. FLT3 mutations in acute myeloid leukemia: What is the best approach in 2013? Hematol. Am. Soc. Hematol. Educ. Program. 2013, 2013, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Hayakawa, F.; Miyata, Y.; Watamoto, K.; Emi, N.; Abe, A.; Kiyoi, H.; Towatari, M.; Naoe, T. Lyn is an important component of the signal transduction pathway specific to FLT3/ITD and can be a therapeutic target in the treatment of AML with FLT3/ITD. Leukemia 2007, 21, 403–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malaise, M.; Steinbach, D.; Corbacioglu, S. Clinical implications of c-Kit mutations in acute myelogenous leukemia. Curr. Hematol. Malig. Rep. 2009, 4, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Linnekin, D.; DeBerry, C.S.; Mou, S. Lyn associates with the juxtamembrane region of c-Kit and is activated by stem cell factor in hematopoietic cell lines and normal progenitor cells. J. Biol. Chem. 1997, 272, 27450–27455. [Google Scholar] [CrossRef] [PubMed]
- Brobeil, A.; Bobrich, M.; Graf, M.; Kruchten, A.; Blau, W.; Rummel, M.; Oeschger, S.; Steger, K.; Wimmer, M. PTPIP51 is phosphorylated by Lyn and c-Src kinases lacking dephosphorylation by PTP1B in acute myeloid leukemia. Leukemia Res. 2011, 35, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Okabe, S.; Tauchi, T.; Tanaka, Y.; Ohyashiki, K. Dasatinib preferentially induces apoptosis by inhibiting Lyn kinase in nilotinib-resistant chronic myeloid leukemia cell line. J. Hematol. Oncol. 2011, 4, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, S.-K.; Noh, E.-K.; Kim, J.Y.; Jeong, Y.K.; Jo, J.-C.; Choi, Y.; Koh, S.; Baek, J.H.; Min, Y.J.; Kim, H. Targeting c-KIT (CD117) by dasatinib and radotinib promotes acute myeloid leukemia cell death. Sci. Rep. 2017, 7, 15278. [Google Scholar] [CrossRef] [PubMed]
- Garbett, D.; Bretscher, A. The surprising dynamics of scaffolding proteins. Mol. Biol. Cell 2014, 25, 2315–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | PTPIP51-Related Mechanisms | Role of PTPIP51 as Diagnostic Biomarker | Targetable Molecule |
|---|---|---|---|
| Insulin Resistance | Transcriptional regulation of the IR via MAPK activation through the formation of the PTPIP51/14-3-3β/Raf-1 complex | expression of PTPIP51 negatively correlates with the grade of insulin sensitivity in mice | shifting PTPIP51 into MAPK signaling could enhance the transcription of IR and thus the insulin sensitivity |
| Breast Cancer | Sensitivity to EGFR/Her2 targeted TKIs depends on the formation of the Her2/c-Src/PTPIP51 complex | PTPIP51/PTP1B interaction positively correlates with the grading | Targeting the formation of the Her2/c-Src/PTPIP51 complex could overcome anti-EGFR/Her2 therapy resistances |
| Glioblastoma Multiforme | EGFR-targeted therapies are potentially bypassed via an enhanced interaction of c-Src and PTPIP51 | PTPIP51 mRNA expression positively correlates with the grading of glioma | Targeting the PTPIP51/c-Src interaction could overcome anti-EGFR therapy resistances. Inhibition of the PTPIP51/14-3-3β interaction could unveil the TMD of PTPIP51 and promote the apoptosis-inducing functions of PTPIP51 |
| Melanoma | Modulation of the serine and tyrosine phosphorylation of PTPIP51 via PKA and PTP1B induces a disease-stage-dependent alteration of the formation of the PTPIP51/14-3-3β/Raf-1 complex and thus the MAPK pathway activation | Phosphorylation and interaction profile of PTPIP51 is altered stage-dependently | Inhibition of PKA and PTP1B could reduce the interaction of PTPIP51 and BRAF and thus the MAPK stimulating effect of PTPIP51 |
| Acute Myeloid Leukemia | Loss of the TMD of PTPIP51 inhibits the apoptosis-inducing functions of PTPIP51. Phosphorylation of PTPIP51 via Lyn and c-Kit prevents the PTPIP51-induced MAPK pathway activation | PTPIP51 is expressed in a disease-related isoform without TMD |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dietel, E.; Brobeil, A.; Gattenlöhner, S.; Wimmer, M. The Importance of the Right Framework: Mitogen-Activated Protein Kinase Pathway and the Scaffolding Protein PTPIP51. Int. J. Mol. Sci. 2018, 19, 3282. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103282
Dietel E, Brobeil A, Gattenlöhner S, Wimmer M. The Importance of the Right Framework: Mitogen-Activated Protein Kinase Pathway and the Scaffolding Protein PTPIP51. International Journal of Molecular Sciences. 2018; 19(10):3282. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103282
Chicago/Turabian StyleDietel, Eric, Alexander Brobeil, Stefan Gattenlöhner, and Monika Wimmer. 2018. "The Importance of the Right Framework: Mitogen-Activated Protein Kinase Pathway and the Scaffolding Protein PTPIP51" International Journal of Molecular Sciences 19, no. 10: 3282. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103282