Many Cells Make Life Work—Multicellularity in Stem Cell-Based Cardiac Disease Modelling


Abstract
:
1. Introduction
2. Heart Disease Models
Stem Cells
3. The Composition of the Healthy Myocardium
3.1. Heart Disease Remodelling and Its Consequences
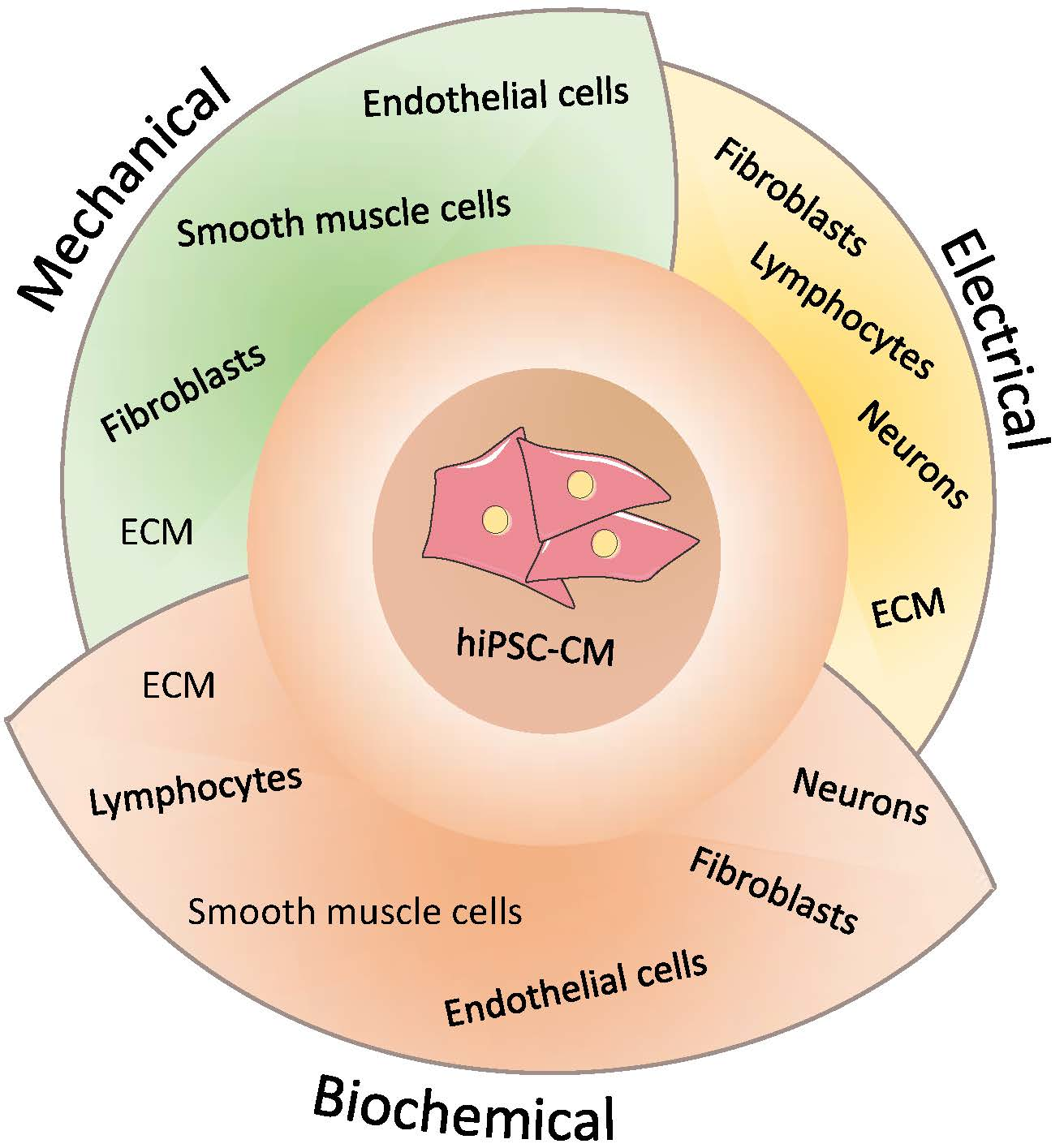
3.2. Multicellularity
3.2.1. Extracellular Matrix (ECM)
3.2.2. Fibroblasts
3.2.3. Endothelial Cells
3.2.4. Vascular Smooth Muscle Cells
3.2.5. Lymphocytes
3.2.6. Neurons
4. Conclusions and Future Prospects
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| ALDH-2 | Aldehyde dehydrogenase 2 |
| CICR | Calcium induced calcium release |
| CM | Cardiomyocyte |
| CPVT | Catecholaminergic polymorphic ventricular tachycardia |
| CHO | Chinese hamster ovary |
| EC | Endothelial cell |
| ECM | Extracellular matrix |
| ELN | Elastin |
| HEK | Human embryonic kidney |
| HiPSC | Human induced pluripotent stem cell |
| LQTS | Long-QT syndrome |
| LQT8 | Timothy Syndrome |
| RBC | Red blood cell |
| SMC | Smooth muscle cell |
| TAZ | Tafazzin |
References
- British Heart Foundation. Cardiovascular Disease Statistics 2015—BHF. Published 2015. Available online: https://www.bhf.org.uk/publications/statistics/cvd-stats-2015 (accessed on 15 September 2018).
- Edwards, A.G.; Louch, W.E. Species-dependent mechanisms of cardiac arrhythmia: A cellular focus. Clin. Med. Insights Cardiol. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Kofron, C.M.; Mende, U. In vitro models of the cardiac microenvironment to study myocyte and non-myocyte crosstalk: Bioinspired approaches beyond the polystyrene dish. J. Physiol. 2017, 595, 3891–3905. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Bernitz, J.M.; Lee, D.-F.; Lemischka, I.R. Genomic Editing Tools to Model Human Diseases with Isogenic Pluripotent Stem Cells. Stem Cells Dev. 2014, 23, 2673–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onakpoya, I.J.; Heneghan, C.J.; Aronson, J.K. Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: A systematic review of the world literature. BMC Med. 2016, 14, 191. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, S.; Kumar, S.; Verma, R.S.; Sharma, R.K. Cardiomyocyte culture—An update on the in vitro cardiovascular model and future challenges. Can. J. Physiol. Pharmacol. 2013, 91, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, J.; Bai, Y.; Zhao, X.; Liu, L. Isolation and Culture of Adult Mouse Cardiomyocytes for Cell Signaling and in vitro Cardiac Hypertrophy. J. Vis. Exp. 2014, 2–9. [Google Scholar] [CrossRef]
- Henrique Franco, N. Animal experiments in biomedical research: A historical perspective. Animals 2013, 3, 238–273. [Google Scholar] [CrossRef] [PubMed]
- Ericsson, A.C.; Crim, M.J.; Franklin, C.L. A brief history of animal modeling. Mo. Med. 2008, 110, 201–205. [Google Scholar] [CrossRef]
- Guénet, J.-L. Animal models of human genetic diseases: Do they need to be faithful to be useful? Mol. Genet. Genom. 2011, 286, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Smithies, O. Animal models of human genetic diseases. Trends Genet. 1993, 9, 112–116. [Google Scholar] [CrossRef]
- Erickson, R.P. Minireview: Creating Animal Models of Genetic Disease. Am. J. Hum. Genet. 1988, 43, 582–586. [Google Scholar] [PubMed]
- Whitelaw, C.B.A.; Sheets, T.P.; Lillico, S.G.; Telugu, B.P. Engineering large animal models of human disease. J. Pathol. 2016, 238, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Olson, H.; Betton, G.; Robinson, D.; Thomas, K.; Monro, A.; Kolaja, G.; Lilly, G.; Sanders, J.; Sipes, G.; Bracken, W.; et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul. Toxicol. Pharmacol. 2000, 32, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.K.; Liu, F.; Vaidyanathan, R.; Eckhardt, L.L.; Trudeau, M.C.; Robertson, G.A. hERG 1b is critical for human cardiac repolarization. Proc. Natl. Acad. Sci. USA 2014, 2014, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Physiol, C. Ion Channels in the Heart. Compr. Physiol. 2016, 5, 1423–1464. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, T.S.; Lee, S.T.; Wawrowsky, K.A.; Cheng, K.; Galang, G.; Malliaras, K.; Abraham, M.R.; Wang, C.; Marbán, E. Dedifferentiation and proliferation of mammalian cardiomyocytes. PLoS ONE 2010, 5, e12559. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.E.; Li, L.; Xia, X.; Fu, W.; Liao, Q.; Lan, C.; Yang, D.; Chen, H.; Yue, R.; Zeng, C.; et al. Dedifferentiation, proliferation, and redifferentiation of adult mammalian cardiomyocytes after ischemic injury. Circulation 2017, 136, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Dispersyn, G.D.; Geuens, E.; Ver Donck, L.; Ramaekers, F.C.S.; Borgers, M. Adult rabbit cardiomyocytes undergo hibernation-like dedifferentiation when co-cultured with cardiac fibroblasts. Cardiovasc. Res. 2001, 51, 230–240. [Google Scholar] [CrossRef] [Green Version]
- Pinz, I.; Zhu, M.; Mende, U.; Ingwall, J.S. An Improved Isolation Procedure for Adult Mouse Cardiomyocytes. Cell Biochem. Biophys. 2011, 61, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.; Nguyen, W.; Nguyenton, B.; Ratchada, P.; Page, G.; Miller, P.E.; Ghetti, A.; Abi-Gerges, N. Adult human primary cardiomyocyte-based model for the simultaneous prediction of drug-induced inotropic and pro-arrhythmia risk. Front. Physiol. 2017, 8, 1073. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Heo, I.; Clevers, H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol. Med. 2017, 23, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.; Pek, N.; Soh, B.-S. Disease Modeling Using 3D Organoids Derived from Human Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2018, 19, 936. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson-Bouchard, K.; Ma, S.P.; Yeager, K.; Chen, T.; Song, L.; Sirabella, D.; Morikawa, K.; Teles, D.; Yazawa, M.; Vunjak-Novakovic, G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 2018, 556, 239. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.J.; Brougham, C.M.; Garciarena, C.D.; Kerrigan, S.W.; O’Brien, F.J. Towards 3D in vitro models for the study of cardiovascular tissues and disease. Drug Discov. Today 2016, 21, 1437–1445. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Ma, Z.; Loskill, P.; Jeeawoody, S.; Healy, K.E. In vitro cardiac tissue models: Current status and future prospects. Adv. Drug Deliv. Rev. 2016, 96, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelton, M.; Kocharyan, A.; Liu, J.; Skerjanc, I.S.; Stanford, W.L. Robust generation and expansion of skeletal muscle progenitors and myocytes from human pluripotent stem cells. Methods 2016, 101, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Zwi, L.; Caspi, O.; Arbel, G.; Huber, I.; Gepstein, A.; Park, I.H.; Gepstein, L. Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation 2009, 120, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. PNAS Plus: Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1848–E1857. [Google Scholar] [CrossRef] [PubMed]
- Tse, H.-F.; Ho, J.C.Y.; Choi, S.-W.; Lee, Y.K.; Butler, A.W.; Ng, K.M.; Siu, C.-W.; Simpson, M.A.; Lai, W.-H.; Chan, Y.-C.; et al. Patient-specific Induced Pluripotent Stem Cells Derived Cardiomycoytes Recapitulates the Pathogenic Phenotypes of Dilated Cardiomyopathy due to a Novel DES Mutation Identified by Whole Exome Sequencing. Hum. Mol. Genet. 2013, 22, 1395–1403. Available online: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=23300193&retmode=ref&cmd=prlinks\npapers2://publication/doi/10.1093/hmg/dds556 (accessed on 22 September 2018).
- Veerman, C.C.; Mengarelli, I.; Guan, K.; Stauske, M.; Barc, J.; Tan, H.L.; Wilde, A.A.M.; Verkerk, A.O.; Bezzina, C.R. HiPSC-derived cardiomyocytes from Brugada Syndrome patients without identified mutations do not exhibit clear cellular electrophysiological abnormalities. Sci. Rep. 2016, 6, 30967. [Google Scholar] [CrossRef] [PubMed]
- Kehl, D.; Weber, B.; Hoerstrup, S.P. Bioengineered living cardiac and venous valve replacements: Current status and future prospects. Cardiovasc. Pathol. 2016, 25, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.-Y.; Wei, C.-Y.; Shah, K.; Wong, J.; Wang, C.; Chen, H.-S.V. Maturation-Based Model of Arrhythmogenic Right Ventricular Dysplasia Using Patient-Specific Induced Pluripotent Stem Cells. Circ. J. 2015, 79, 1402–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freund, C.; Davis, R.P.; Gkatzis, K.; Ward-van Oostwaard, D.; Mummery, C.L. The first reported generation of human induced pluripotent stem cells (iPS cells) and iPS cell-derived cardiomyocytes in The Netherlands. Neth. Heart J. 2010, 18, 51–54. [Google Scholar] [PubMed]
- Zwi-Dantsis, L.; Huber, I.; Habib, M.; Winterstern, A.; Gepstein, A.; Arbel, G.; Gepstein, L. Derivation and cardiomyocyte differentiation of induced pluripotent stem cells from heart failure patients. Eur. Heart J. 2013, 34, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Karakikes, I.; Ameen, M.; Termglinchan, V.; Wu, J.C. Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Insights into Molecular, Cellular, and Functional Phenotypes. Circ. Res. 2015, 117, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Adams, W.J.; Zhang, Y.; Cloutier, J.; Kuchimanchi, P.; Newton, G.; Sehrawat, S.; Aird, W.C.; Mayadas, T.N.; Luscinskas, F.W.; García-Cardeña, G. Functional Vascular Endothelium Derived from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2013, 1, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.F.; et al. A human iPSC model of hutchinson gilford progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011, 8, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Ren, Y.; Bartulos, O.; Lee, M.Y.; Yue, Z.; Kim, K.Y.; Li, W.; Amos, P.J.; Bozkulak, E.C.; Iyer, A.; et al. Modeling supravalvular aortic stenosis syndrome with human induced pluripotent stem cells. Circulation 2012, 126, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- Granata, A.; Serrano, F.; Bernard, W.G.; McNamara, M.; Low, L.; Sastry, P.; Sinha, S. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat. Genet. 2017, 49, 97–109. [Google Scholar] [CrossRef] [PubMed]
- French, A.; Yang, C.-T.; Taylor, S.; Watt, S.M.; Carpenter, L. Human Induced Pluripotent Stem Cell-Derived B Lymphocytes Express sIgM and Can Be Generated via a Hemogenic Endothelium Intermediate. Stem Cells Dev. 2015, 24, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Yu, Y.; Sun, L.; Wang, S.; Wang, R.; Zhang, L.; Li, C.; Wang, D. Induction of Pluripotent Stem Cell-Derived Cardiomyocyte Toxicity by Supernatant of Long Term-Stored Red Blood Cells in Vitro. Cell Physiol. Biochem. 2018, 46, 1230–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Habibollah, S.; Tilgner, K.; Collin, J.; Barta, T.; Al-Aama, J.Y.; Tesarov, L.; Hussain, R.; Trafford, A.W.; Kirkwood, G.; et al. An Induced Pluripotent Stem Cell Model of Hypoplastic Left Heart Syndrome (HLHS) Reveals Multiple Expression and Functional Differences in HLHS-Derived Cardiac Myocytes. Stem Cells Transl. Med. 2014, 3, 416–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Wong, J.; Wen, J.; Wang, S.; Wang, C.; Spiering, S.; Kan, N.G.; Forcales, S.; Puri, P.L.; Leone, T.C.; et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 2013, 494, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, F.; Lee, A.S.S.; Liang, P.; Sanchez-Freire, V.; Nguyen, P.K.K.; Wang, L.; Han, L.; Yen, M.; Wang, Y.; Sun, N.; et al. Abnormal Calcium Handling Properties Underlie Familial Hypertrophic Cardiomyopathy Pathology in Patient-Specific Induced Pluripotent Stem Cells. Cell Stem Cell 2013, 12, 101–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvajal-Vergara, X.; Sevilla, A.; Dsouza, S.L.; Ang, Y.S.; Schaniel, C.; Lee, D.F.; Yang, L.; Kaplan, A.D.; Adler, E.D.; Rozov, R.; et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 2010, 465, 808–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hick, A.; Wattenhofer-Donze, M.; Chintawar, S.; Tropel, P.; Simard, J.P.; Vaucamps, N.; Gall, D.; Lambot, L.; Andre, C.; Reutenauer, L.; et al. Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in Friedreich’s ataxia. Dis. Model Mech. 2013, 6, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Fatima, A.; Xu, G.; Shao, K.; Papadopoulos, S.; Lehmann, M.; Arnáiz-Cot, J.J.; Rosa, A.O.; Nguemo, F.; Matzkies, M.; Dittmann, S.; et al. Cellular Physiology Cellular Physiology Cellular Physiology Cellular Physiology Cellular Physiology In vitro Modeling of Ryanodine Receptor 2 Dysfunction Using Human Induced Pluripotent Stem Cells. Cell Physiol. Biochem. 2011, 28, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.; Barad, L.; Zeevi-Levin, N.; Shick, R.; Shtrichman, R.; Lorber, A.; Itskovitz-Eldor, J.; Binah, O. Cardiomyocytes generated from CPVT D307H patients are arrhythmogenic in response to β-adrenergic stimulation. J. Cell. Mol. Med. 2012, 16, 468–482. [Google Scholar] [CrossRef] [PubMed]
- Lahti, A.L.; Kujala, V.J.; Chapman, H.; Koivisto, A.-P.; Pekkanen-Mattila, M.; Kerkela, E.; Hyttinen, J.; Kontula, K.; Swan, H.; Conklin, B.R.; et al. Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis. Model Mech. 2012, 5, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.P.; Casini, S.; Van Den Berg, C.W.; Hoekstra, M.; Remme, C.A.; Dambrot, C.; Salvatori, D.; Oostwaard, D.W.; Wilde, A.A.; Bezzina, C.R.; et al. Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation 2012, 125, 3079–3091. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flügel, L.; Dorn, T.; Goedel, A.; Höhnke, C.; Hofmann, F.; et al. Patient-Specific Induced Pluripotent Stem-Cell Models for Long-QT Syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, M.; Hsueh, B.; Jia, X.; Pasca, A.M.; Bernstein, J.A.; Hallmayer, J.; Dolmetsch, R.E. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 2011, 471, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Rocchetti, M.; Sala, L.; Dreizehnter, L.; Crotti, L.; Sinnecker, D.; Mura, M.; Pane, L.S.; Altomare, C.; Torre, E.; Mostacciuolo, G.; et al. Elucidating arrhythmogenic mechanisms of long-QT syndrome CALM1-F142L mutation in patient-specific induced pluripotent stem cell-derived cardiomyocytes. Cardiovasc. Res. 2017, 113, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Limpitikul, W.B.; Dick, I.E.; Tester, D.J.; Boczek, N.J.; Limphong, P.; Yang, W.; Choi, M.H.; Babich, J.; Disilvestre, D.; Kanter, R.J.; et al. A Precision Medicine Approach to the Rescue of Function on Malignant Calmodulinopathic Long-QT Syndrome. Circ. Res. 2017, 120, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, A.D.; Kodo, K.; Liang, P.; Wu, H.; Huber, B.C.; Riegler, J.; Churko, J.; Lee, J.; de Almeida, P.; Lan, F.; et al. Characterization of the molecular mechanisms underlying increased ischemic damage in the aldehyde dehydrogenase 2 genetic polymorphism using a human induced pluripotent stem cell model system. Sci. Transl. Med. 2014, 6, 255ra130. [Google Scholar] [CrossRef] [PubMed]
- Kane, C.; Couch, L.; Terracciano, C.M. Excitation–contraction coupling of human induced pluripotent stem cell-derived cardiomyocytes. Front. Cell Dev. Biol. 2015, 3, 59. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Rollins, A.; Polo-Parada, L.; Ma, P.; Gu, S.; Jenkins, M. Probing the Electrophysiology of the Developing Heart. J. Cardiovasc. Dev. Dis. 2016, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Gwathmey, J.K.; Slawsky, M.T.; Hajjar, R.J.; Briggs, G.M.; Morgan, J.P. Role of intracellular calcium handling in force-interval relationships of human ventricular myocardium. J. Clin. Investig. 1990, 85, 1599–1613. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.L.; Piper, H.M. Cell cultures of adult cardiomyocytes as models of the myocardium. J. Mol. Cell. Cardiol. 1986, 18, 661–678. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouadon, E.; Moore-Morris, T.; Smit, N.W.; Chatenoud, L.; Coronel, R.; Harding, S.E.; Jourdon, P.; Lambert, V.; Rucker-Martin, C.; Pucéat, M. Concise Review: Pluripotent Stem Cell-Derived Cardiac Cells, A Promising Cell Source for Therapy of Heart Failure: Where Do We Stand? Stem Cells 2016, 34, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Beuckelmann, D.J.; Nabauer, M.; Erdmann, E. Intracellular Calcium Handling in Isolated Ventricular Myocytes from Patients with Terminal Heart Failure. Am. Heart J. 1992, 85, 1046–1055. [Google Scholar] [CrossRef]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Kraev, A.; Chumakov, I.; Carafoli, E. The organization of the human gene NCX1 encoding the sodium-calcium exchanger. Genomics 1996, 37, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Osafune, K.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Chen, S.; Muhlestein, W.; Melton, D.A. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat. Biotechnol. 2008, 26, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Olson, E.N.; Bassel-Duby, R. Mending broken hearts: Cardiac development as a basis for adult heart regeneration and repair. Nat. Rev. Mol. Cell Biol. 2013, 14, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Grubb, S.; Calloe, K.; Thomsen, M.B. Impact of KChiP2 on cardiac electrophysiology and the progression of heart failure. Front. Physiol. 2012, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Deschenes, I. Regulation of Kv4.3 Current by KChIP2 Splice Variants: A Component of Native Cardiac Ito? Circulation 2002, 106, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Razeghi, P.; Young, M.E.; Alcorn, J.L.; Moravec, C.S.; Frazier, O.H.; Taegtmeyer, H. Metabolic Gene Expression in Fetal and Failing Human Heart. Circulation 2001, 104, 2923–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Gros, D.B.; Jongsma, H.J. Connexins in mammalian heart function. Bioessays 1996, 18, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Dick, E.; Rajamohan, D.; Ronksley, J.; Denning, C. Evaluating the utility of cardiomyocytes from human pluripotent stem cells for drug screening. Biochem. Soc. Trans. 2010, 38, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Gaborit, N.; Le Bouter, S.; Szuts, V.; Varro, A.; Escande, D.; Nattel, S.; Demolombe, S. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J. Physiol. 2007, 582, 675–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Berg, C.W.; Okawa, S.; Chuva de Sousa Lopes, S.M.; van Iperen, L.; Passier, R.; Braam, S.R.; Tertoolen, L.G.; Del Sol, A.; Davis, R.P.; Mummery, C.L. Transcriptome of human foetal heart compared with cardiomyocytes from pluripotent stem cells. Development 2015, 142, 3231–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, H.S.; Kryshtal, D.O.; Feaster, T.K.; Sánchez-Freire, V.; Zhang, J.; Kamp, T.J.; Hong, C.C.; Wu, J.C.; Knollmann, B.C. Comparable calcium handling of human iPSC-derived cardiomyocytes generated by multiple laboratories. J. Mol. Cell. Cardiol. 2015, 85, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Bizy, A.; Guerrero-Serna, G.; Hu, B.; Ponce-Balbuena, D.; Willis, B.C.; Zarzoso, M.; Ramirez, R.J.; Sener, M.F.; Mundada, L.V.; Klos, M.; et al. Myosin light chain 2-based selection of human iPSC-derived early ventricular cardiac myocytes. Stem Cell Res. 2013, 11, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Miyagawa, S.; Pearson, J.T.; Fukushima, S.; Saito, A.; Tsuchimochi, H.; Sonobe, T.; Fujii, Y.; Yagi, N.; Astolfo, A.; et al. Functional and Electrical Integration of Induced Pluripotent Stem Cell-Derived Cardiomyocytes in a Myocardial Infarction Rat Heart. Cell Transplant. 2015, 24, 2479–2489. [Google Scholar] [CrossRef] [PubMed]
- Du, D.T.M.; Hellen, N.; Kane, C.; Terracciano, C.M.N. Action potential morphology of human induced pluripotent stem cell-derived cardiomyocytes does not predict cardiac chamber specificity and is dependent on cell density. Biophys. J. 2015, 108, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Sallam, K.; Li, Y.; Sager, P.T.; Houser, S.R.; Wu, J.C. Finding the Rhythm of Sudden Cardiac Death: New Opportunities Using Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. Res. 2015, 116, 1989–2004. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Cheng, H.; Tomaselli, G.F.; Li, R.A. Mechanistic basis of excitation-contraction coupling in human pluripotent stem cell-derived ventricular cardiomyocytes revealed by Ca2+ spark characteristics: Direct evidence of functional Ca2+-induced Ca2+ release. Heart Rhythm. 2014, 11, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.; Aylott, M.; Cui, Y.; Louttit, J.B.; McMahon, N.C.; Sridhar, A. Comparison of Electrophysiological Data From Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes to Functional Preclinical Safety Assays. Toxicol. Sci. 2013, 134, 412–426. [Google Scholar] [CrossRef] [PubMed]
- Herron, T.J.; Lee, P.; Jalife, J. Optical Imaging of Voltage and Calcium in Cardiac Cells & Tissues. Circ. Res. 2012, 110, 609–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Pabon, L.; Murry, C.E. Engineering Adolescence: Maturation of Human Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. Res. 2014, 114, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokushalov, E.; Romanov, A.; Steinberg, J.S. Stem Cell Therapy for Electrophysiological Disorders. Curr. Cardiol. Rep. 2013, 15, 408. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Holmström, A.; Wu, J.C. Chemically Defined Culture and Cardiomyocyte Differentiation of Human Pluripotent Stem Cells. In Current Protocols in Human Genetics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015. [Google Scholar]
- Liu, J.; Laksman, Z.; Backx, P.H. The electrophysiological development of cardiomyocytes. Adv. Drug Deliv. Rev. 2016, 96, 253–273. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Verma, V.; Nandihalli, M.; Ramachandra, C.J.A.; Sequiera, G.L.; Sudibyo, Y.; Chung, Y.; Sun, W.; Shim, W. A systemic evaluation of cardiac differentiation from mRNA reprogrammed human induced pluripotent stem cells. PLoS ONE 2014, 9, e103485. [Google Scholar] [CrossRef] [PubMed]
- Argenziano, M.; Lambers, E.; Hong, L.; Sridhar, A.; Zhang, M.; Chalazan, B.; Menon, A.; Savio-Galimberti, E.; Wu, J.C.; Rehman, J.; et al. Electrophysiologic Characterization of Calcium Handling in Human Induced Pluripotent Stem Cell-Derived Atrial Cardiomyocytes. Stem Cell Rep. 2018, 10, 1867–1878. [Google Scholar] [CrossRef] [PubMed]
- Marcu, I.C.; Illaste, A.; Heuking, P.; Jaconi, M.E.; Ullrich, N.D. Functional characterization and comparison of intercellular communication in stem cell-derived cardiomyocytes. Stem Cells 2015, 33, 2208–2218. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Pu, W.T. Recounting Cardiac Cellular Composition. Circ. Res. 2016, 118, 368–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vedin, O.; Lam, C.S.P.; Koh, A.S.; enson, L.; Teng, T.H.K.; Tay, W.T.; Braun, O.O.; Savarese, G.; Dahlström, U.; Lund, L.H. Significance of Ischemic Heart Disease in Patients with Heart Failure and Preserved, Midrange, and Reduced Ejection Fraction: A Nationwide Cohort Study. Circ. Heart Fail. 2017, 10, e003875. [Google Scholar] [CrossRef] [PubMed]
- Shirazi, L.F.; Bissett, J.; Romeo, F.; Mehta, J.L. Role of Inflammation in Heart Failure. Curr. Atheroscler. Rep. 2017, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Tuzun, E.; Bick, R.; Kadipasaoglu, C.; Conger, J.L.; Poindexter, B.J.; Gregoric, I.D.; Frazier, O.H.; Towbin, J.A.; Radovancevic, B. Modification of a volume-overload heart failure model to track myocardial remodeling and device-related reverse remodeling. ISRN Cardiol. 2011, 2011, 831062. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, V.; Pirozzi, F.; Lazzarini, E.; Marone, G.; Rizzo, P.; Agnetti, G.; Tocchetti, C.G.; Ghigo, A.; Ameri, P. Models of Heart Failure Based on the Cardiotoxicity of Anticancer Drugs. J. Card. Fail. 2016, 22, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, P.S.; Polegato, B.F.; Minicucci, M.F.; Paiva, S.A.R.; Zornoff, L.A.M. Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arq. Bras. Cardiol. 2016, 106, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Li, A.H.; Liu, P.P.; Villarreal, F.J.; Garcia, R.A. Dynamic changes in myocardial matrix and relevance to disease: Translational perspectives. Circ. Res. 2014, 114, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, A.M.; Kellerman, S.E.; Moore, J.A.; Muffly, K.E.; Clark, L.C.; Reaves, P.Y.; Malec, K.B.; McKeown, P.P.; Schocken, D.D. Structural remodeling of cardiac myocytes in patients with ischemic cardiomyopathy. Circulation 1992, 86, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Peter, A.K.; Bjerke, M.A.; Leinwand, L.A. Biology of the cardiac myocyte in heart disease. Mol. Biol. Cell 2016, 27, 2149–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, C.M.; Baudino, T.A. Dynamic cell-cell and cell-ECM interactions in the heart. J. Mol. Cell. Cardiol. 2014, 70, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Doppler, S.A.; Carvalho, C.; Lahm, H.; Deutsch, M.-A.; Dreßen, M.; Puluca, N.; Lange, R.; Krane, M. Cardiac fibroblasts: More than mechanical support. J. Thorac. Dis. 2017, 9, S36–S51. [Google Scholar] [CrossRef] [PubMed]
- Segers, V.F.M.; Brutsaert, D.L.; De Keulenaer, G.W. Cardiac remodeling: Endothelial cells have more to say than just NO. Front. Physiol. 2018, 9, 382. [Google Scholar] [CrossRef] [PubMed]
- Talman, V.; Kivelä, R. Cardiomyocyte—Endothelial Cell Interactions in Cardiac Remodeling and Regeneration. Front. Cardiovasc. 2018, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Furtado, M.B.; Rosenthal, N. The interstitium in cardiac repair: Role of the immune–stromal cell interplay. Nat. Rev. Cardiol. 2018, 15, 601–616. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tu, H.; Wang, C.; Cao, L.; Muelleman, R.L.; Wadman, M.C.; Li, Y.L. Correlation of ventricular arrhythmogenesis with neuronal remodeling of cardiac postganglionic parasympathetic neurons in the late stage of heart failure after myocardial infarction. Front. Neurosci. 2017, 11, 252. [Google Scholar] [CrossRef] [PubMed]
- Joki, Y.; Ohashi, K.; Yuasa, D.; Shibata, R.; Kataoka, Y.; Kambara, T.; Uemura, Y.; Matsuo, K.; Hayakawa, S.; Hiramatsu-Ito, M.; et al. Neuron-derived neurotrophic factor ameliorates adverse cardiac remodeling after experimental myocardial infarction. Circ. Heart Fail. 2015, 8, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Takawale, A.; Lee, J.; Kassiri, Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 2012, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Endemann, D.H.; Schiffrin, E.L. Endothelial dysfunction. J. Am. Soc. Nephrol. 2004, 15, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Liu, P.P.; Mann, D.L. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat. Rev. Immunol. 2015, 15, 117–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartledge, J.E.; Kane, C.; Dias, P.; Tesfom, M.; Clarke, L.; Mckee, B.; Al Ayoubi, S.; Chester, A.; Yacoub, M.H.; Camelliti, P.; et al. Functional crosstalk between cardiac fibroblasts and adult cardiomyocytes by solublemediators. Cardiovasc. Res. 2015, 105, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, M.; Watari, K.; Tajima, M.; Nakaya, T.; Matsuda, S.; Ohara, H.; Nishihara, H.; Yamaguchi, H.; Hashimoto, A.; Nishida, M.; et al. Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J. Clin. Investig. 2017, 127, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Snider, P.; Standley, K.N.; Wang, J.; Azhar, M.; Doetschman, T.; Conway, S.J. Origin of Cardiac Fibroblasts and the Role of Periostin. Circ. Res. 2009, 105, 934–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourdan-LeSaux, C.; Zhang, J.; Lindsey, M.L. Extracellular matrix roles during cardiac repair. Life Sci. 2010, 87, 391–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Huang, X.; Kanisicak, O.; Li, Y.; Wang, Y.; Li, Y.; Pu, W.; Liu, Q.; Zhang, H.; Tian, X.; et al. Preexisting endothelial cells mediate cardiac neovascularization after injury. J. Clin. Investig. 2017, 127, 2968–2981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Zhang, H.; Liu, Y.; Adams, S.; Eilken, H.; Stehling, M.; Corada, M.; Dejana, E.; Zhou, B.; Adams, R.H. Endothelial cells are progenitors of cardiac pericytes and vascular smooth muscle cells. Nat. Commun. 2016, 7, 12422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, C.; Bernardo, A.S.; Trotter, M.W.B.; Pedersen, R.A.; Sinha, S. Generation of human vascular smooth muscle subtypes provides insight into embryological origin–dependent disease susceptibility. Nat. Biotechnol. 2012, 30, 165–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, C.I.; Jansen, M.A.; McGregor, K.; Mylonas, K.J.; Richardson, R.V.; Thomson, A.; Moran, C.M.; Seckl, J.R.; Walker, B.R.; Chapman, K.E.; et al. Cardiomyocyte and vascular smooth muscle-independent 11β-hydroxysteroid dehydrogenase 1 amplifies infarct expansion, hypertrophy, and the development of heart failure after myocardial infarction in male mice. Endocrinology 2016, 157, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Ardell, J.L.; Andresen, M.C.; Armour, J.A.; Billman, G.E.; Chen, P.S.; Foreman, R.D.; Herring, N.; O’Leary, D.S.; Sabbah, H.N.; Schultz, H.D.; et al. Translational neurocardiology: Preclinical models and cardioneural integrative aspects. J. Physiol. 2016, 594, 3877–3909. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.E.; Connelly, K.; Kelly, D.J.; Pollock, C.A.; Krum, H. Heart failure and nephropathy: Catastrophic and interrelated complications of diabetes. Clin. J. Am. Soc. Nephrol. 2006, 1, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Margariti, A. Peripheral neuropathy may be a potential risk of cardiovascular disease in diabetes mellitus. Heart 2014, 100, 1823–1824. [Google Scholar] [CrossRef] [PubMed]
- Akar, F.G.; Nass, R.D.; Hahn, S.; Cingolani, E.; Shah, M.; Hesketh, G.G.; DiSilvestre, D.; Tunin, R.S.; Kass, D.A.; Tomaselli, G.F. Dynamic changes in conduction velocity and gap junction properties during development of pacing-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1223–H1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, U.; Frantz, S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ. Res. 2015, 116, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Tejada, T.; Tan, L.; Torres, R.A.; Calvert, J.W.; Lambert, J.P.; Zaidi, M.; Husain, M.; Berce, M.D.; Naib, H.; Pejler, G.; et al. IGF-1 degradation by mouse mast cell protease 4 promotes cell death and adverse cardiac remodeling days after a myocardial infarction. Proc. Natl. Acad. Sci. USA 2016, 113, 6949–6954. [Google Scholar] [CrossRef] [PubMed]
- Marino, A.; Martelli, A.; Citi, V.; Fu, M.; Wang, R.; Calderone, V.; Levi, R. The novel H2S donor 4-carboxy-phenyl isothiocyanate inhibits mast cell degranulation and renin release by decreasing intracellular calcium. Br. J. Pharmacol. 2016, 173, 3222–3234. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wülfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; Michelle, L.; Antoni, D.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K. Revisiting Cardiac Cellular Composition. Circ. Res. 2017, 118, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Tracy, L.E.; Minasian, R.A.; Caterson, E.J. Extracellular Matrix and Dermal Fibroblast Function in the Healing Wound. Adv. Wound Care 2016, 5, 119–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, C.; Terracciano, C.M. Human Cardiac Fibroblasts Engage the Sarcoplasmic Reticulum in Induced Pluripotent Stem Cell-Derived Cardiomyocyte Excitation–Contraction Coupling. J. Am. Coll. Cardiol. 2018, 72, 1061–1063. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.C.H.; Davis, M.E.; Lisowski, L.K.; Lee, R.T. Endothelial-cardiomyocyte interactions in cardiac development and repair. Annu. Rev. Physiol. 2006, 68, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Challet Meylan, L.; Challet Meylan, L.; Patsch, C.; Thoma, E. Endothelial cells differentiation from hPSCs. Protocol. Exchang. 2015. [Google Scholar] [CrossRef] [Green Version]
- Tulloch, N.L.; Muskheli, V.; Razumova, M.V.; Korte, F.S.; Regnier, M.; Hauch, K.D.; Pabon, L.; Reinecke, H.; Murry, C.E. Growth of engineered human myocardium with mechanical loading and vascular coculture. Circ. Res. 2011, 109, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Clayton, Z.E.; Sadeghipour, S.; Patel, S. Generating induced pluripotent stem cell derived endothelial cells and induced endothelial cells for cardiovascular disease modelling and therapeutic angiogenesis. Int. J. Cardiol. 2015, 197, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Margariti, A.; Winkler, B.; Karamariti, E.; Zampetaki, A.; Tsai, T.-N.; Baban, D.; Ragoussis, J.; Huang, Y.; Han, J.-D.J.; Zeng, L.; et al. Direct reprogramming of fibroblasts into endothelial cells capable of angiogenesis and reendothelialization in tissue-engineered vessels. Proc. Natl. Acad. Sci. USA 2012, 109, 13793–13798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, B.C.; Jiang, Z.; Suh, C.; Qyang, Y. Induced pluripotent stem cell-derived vascular smooth muscle cells: Methods and application. Biochem. J. 2015, 465, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Bansal, S.S.; Ismahil, M.A.; Goel, M.; Patel, B.; Hamid, T.; Rokosh, G.; Prabhu, S.D. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ. Heart Fail. 2017, 10, e003688. [Google Scholar] [CrossRef] [PubMed]
- Ke, D.; Fang, J.; Fan, L.; Chen, Z.; Chen, L. Regulatory T cells contribute to rosuvastatin-induced cardioprotection against ischemia-reperfusion injury. Coronary Artery Dis. 2013, 24, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Cho, G.-S.; Li, Z.; Hong, I.; Zhu, R.; Kim, M.-J.; Kim, Y.J.; Tampakakis, E.; Tung, L.; Huganir, R.; et al. Functional Coupling with Cardiac Muscle Promotes Maturation of hPSC-Derived Sympathetic Neurons. Cell Stem Cell 2016, 19, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Rother, J.; Richter, C.; Turco, L.; Knoch, F.; Mey, I.; Luther, S.; Janshoff, A.; Bodenschatz, E.; Tarantola, M. Crosstalk of cardiomyocytes and fibroblasts in co-cultures. Open Biol. 2015, 5, 150038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mozaffari, M.S.; Liu, J.Y.; Abebe, W.; Baban, B. Mechanisms of load dependency of myocardial ischemia reperfusion injury. Am. J. Cardiovasc. Dis. 2013, 3, 180–196. [Google Scholar] [PubMed]
- Jahng, J.W.S.; Song, E.; Sweeney, G. Crosstalk between the heart and peripheral organs in heart failure. Exp. Mol. Med. 2016, 48, e217. [Google Scholar] [CrossRef] [PubMed]
- Ismahil, M.A.; Hamid, T.; Bansal, S.S.; Patel, B.; Kingery, J.R.; Prabhu, S.D. Remodeling of the Mononuclear Phagocyte Network Underlies Chronic Inflammation and Disease Progression in Heart Failure: Critical Importance of the Cardiosplenic Axis. Circ. Res. 2014, 114, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Smolina, K.; Wright, F.L.; Rayner, M.; Goldacre, M.J. Determinants of the decline in mortality from acute myocardial infarction in England between 2002 and 2010: Linked national database study. BMJ 2012, 344, d8059. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Muntner, P.; Lloyd, A.; Manns, B.J.; Klarenbach, S.; Pannu, N.; James, M.T.; Hemmelgarn, B.R. Risk of coronary events in people with chronic kidney disease compared with those with diabetes: A population-level cohort study. Lancet 2012, 380, 807–814. [Google Scholar] [CrossRef]
- Szummer, K.; Lundman, P.; Jacobson, S.H.; Schön, S.; Lindbäck, J.; Stenestrand, U.; Wallentin, L.; Jernberg, T. Influence of renal function on the effects of early revascularization in non-st-elevation myocardial infarction: Data from the swedish web-system for enhancement and development of evidence-based care in heart disease evaluated according to recommended th. Circulation 2009, 120, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanjare, M.; Kuo, F.; Gerecht, S. Derivation and maturation of synthetic and contractile vascular smooth muscle cells from human pluripotent stem cells. Cardiovasc. Res. 2013, 97, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Patsch, C.; Challet-Meylan, L.; Thoma, E.C.; Urich, E.; Heckel, T.; O’Sullivan, J.F.; Grainger, S.J.; Kapp, F.G.; Sun, L.; Christensen, K.; et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat. Cell Biol. 2015, 17, 994–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, L.C.; Keeney, P.M.; Bennett, J.P. Differentiation of Human Neural Stem Cells into Motor Neurons Stimulates Mitochondrial Biogenesis and Decreases Glycolytic Flux. Stem Cells Dev. 2015, 24, 1984–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, A.J.; Kaller, M.S.; Galino, J.; Willison, H.J.; Rinaldi, S.; Bennett, D.L.H. Co-cultures with stem cell-derived human sensory neurons reveal regulators of peripheral myelination. Brain 2017, 140, 898–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, B.; Carusi, A.; Abi-Gerges, N.; Ariga, R.; Britton, O.; Bub, G.; Bueno-Orovio, A.; Burton, R.A.B.; Carapella, V.; Cardone-Noott, L.; et al. Human-based approaches to pharmacology and cardiology: An interdisciplinary and intersectorial workshop. Europace 2016, 18, 1287–1298. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Model Type | Description | Benefits | Limitations | Ref. |
|---|---|---|---|---|
| Animal | Animals with defined genetic background or subject to acute intervention (e.g., coronary obstruction) to mimic discrete time points. | Small animals: delineate molecular pathways in early- or late-stage heart failure, aiding the identification of biomarkers and therapeutic targets. Large animals: preclinical proof of concept for novel therapies before clinical trials. | Gene expression-silencing or drug-induced pathogenesis does not recapitulate the disease initiation in humans. Many human diseases are human-specific. Differences in physiology (circulation, etc.), cardiac output requirements, myocardial composition (vascular supply, etc.), biochemical absorption, distribution, metabolism and immunoresponses. | [8,9,10,11,12,13,14] |
| Human-specific expression in Chinese hamster ovary (CHO) and human embryonic kidney (HEK) cells | Model protein force expression, e.g., tests the off-target effects to ion channels prolonging the QT interval. | Expression of human ion channels. Avoids the expense of whole animal studies. Reproducibility in cryogenically freezing and thawing cell lines for the stable expression of the desired channels. | Single ion channel does not recapitulate diseases in humans. Does not negate species mismatch. | [15,16] |
| Adult human cardiomyocytes | Isolated from diseased or non-diseased patients during surgery. | Human genome so we can map the response in humans to cardiac disease. | Limited quantities (e.g., ethical limitations). Large variability in phenotype and rapid dedifferentiation. | [17,18,19,20,21] |
| Organoid | 3D in vitro culture systems derived from self-organizing stem cells and extracellular matrix (ECM) proteins secreted from the cells. | Higher complexity compared to the 2D models, with more extensive cell-ECM interactions and possible vessel formation. | Expensive and technically challenging setup, resulting in poor reproducibility. | [22,23] |
| 3D cardiac tissue | 3D in vitro culture systems with natural and/or synthetic ECM structural support. | Ability to manipulate ECM components enables a greater control of the scaffold composition and more complex cell-ECM interactions. Decellularized scaffold for cell adhesion mimics the naturally occurring macro and microstructures. | Limited information on cost, reproducibility, and performance. | [24,25,26] |
| Pathology | Cell Type Involved | Mutation | (Drug/Treatment) Test | Ref. |
|---|---|---|---|---|
| Endothelial | ||||
| Healthy | EC | N/A | Flow-induced disease and simvastatin | [39] |
| Hutchison-Gilford Progeria Syndrome | EC | Patient-derived | N/A | [40] |
| Smooth muscle cells | ||||
| Supravalvular aortic stenosis | SMC | Elastin (ELN) | Elastin recombinant protein | [41] |
| Marfan syndrome | SMC | FBN1 | Gene editing and drugs | [42] |
| Lymphocytes | ||||
| Healthy | B-cell lymphoid lineage | N/A | N/A | [43] |
| Red Blood cell (RBC) | ||||
| Healthy | CM and RBC | N/A | Toxicity of RBC | [44] |
| CM | ||||
| Hypoplastic left heart syndrome | CM | Patient-derived (GM12601) | Isoproterenol | [45] |
| Arrhythmogenic right ventricular dysplasia | CM | Plakoglobin, plakophilin-2 | Metabolism induced onset | [46] |
| Familial hypertrophic cardiomyopathy | CM | MYH7 Arg663His | Verapamil, Diltiazem, Mexiletine among 15 drugs | [47] |
| LEOPARD syndrome | CM and all three germ layers | PTPN11 | N/A | [48] |
| Friedreich’s ataxia | Neurons and CM | GAA triplet repeat expansion within the first intron of the frataxin gene | N/A | [49] |
| Catecholaminergic polymorphic ventricular tachycardia type 1 | CM | Ryanodine Receptor 2 (RYR2) | Isoproterenol | [50,51] |
| LQT1,2,3,5,8,14 | CM | Patient-derived | Common drugs | [52,53,54,55,56,57] |
| Barth syndrome | CM | Tafazzin (TAZ) | Genetic rescue | [58] |
| Ischemic heart damage | CM | Aldehyde dehydrogenase 2 (ALDH-2) deficiency | siRNA knockdown | [59] |
| Brugada syndrome | CM | SCN5A-1795insD mutation | N/A | [33] |
| Structure | ||||
| hiPSC-CM | Atrial | Ventricular | ||
| Shape | Any, not defined | Cylindrical | Cylindrical and bifurcated | |
| Volume | Small | Large | Very large | |
| Sarcomere Organization | Random | Orderly and aligned | Orderly and aligned | |
| Mitochondria population | Few | Abundant | Abundant | |
| T-tubule organization | Absent | Scarce | Abundant | |
| Glucose Metabolism | High | Low | Low | |
| Nucleus morphology | Mono | Mono, bi, multi | Mono, bi, multi | |
| Electrophysiology | ||||
| Spontaneous activity | Very frequent | Absent | Absent | |
| Maximum diastolic potential | −60 mV | −70 mV | −80 mV | |
| Maximum upstroke velocity | 44–187 V/s | 200 V/s | 200 V/s | |
| Action potential amplitude | 94–113 mV | 80–130 mV | 100 mV | |
| * Action potential duration at 50% | 60–400 ms | 200 ms | 200–300 ms | |
| * Action potential duration at 90% | 80–500 ms | 200–400 ms | 250–400 ms | |
| Force Generation | 100–150 Pa for a single cell | Myocardium tensile force ≈ 56 kPa | Myocardium tensile force ≈ 56 kPa | |
| Elastic modulus | 466 Pa | 22–55 kPa | 22–55 kPa | |
| Molecular Marker | ||||
| Gap junction | Cx40 | + | + | - |
| Cx43 | + | + | + | |
| Cx45 | + | - | - | |
| Ion channel | KCNA5 | + | + | - |
| NCX1 | + | + | + | |
| SERCA2a | + | + | + | |
| RYR2 | + | + | + | |
| Cav 1.2 | + | + | + | |
| Kir 2.1 | + | + | + | |
| Kv 4.3 | + | + | + | |
| KChip 2 | + | + | + | |
| KCNH2 (HERG) | + | + | + | |
| Structural protein | TNNT2 | + | + | + |
| ACTN2 | + | + | + | |
| MLC2A | + | + | + | |
| MLC2V | + | - | + | |
| MYL2 | + | + | + | |
| MYH6 | + | + | + | |
| Master gene | NKX2.5 | + | ± | ± |
| Cell. | Healthy | Disease | Notes |
|---|---|---|---|
| Fibroblasts | ● ECM turnover, maintaining a balance between the synthesis and degradation of the matrix | ● Scar formation (fibrosis) ● Increase ECM protein ● Phagocytose apoptotic cells ● Crosstalk to EC and macrophage for angiogenesis and matrix synthesis | [105,111,115,116] |
| ECM | ● Periostin, laminin, vimentin, fibronectin, and collagen types I (90%), III, V, and VI ● Alignment ● Mechanical support | ● Increase in collagen I, III, IV, V, and VI ● laminin, fibronectin, thrombospondin, and tenascin | [104,111,117,118,119] |
| Endothelial cells | ● Structural support ● Vasculature homeostasis ● Biochemical factors such as nitric oxide, endothelin-1, IL-6 ● Progenitor of cardiac pericytes and vascular smooth muscle cells | ● Inflammation (hypertrophy, inotropy, apoptosis, mitosis) ● Neovascularization increase the density of peri-infarct vessels ● Paracrine | [112,120,121] |
| SMCs | ● Mechanical support of vasculature: contractile or synthetic (proliferative) mode | ● Loss of elasticity ● Reduced contractile ● Increased proliferation | [122,123] |
| Neuronal cells | ● Conduction fibre and pacemaker (AV, SA, Purkinje) ● Control of rhythmic beating | ● Block, slow down conduction ● Essential component for embryo development | [124,125,126,127] |
| Lymphocytes | ● Few residents ● Mast cells act as inflammatory mediator storage and activating the local renin-angiotensin system ● Macrophage performs a janitorial homeostasis and facilitates electrical conduction | ● Macrophage has a role in ECM turnover/cell death, scar formation, neutrophil recruitment, and vascularization support | [128,129,130,131] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.X.; Kit-Anan, W.; Terracciano, C.M.N. Many Cells Make Life Work—Multicellularity in Stem Cell-Based Cardiac Disease Modelling. Int. J. Mol. Sci. 2018, 19, 3361. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113361
Wang BX, Kit-Anan W, Terracciano CMN. Many Cells Make Life Work—Multicellularity in Stem Cell-Based Cardiac Disease Modelling. International Journal of Molecular Sciences. 2018; 19(11):3361. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113361
Chicago/Turabian StyleWang, Brian X., Worrapong Kit-Anan, and Cesare M. N. Terracciano. 2018. "Many Cells Make Life Work—Multicellularity in Stem Cell-Based Cardiac Disease Modelling" International Journal of Molecular Sciences 19, no. 11: 3361. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113361