Non-Coding Micro RNAs and Hypoxia-Inducible Factors Are Selenium Targets for Development of a Mechanism-Based Combination Strategy in Clear-Cell Renal Cell Carcinoma—Bench-to-Bedside Therapy

Abstract
:1. Introduction
2. Results
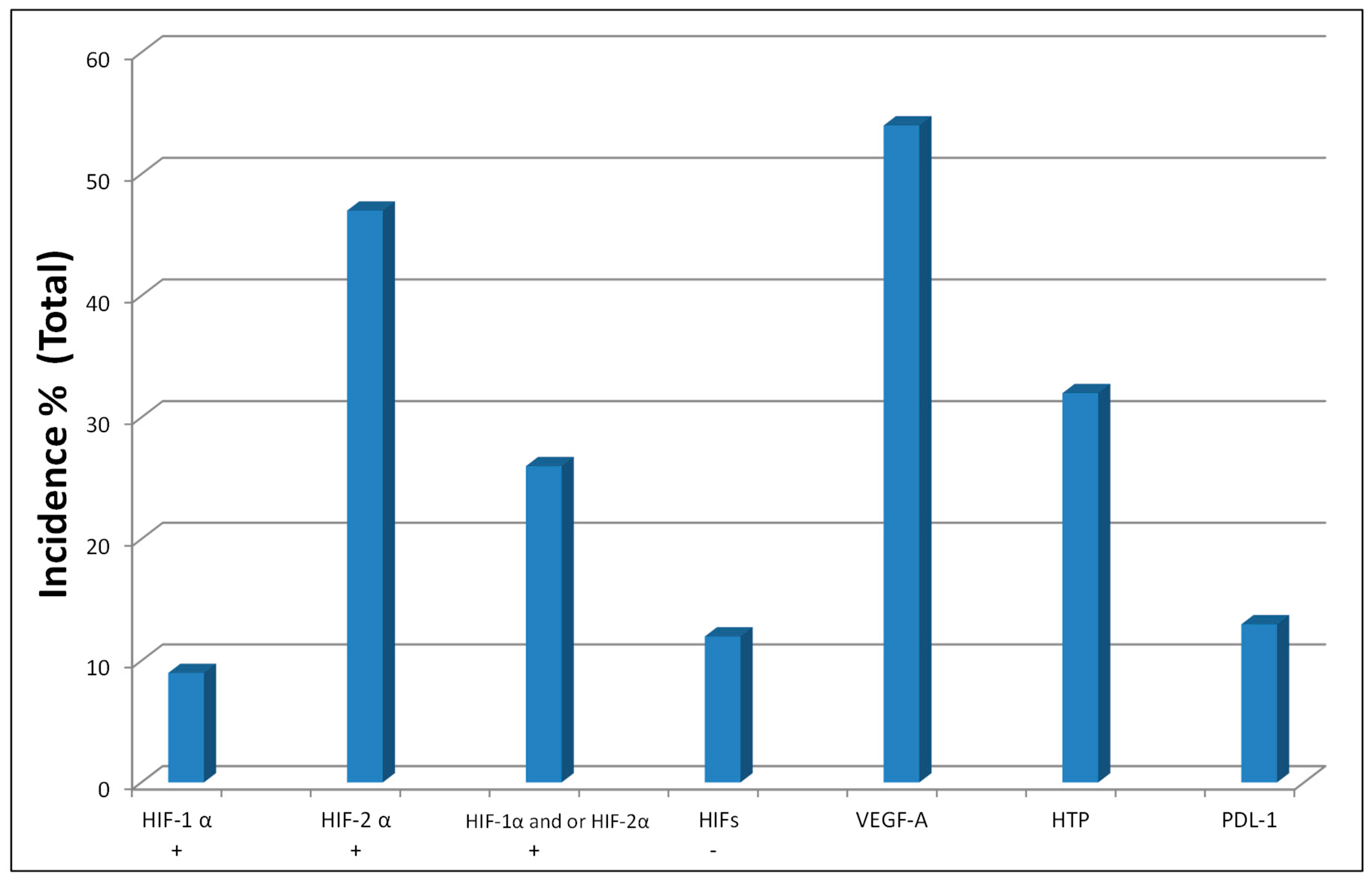
2.1. Hypoxia-Inducible Factors 1α and 2α (HIFs) and VHL Tumor Suppressor Gene
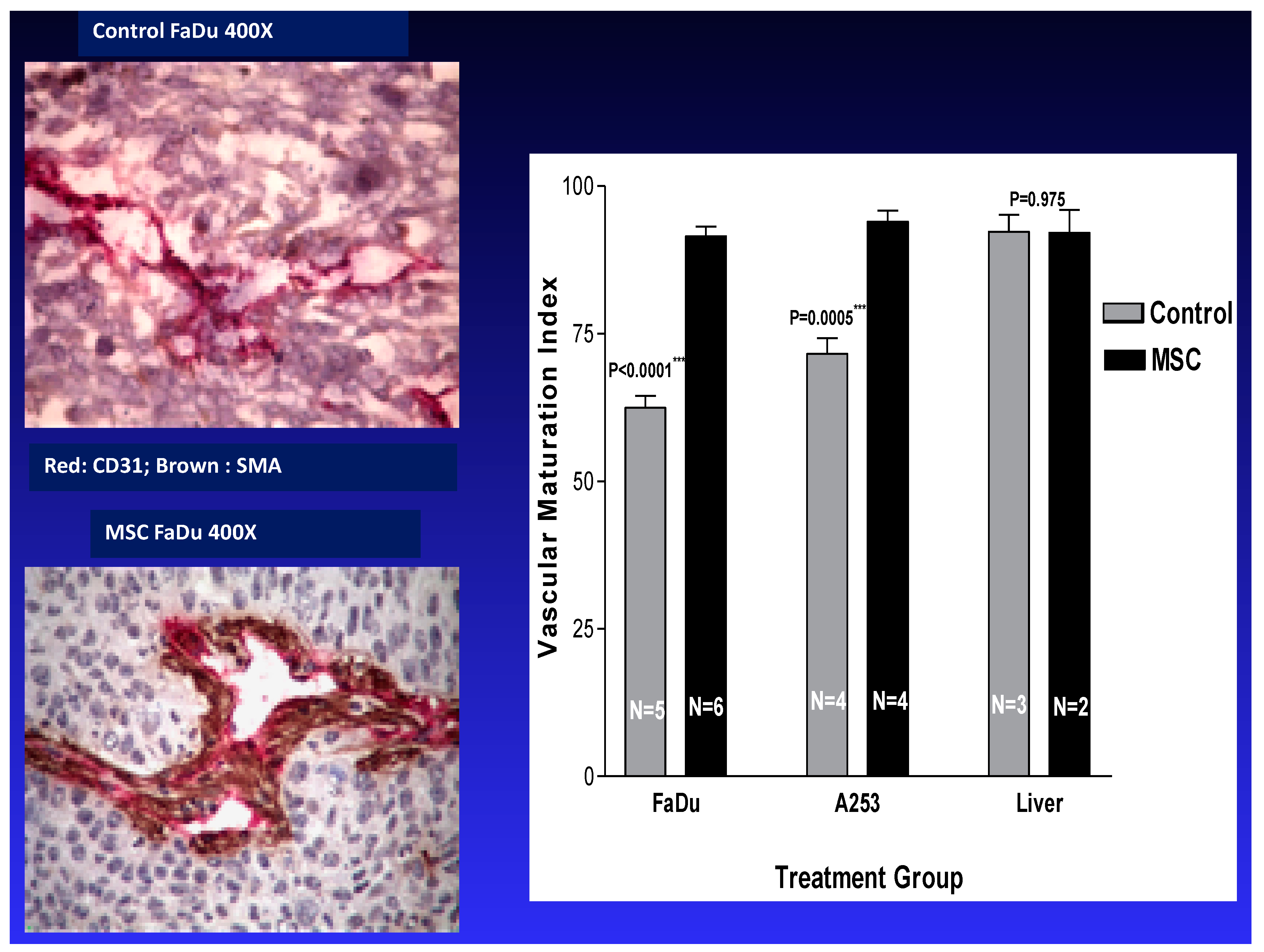
2.2. Tumor Vasculature
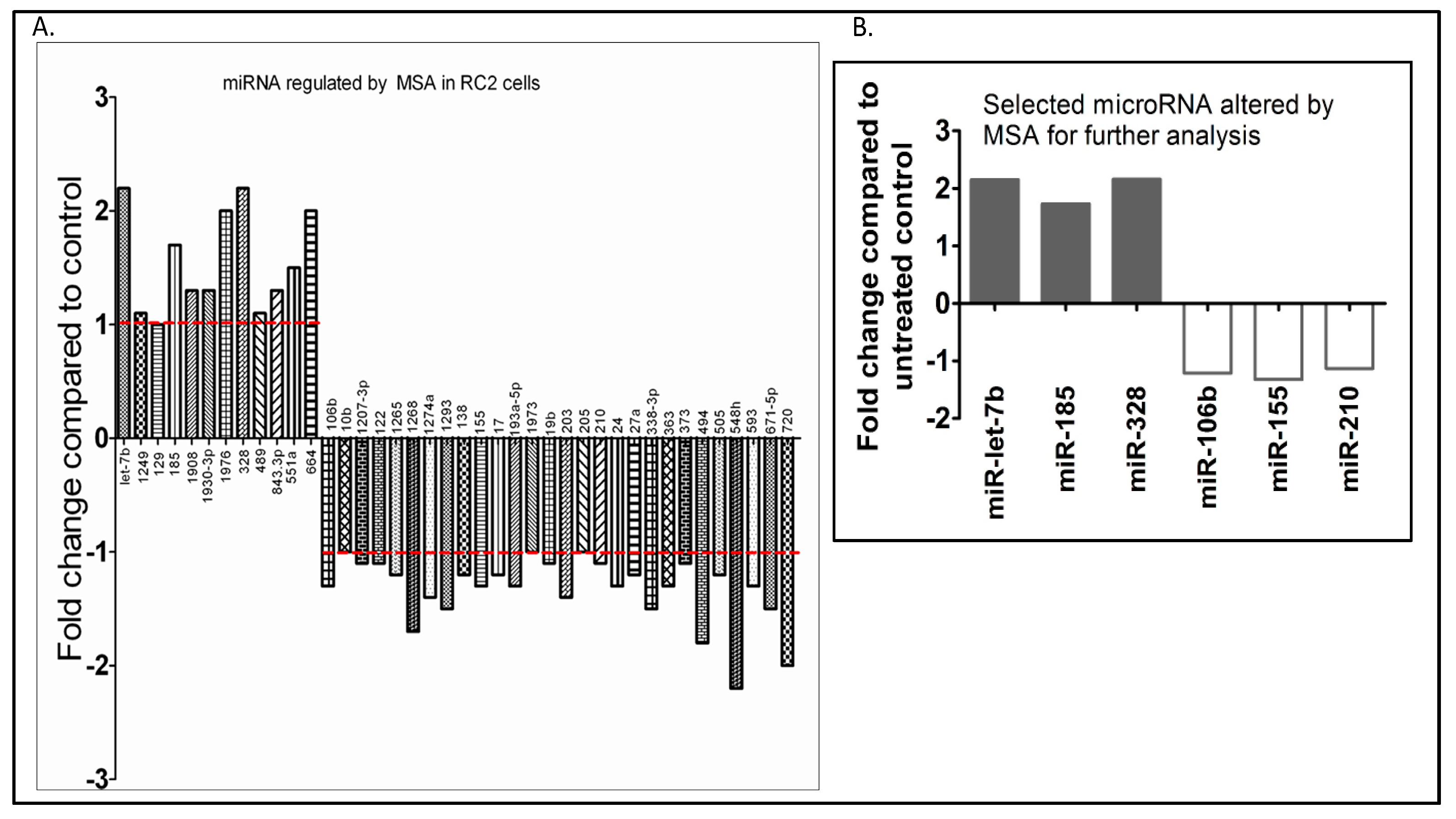
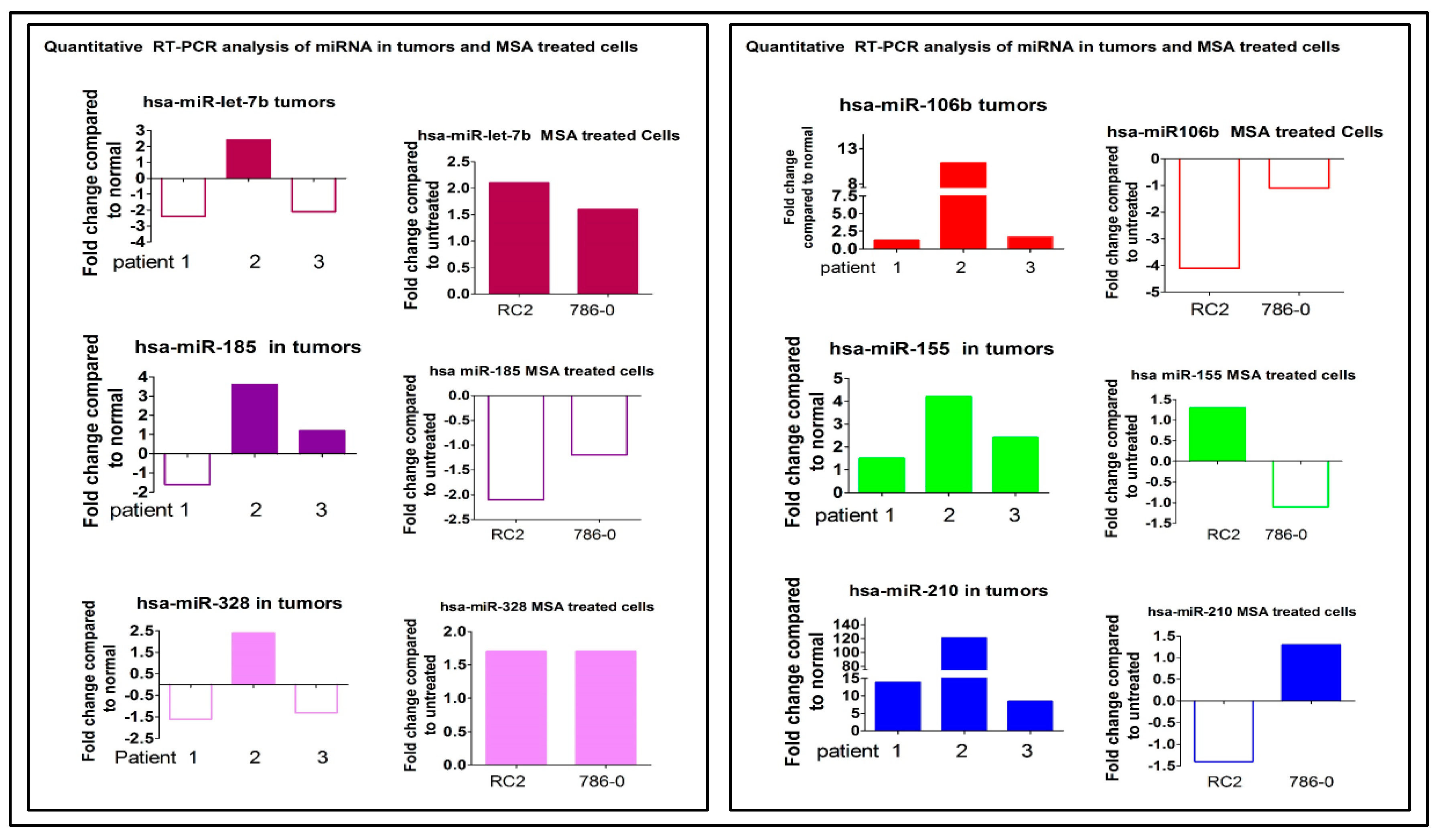
2.3. Oncogenic miRNA-155 and miRNA-210
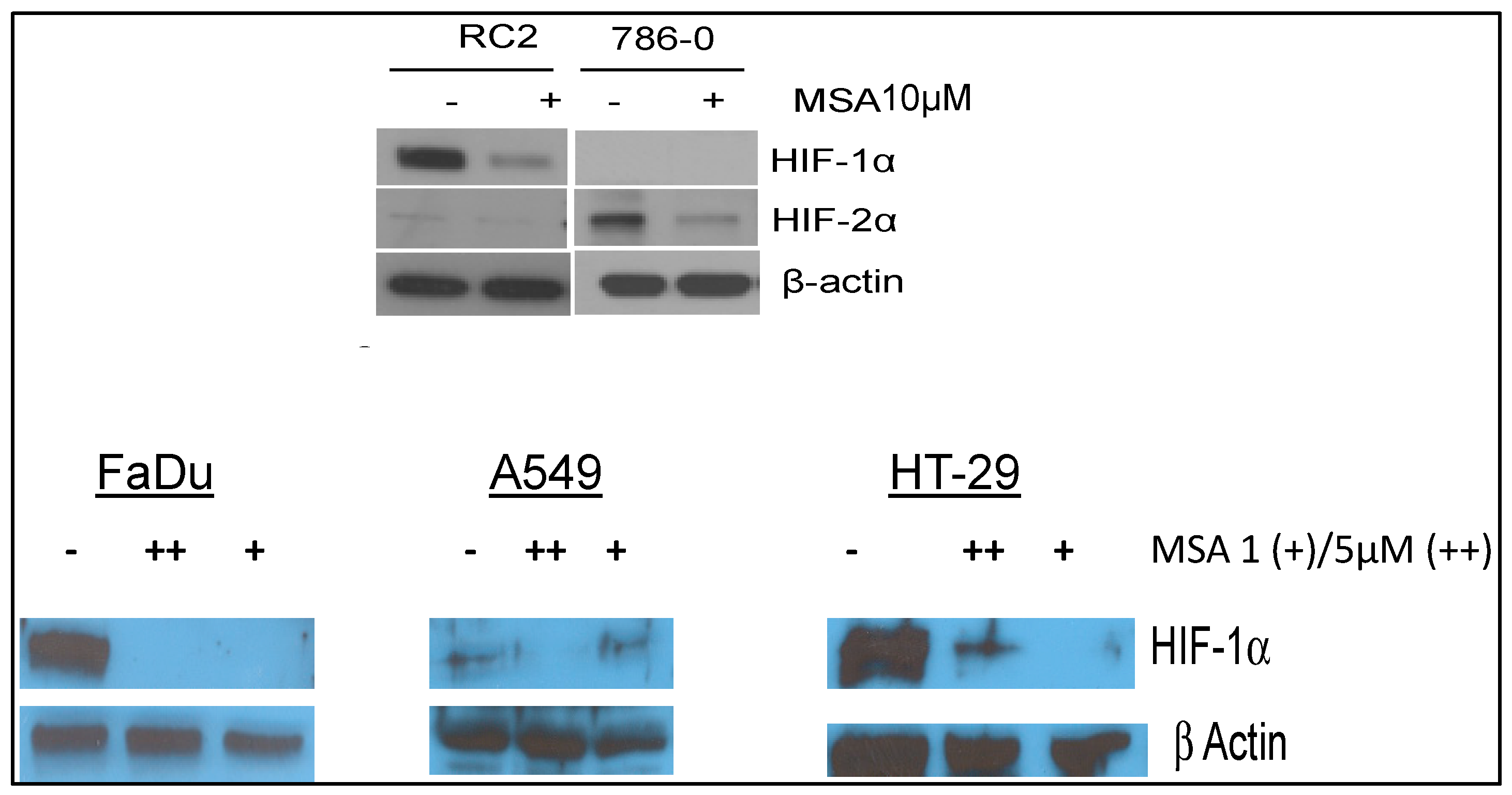
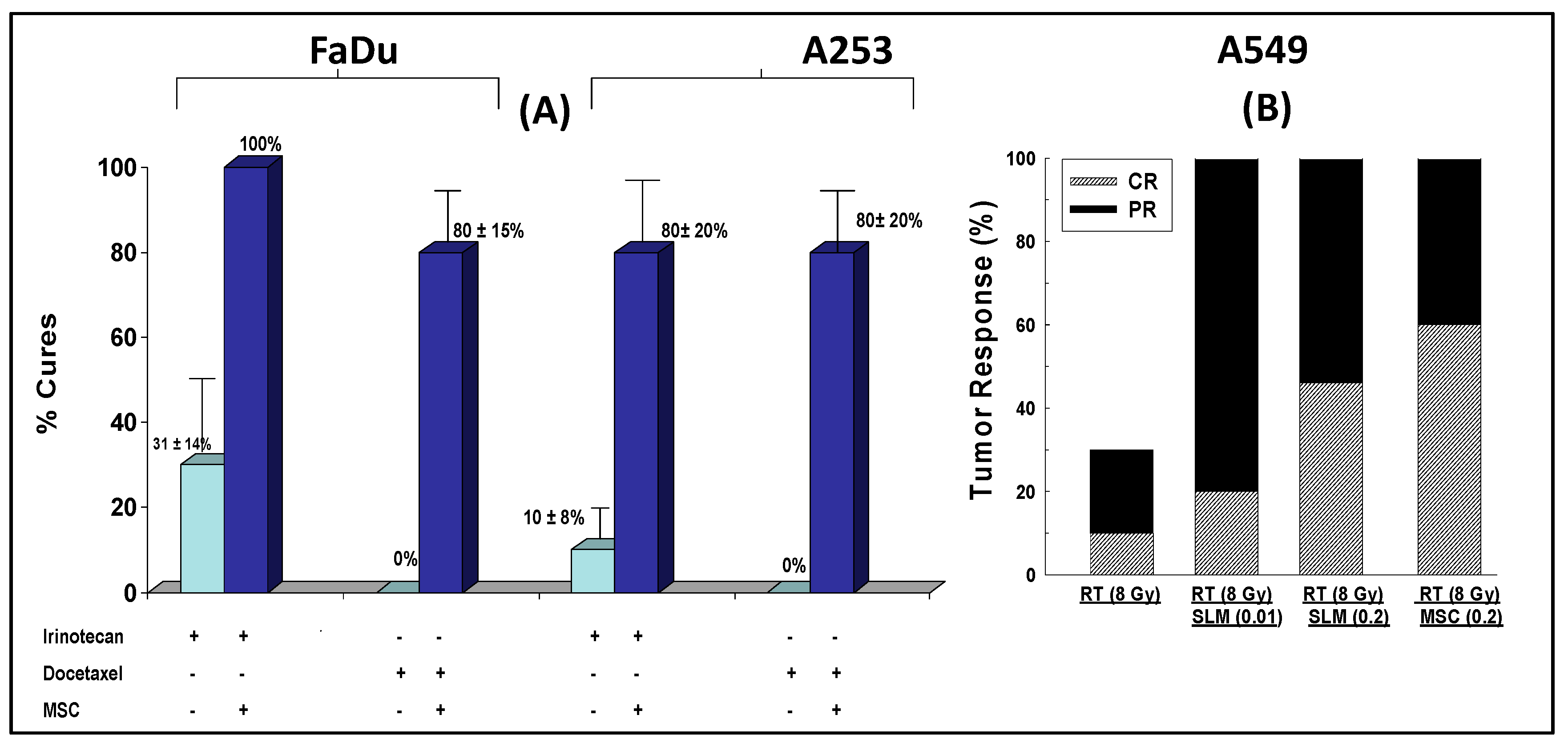
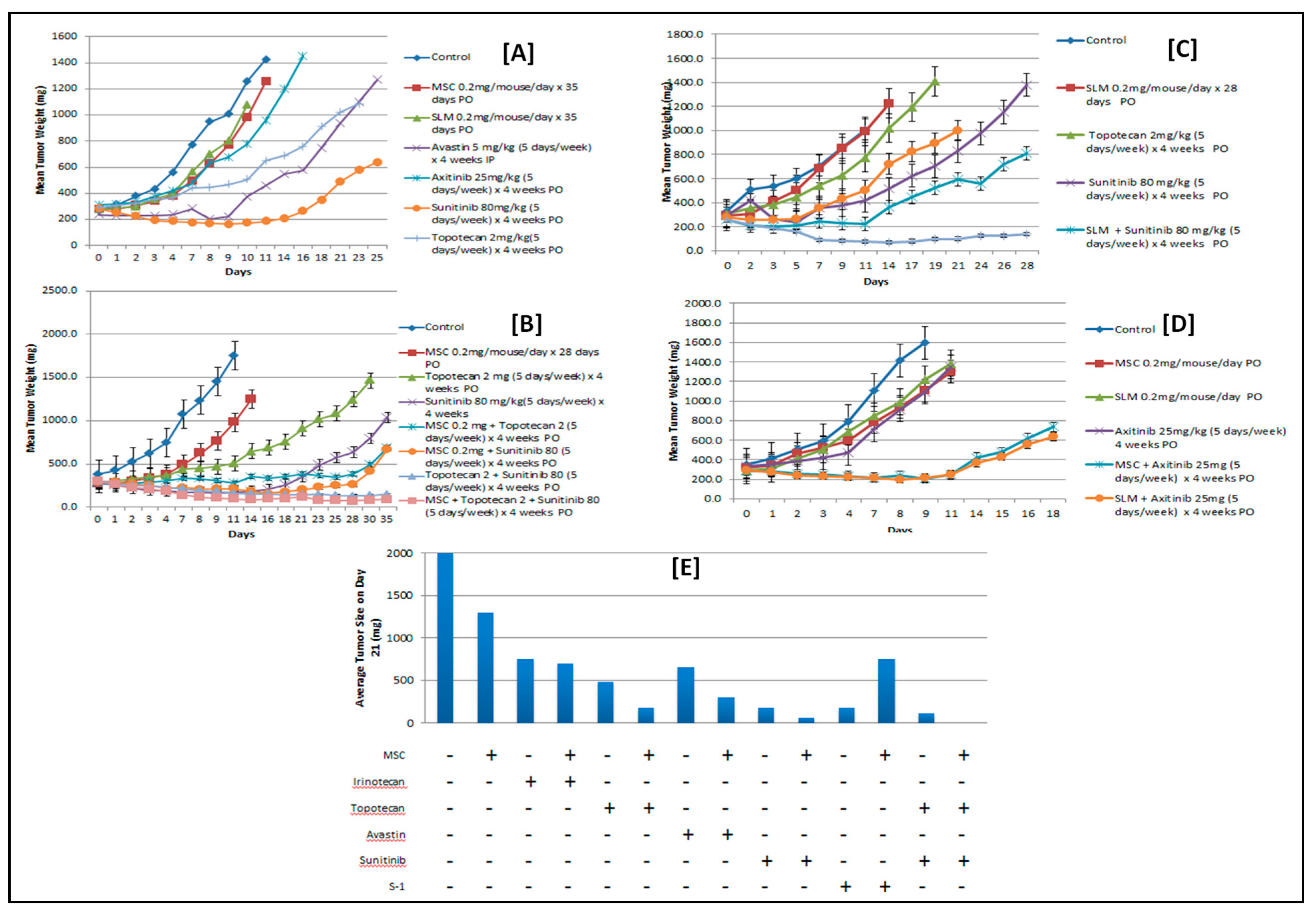
2.4. Selenium: A Selective Modulator of Anticancer Therapies
2.4.1. Nude Mice Bearing HIF1α
2.4.2. Nude Mice Bearing Tumor Xenografts That Constitutively Expressed HIF2α
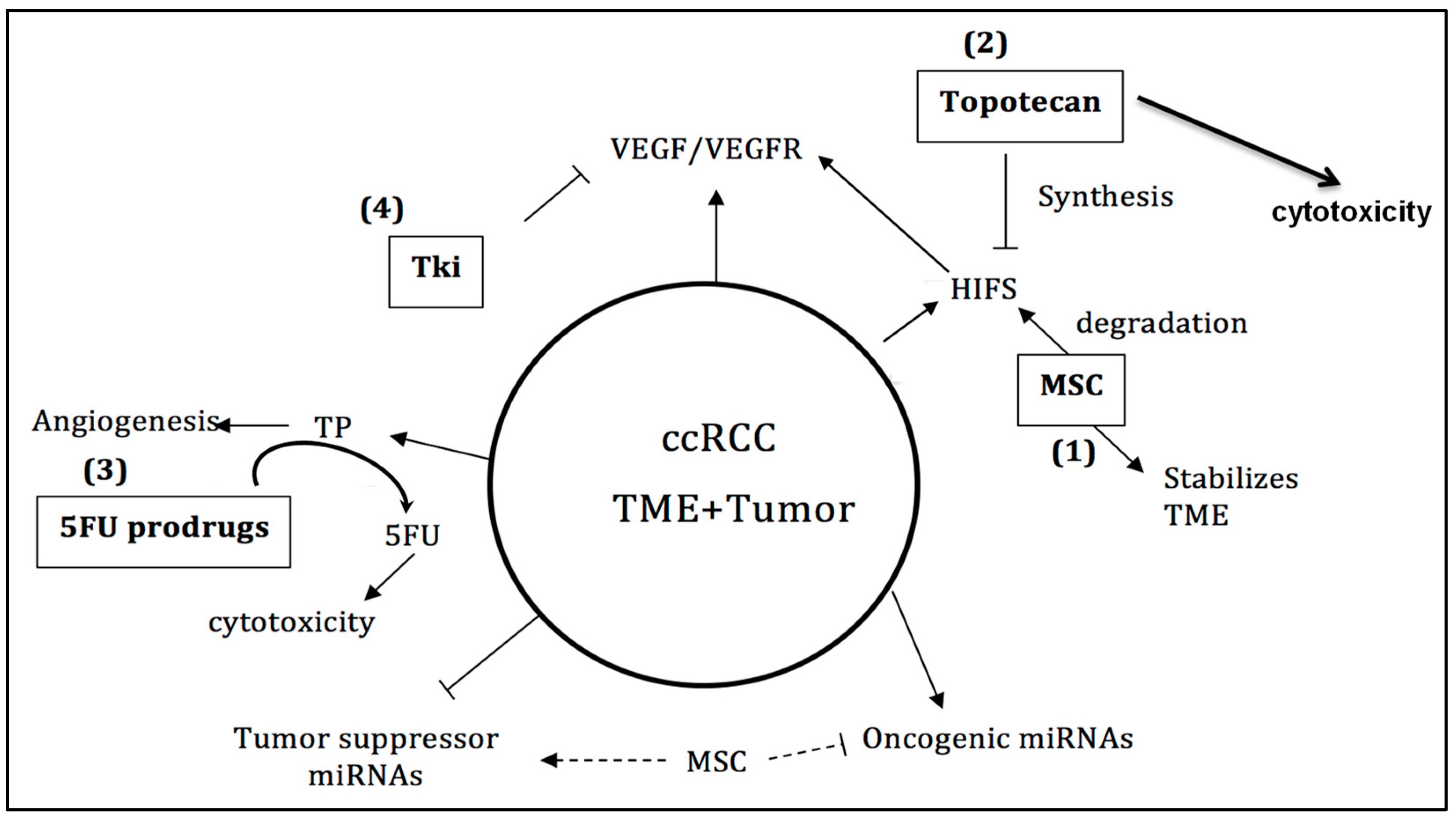
3. Discussion
4. Conclusions and Future Perspectives
5. Material and Methods
5.1. Cell Culture and Drug Treatments
5.2. Animals
Tumor Xenografts
5.3. Drugs
5.4. Total RNA Isolation from ccRCC Cells Treated with and without MSA
5.5. Total RNA from ccRCC Patient Tumors and Their Matched Normal Tissues
5.6. Reverse Transcription (RT) and miRNA qPCR
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ccRCC | Clear-cell renal cell carcinoma |
| HIF | Hypoxia-inducible factor |
| HIG-2 | Hypoxia-inducible protein 2 |
| IFN | Interferon |
| MSC | Se-methylselenocysteine |
| PD-1 | Programmed death 1 receptor |
| PD-L1 | Program death ligand 1 |
| SLM | Seleno-l-methionine |
| TP | Thymidine phosphorylase |
| TNF | Tumor necrosis factor |
| TKI | Tyrosine kinase inhibitor |
| TIL | Tumor-infiltrating lymphocyte |
| VEGF | Vascular endothelial growth factor |
| VHL | von Hippel–Lindau |
References
- Albiges, L.; Choueiri, T.K. Renal-cell carcinoma in 2016: Advances in treatment—Jostling for pole position. Nat. Rev. Clin. Oncol. 2017, 14, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Halabi, S.; Eisen, T.; Broderick, S.; Stadler, W.M.; Jones, R.J.; Garcia, J.A.; Vaishampayan, U.N.; Picus, J.; Hawkins, R.E.; et al. Everolimus versus sunitinib for patients with metastatic non-clear cell renal cell carcinoma (ASPEN): A multicentre, open-label, randomised phase 2 trial. Lancet Oncol. 2016, 17, 378–388. [Google Scholar] [CrossRef]
- Escudier, B.; Powles, T.; Motzer, R.J.; Olencki, T.; Aren Frontera, O.; Oudard, S.; Rolland, F.; Tomczak, P.; Castellano, D.; Appleman, L.J.; et al. Cabozantinib, a New Standard of Care for Patients With Advanced Renal Cell Carcinoma and Bone Metastases? Subgroup Analysis of the METEOR Trial. J. Clin. Oncol. 2018, 36, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Pello, S.; Hofmann, F.; Tahbaz, R.; Marconi, L.; Lam, T.B.; Albiges, L.; Bensalah, K.; Canfield, S.E.; Dabestani, S.; Giles, R.H.; et al. A Systematic Review and Meta-analysis Comparing the Effectiveness and Adverse Effects of Different Systemic Treatments for Non-clear Cell Renal Cell Carcinoma. Eur. Urol. 2017, 71, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Greef, B.; Eisen, T. Medical treatment of renal cancer: New horizons. Br. J. Cancer 2016, 115, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Chen, D.; Wang, P.I.; Marker, M.; Redzematovic, A.; Chen, Y.B.; Selcuklu, S.D.; Weinhold, N.; Bouvier, N.; Huberman, K.H.; et al. Genomic Biomarkers of a Randomized Trial Comparing First-line Everolimus and Sunitinib in Patients with Metastatic Renal Cell Carcinoma. Eur. Urol. 2017, 71, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Hodi, F.S.; Wolchok, J.D. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 1270–1271. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powles, T.; Albiges, L.; Staehler, M.; Bensalah, K.; Dabestani, S.; Giles, R.H.; Hofmann, F.; Hora, M.; Kuczyk, M.A.; Lam, T.B.; et al. Updated European Association of Urology Guidelines Recommendations for the Treatment of First-line Metastatic Clear Cell Renal Cancer. Eur. Urol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Tannir, N.M.; Jonasch, E.; Albiges, L.; Altinmakas, E.; Ng, C.S.; Matin, S.F.; Wang, X.; Qiao, W.; Dubauskas Lim, Z.; Tamboli, P.; et al. Everolimus Versus Sunitinib Prospective Evaluation in Metastatic Non-Clear Cell Renal Cell Carcinoma (ESPN): A Randomized Multicenter Phase 2 Trial. Eur. Urol. 2016, 69, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.Y.; Wu, S. Update on the treatment of metastatic clear cell and non-clear cell renal cell carcinoma. Biomark Res. 2015, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Figueroa, D.J.; Fay, A.P.; Signoretti, S.; Liu, Y.; Gagnon, R.; Deen, K.; Carpenter, C.; Benson, P.; Ho, T.H.; et al. Correlation of PD-L1 tumor expression and treatment outcomes in patients with renal cell carcinoma receiving sunitinib or pazopanib: Results from COMPARZ, a randomized controlled trial. Clin. Cancer Res. 2015, 21, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Wallis, C.J.D.; Klaassen, Z.; Bhindi, B.; Ye, X.Y.; Chandrasekar, T.; Farrell, A.M.; Goldberg, H.; Boorjian, S.A.; Leibovich, B.; Kulkarni, G.S.; et al. First-line Systemic Therapy for Metastatic Renal Cell Carcinoma: A Systematic Review and Network Meta-analysis. Eur. Urol. 2018, 74, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Kammerer-Jacquet, S.F.; Brunot, A.; Pladys, A.; Bouzille, G.; Dagher, J.; Medane, S.; Peyronnet, B.; Mathieu, R.; Verhoest, G.; Bensalah, K.; et al. Synchronous Metastatic Clear-Cell Renal Cell Carcinoma: A Distinct Morphologic, Immunohistochemical, and Molecular Phenotype. Clin. Genitourin Cancer 2017, 15, e1–e7. [Google Scholar] [CrossRef] [PubMed]
- Kammerer-Jacquet, S.F.; Crouzet, L.; Brunot, A.; Dagher, J.; Pladys, A.; Edeline, J.; Laguerre, B.; Peyronnet, B.; Mathieu, R.; Verhoest, G.; et al. Independent association of PD-L1 expression with noninactivated VHL clear cell renal cell carcinoma-A finding with therapeutic potential. Int. J. Cancer 2017, 140, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; He, L.; Richards, E.J.; Challa, S.; Xu, C.X.; Permuth-Wey, J.; Lancaster, J.M.; Coppola, D.; Sellers, T.A.; Djeu, J.Y.; et al. Upregulation of miRNA-155 promotes tumour angiogenesis by targeting VHL and is associated with poor prognosis and triple-negative breast cancer. Oncogene 2014, 33, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, M.L.; Jaeger, E.; Shi, Y.; Durocher, J.A.; Mahurkar, S.; Zaridze, D.; Matveev, V.; Janout, V.; Kollarova, H.; Bencko, V.; et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin. Cancer Res. 2008, 14, 4726–4734. [Google Scholar] [CrossRef] [PubMed]
- Melendez-Rodriguez, F.; Roche, O.; Sanchez-Prieto, R.; Aragones, J. Hypoxia-Inducible Factor 2-Dependent Pathways Driving Von Hippel-Lindau-Deficient Renal Cancer. Front. Oncol. 2018, 8, 214. [Google Scholar] [CrossRef] [PubMed]
- Chintala, S.; Najrana, T.; Toth, K.; Cao, S.; Durrani, F.A.; Pili, R.; Rustum, Y.M. Prolyl hydroxylase 2 dependent and Von-Hippel-Lindau independent degradation of Hypoxia-inducible factor 1 and 2α by selenium in clear cell renal cell carcinoma leads to tumor growth inhibition. BMC Cancer 2012, 12, 293. [Google Scholar] [CrossRef] [PubMed]
- Schodel, J.; Grampp, S.; Maher, E.R.; Moch, H.; Ratcliffe, P.J.; Russo, P.; Mole, D.R. Hypoxia, Hypoxia-inducible Transcription Factors, and Renal Cancer. Eur. Urol. 2016, 69, 646–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth, K.; Chintala, S.; Rustum, Y.M. Constitutive expression of HIF-alpha plays a major role in generation of clear-cell phenotype in human primary and metastatic renal carcinoma. Appl. Immunohistochem. Mol. Morphol. 2014, 22, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.B.; Li, Z.R.; Toth, K.; Cao, S.; Durrani, F.A.; Hapke, G.; Bhattacharya, A.; Azrak, R.G.; Frank, C.; Rustum, Y.M. Potentiation of irinotecan sensitivity by Se-methylselenocysteine in an in vivo tumor model is associated with downregulation of cyclooxygenase-2, inducible nitric oxide synthase, and hypoxia-inducible factor 1-alpha expression, resulting in reduced angiogenesis. Oncogene 2006, 25, 2509–2519. [Google Scholar] [PubMed]
- Kondo, K.; Klco, J.; Nakamura, E.; Lechpammer, M.; Kaelin, W.G., Jr. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell 2002, 1, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Raval, R.R.; Lau, K.W.; Tran, M.G.; Sowter, H.M.; Mandriota, S.J.; Li, J.L.; Pugh, C.W.; Maxwell, P.H.; Harris, A.L.; Ratcliffe, P.J. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol. Cell. Boil. 2005, 25, 5675–5686. [Google Scholar] [CrossRef] [PubMed]
- del Peso, L.; Castellanos, M.C.; Temes, E.; Martin-Puig, S.; Cuevas, Y.; Olmos, G.; Landazuri, M.O. The von Hippel Lindau/hypoxia-inducible factor (HIF) pathway regulates the transcription of the HIF-proline hydroxylase genes in response to low oxygen. J. Boil. Chem. 2003, 278, 48690–48695. [Google Scholar] [CrossRef] [PubMed]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat. Rev. Cancer 2002, 2, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Linehan, W.M.; Lerman, M.I.; Zbar, B. Identification of the von Hippel-Lindau (VHL) gene. Its role in renal cancer. JAMA 1995, 273, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Tan, J.; Lim, K.J.; Koh, J.; Ooi, W.F.; Li, Z.; Huang, D.; Xing, M.; Chan, Y.S.; Qu, J.Z.; et al. VHL Deficiency Drives Enhancer Activation of Oncogenes in Clear Cell Renal Cell Carcinoma. Cancer Discov. 2017, 7, 1284–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razafinjatovo, C.; Bihr, S.; Mischo, A.; Vogl, U.; Schmidinger, M.; Moch, H.; Schraml, P. Characterization of VHL missense mutations in sporadic clear cell renal cell carcinoma: Hotspots, affected binding domains, functional impact on pVHL and therapeutic relevance. BMC Cancer 2016, 16, 638. [Google Scholar] [CrossRef] [PubMed]
- Chintala, S.; Toth, K.; Cao, S.; Durrani, F.A.; Vaughan, M.M.; Jensen, R.L.; Rustum, Y.M. Se-methylselenocysteine sensitizes hypoxic tumor cells to irinotecan by targeting hypoxia-inducible factor 1 alpha. Cancer Chemother. Pharmacol. 2010, 66, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Wang, L.Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1 alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol. Cell. Boil. 2003, 23, 9361–9374. [Google Scholar] [CrossRef]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [PubMed]
- Forster, J.C.; Harriss-Phillips, W.M.; Douglass, M.J.; Bezak, E. A review of the development of tumor vasculature and its effects on the tumor microenvironment. Hypoxia 2017, 5, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Chouaib, S. Targeting hypoxia at the forefront of anticancer immune responses. Oncoimmunology 2014, 3, e954463. [Google Scholar] [CrossRef] [PubMed]
- Baine, M.K.; Turcu, G.; Zito, C.R.; Adeniran, A.J.; Camp, R.L.; Chen, L.; Kluger, H.M.; Jilaveanu, L.B. Characterization of tumor infiltrating lymphocytes in paired primary and metastatic renal cell carcinoma specimens. Oncotarget 2015, 6, 24990–25002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaaf, M.B.; Garg, A.D.; Agostinis, P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018, 9, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, M.; Steffens, S.; Bellut, M.; Eggers, H.; Grosshennig, A.; Becker, J.U.; Wegener, G.; Schrader, A.J.; Grunwald, V.; Ivanyi, P. Intratumoral expression of programmed death ligand 1 (PD-L1) in patients with clear cell renal cell carcinoma (ccRCC). Med. Oncol. 2016, 33, 80. [Google Scholar] [CrossRef] [PubMed]
- Callea, M.; Albiges, L.; Gupta, M.; Cheng, S.C.; Genega, E.M.; Fay, A.P.; Song, J.; Carvo, I.; Bhatt, R.S.; Atkins, M.B.; et al. Differential Expression of PD-L1 between Primary and Metastatic Sites in Clear-Cell Renal Cell Carcinoma. Cancer Immunol. Res. 2015, 3, 1158–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Jiang, C.C.; Jin, L.; Zhang, X.D. Regulation of PD-L1: A novel role of pro-survival signalling in cancer. Ann. Oncol. 2016, 27, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Fay, A.P.; Gray, K.P.; Callea, M.; Ho, T.H.; Albiges, L.; Bellmunt, J.; Song, J.; Carvo, I.; Lampron, M.; et al. PD-L1 expression in nonclear-cell renal cell carcinoma. Ann. Oncol. 2014, 25, 2178–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jilaveanu, L.B.; Shuch, B.; Zito, C.R.; Parisi, F.; Barr, M.; Kluger, Y.; Chen, L.; Kluger, H.M. PD-L1 Expression in Clear Cell Renal Cell Carcinoma: An Analysis of Nephrectomy and Sites of Metastases. J. Cancer 2014, 5, 166–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, R.W.; Millis, S.Z.; Carballido, E.M.; Bryant, D.; Gatalica, Z.; Reddy, S.; Bryce, A.H.; Vogelzang, N.J.; Stanton, M.L.; Castle, E.P.; et al. PD-1 and PD-L1 Expression in Renal Cell Carcinoma with Sarcomatoid Differentiation. Cancer Immunol. Res. 2015, 3, 1303–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leite, K.R.; Reis, S.T.; Junior, J.P.; Zerati, M.; Gomes Dde, O.; Camara-Lopes, L.H.; Srougi, M. PD-L1 expression in renal cell carcinoma clear cell type is related to unfavorable prognosis. Diagn. Pathol. 2015, 10, 189. [Google Scholar] [CrossRef] [PubMed]
- Messai, Y.; Gad, S.; Noman, M.Z.; Le Teuff, G.; Couve, S.; Janji, B.; Kammerer, S.F.; Rioux-Leclerc, N.; Hasmim, M.; Ferlicot, S.; et al. Renal Cell Carcinoma Programmed Death-ligand 1, a New Direct Target of Hypoxia-inducible Factor-2 Alpha, is Regulated by von Hippel-Lindau Gene Mutation Status. Eur. Urol. 2016, 70, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Ruf, M.; Moch, H.; Schraml, P. PD-L1 expression is regulated by hypoxia inducible factor in clear cell renal cell carcinoma. Int. J. Cancer 2016, 139, 396–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Lastwika, K.J.; Wilson, W., 3rd; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Mimura, K.; Teh, J.L.; Okayama, H.; Shiraishi, K.; Kua, L.F.; Koh, V.; Smoot, D.T.; Ashktorab, H.; Oike, T.; Suzuki, Y.; et al. PD-L1 expression is mainly regulated by interferon gamma associated withJAK-STAT pathway in gastric cance. Cancer Sci. 2018, 109, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, L.; Huang, F.; Zhang, Q.; Liu, S.; Ma, L.; You, Z. Inflammatory cytokines IL-17 and TNF-alpha up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol. Lett. 2017, 184, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Neal, C.S.; Michael, M.Z.; Rawlings, L.H.; Van der Hoek, M.B.; Gleadle, J.M. The VHL-dependent regulation of microRNAs in renal cancer. BMC Med. 2010, 8, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, M.W.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaunitz, G.J.; Cottrell, T.R.; Lilo, M.; Muthappan, V.; Esandrio, J.; Berry, S.; Xu, H.; Ogurtsova, A.; Anders, R.A.; Fischer, A.H.; et al. Melanoma subtypes demonstrate distinct PD-L1 expression profiles. Lab. Investig. 2017, 97, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noman, M.Z.; Janji, B.; Hu, S.; Wu, J.C.; Martelli, F.; Bronte, V.; Chouaib, S. Tumor-Promoting Effects of Myeloid-Derived Suppressor Cells Are Potentiated by Hypoxia-Induced Expression of miR-210. Cancer Res. 2015, 75, 3771–3787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miscoria, M.; Di Loreto, C.; Puglisi, F.; Murray, P.G.; Deroma, L.; Atmadini, M. Thymidine phosphorylase expression in metastatic kidney cancer as a potential predictor of outcome in patients treated with sunitinib. J. Clin. Oncol. 2012. [Google Scholar] [CrossRef]
- Eda, H.; Fujimoto, K.; Watanabe, S.; Ura, M.; Hino, A.; Tanaka, Y.; Wada, K.; Ishitsuka, H. Cytokines induce thymidine phosphorylase expression in tumor cells and make them more susceptible to 5′-deoxy-5-fluorouridine. Cancer Chemother. Pharmacol. 1993, 32, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wang, L.; Chen, Y.; Zheng, X.; Wang, X. Poor Prognosis Associated with High Levels of Thymidine Phosphorylase and Thrombocytosis in Patients with Renal Cell Carcinoma. Urol. Int. 2017, 98, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Lai, H.; Qin, Y.; Chen, J.; Lin, Y. Thymidine phosphorylase and hypoxia-inducible factor 1-alpha expression in clinical stage II/III rectal cancer: Association with response to neoadjuvant chemoradiation therapy and prognosis. Int. J. Clin. Exp. Pathol. 2015, 8, 10680–10688. [Google Scholar] [PubMed]
- Atrih, A.; Mudaliar, M.A.V.; Zakikhani, P.; Lamont, D.J.; Huang, J.T.-J.; Bray, S.E.; Barton, G.; Fleming, S.; Nabi, G. Quantitative proteomics in resected renal cancer tissue for biomarker discovery and profiling. Br. J. Cancer 2014, 110, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Padrik, P.; Saar, H. Thymidine phosphorylase as a prognostic factor in renal cell carcinoma. Int. Urol. Nephrol. 2010, 42, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Takayama, T.; Mugiya, S.; Sugiyama, T.; Aoki, T.; Furuse, H.; Liu, H.; Hirano, Y.; Kai, F.; Ushiyama, T.; Ozono, S. High levels of thymidine phosphorylase as an independent prognostic factor in renal cell carcinoma. Jpn. J. Clin. Oncol. 2006, 36, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Gimm, T.; Wiese, M.; Teschemacher, B.; Deggerich, A.; Schodel, J.; Knaup, K.X.; Hackenbeck, T.; Hellerbrand, C.; Amann, K.; Wiesener, M.S.; et al. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J. 2010, 24, 4443–4458. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I.; et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Q.; Ruan, H.; Wang, K.; Song, Z.; Bao, L.; Xu, T.; Xiao, H.; Wang, C.; Cheng, G.; Tong, J.; et al. Overexpression of PLIN2 is a prognostic marker and attenuate tumor progression in clear cell renal cell carcinoma. Int J. Oncol. 2018, 53, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Azrak, R.G.; Cao, S.; Durrani, F.A.; Toth, K.; Bhattacharya, A.; Rustum, Y.M. Augmented therapeutic efficacy of irinotecan is associated with enhanced drug accumulation. Cancer Lett. 2011, 311, 219–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, A.; Seshadri, M.; Oven, S.D.; Toth, K.; Vaughan, M.M.; Rustum, Y.M. Tumor vascular maturation and improved drug delivery induced by methylselenocysteine leads to therapeutic synergy with anticancer drugs. Clin. Cancer Res. 2008, 14, 3926–3932. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalizing tumor microenvironment to treat cancer: Bench to bedside to biomarkers. J. Clin. Oncol. 2013, 31, 2205–2218. [Google Scholar] [CrossRef] [PubMed]
- Rustum, Y.M.; Toth, K.; Seshadri, M.; Sen, A.; Durrani, F.A.; Stott, E.; Morrison, C.D.; Cao, S.; Bhattacharya, A. Architectural heterogeneity in tumors caused by differentiation alters intratumoral drug distribution and affects therapeutic synergy of antiangiogenic organoselenium compound. J. Oncol. 2010, 2010, 396286. [Google Scholar] [CrossRef] [PubMed]
- Lytle, J.R.; Yario, T.A.; Steitz, J.A. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′ UTR as in the 3′ UTR. Proc. Natl. Acad. Sci. USA 2007, 104, 9667–9672. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rana, T.M. Therapeutic targeting of microRNAs: Current status and future challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, L.F.; Kushlinskiy, N.E. Regulatory mechanisms of microRNA expression. J. Transl. Med. 2016, 14, 143. [Google Scholar] [CrossRef] [PubMed]
- Kuninty, P.R.; Schnittert, J.; Storm, G.; Prakash, J. MicroRNA Targeting to Modulate Tumor Microenvironment. Front. Oncol. 2016, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, Y.; Song, Y.; Bu, R.; Yin, B.; Fei, X.; Guo, Q.; Wu, B. MicroRNAs in renal cell carcinoma: A systematic review of clinical implications (Review). Oncol. Rep. 2015, 33, 1571–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, R.I.; Blick, C.; Ragoussis, J.; Schoedel, J.; Mole, D.R.; Young, A.C.; Selby, P.J.; Banks, R.E.; Harris, A.L. miR-210 is a target of hypoxia-inducible factors 1 and 2 in renal cancer, regulates ISCU and correlates with good prognosis. Br. J. Cancer 2013, 108, 1133–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schanza, L.M.; Seles, M.; Stotz, M.; Fosselteder, J.; Hutterer, G.C.; Pichler, M.; Stiegelbauer, V. MicroRNAs Associated with Von Hippel-Lindau Pathway in Renal Cell Carcinoma: A Comprehensive Review. Int. J. Mol. Sci. 2017, 18, 2495. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Li, X.; Jia, Y.F.; Piazza, G.A.; Xi, Y. Hypoxia-regulated microRNAs in human cancer. Acta Pharmacol. Sin. 2013, 34, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Xu, H. Prognostic value of meta-signature miRNAs in renal cell carcinoma: An integrated miRNA expression profiling analysis. Sci. Rep. 2015, 5, 10272. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lin, W.; Tang, X.; Li, S.; Guo, L.; Lin, Y.; Kwok, H.F. The Roles of microRNAs in Regulating the Expression of PD-1/PD-L1 Immune Checkpoint. Int. J. Mol. Sci. 2017, 18, 2540. [Google Scholar] [CrossRef] [PubMed]
- Yee, D.; Shah, K.M.; Coles, M.C.; Sharp, T.V.; Lagos, D. MicroRNA-155 induction via TNF-alpha and IFN-gamma suppresses expression of programmed death ligand-1 (PD-L1) in human primary cells. J. Boil. Chem. 2017, 292, 20683–20693. [Google Scholar] [CrossRef] [PubMed]
- Durrani, F.; Cao, S.; Park, Y.-M.; Thiompson, G.; Martin, J.; Yang, G.; Kuettel, M.; Rustum, Y.M. Synergistic effect of selenium compounds with radiation therapy in human A549 lung xenografts. Cancer Res. 2007, 67 (Suppl. 9), 750. [Google Scholar]
- Durrani, F.A.; Chintala, S.; Toth, K.; Cao, S.; Rustum, Y.M. Mechanism-based drug combination targeting HIF-2α and VEGF in renal cancer xenografts. Trends Cell Mol. Boil. 2015, 10, 12. [Google Scholar]
- Frost, J.; Galdeano, C.; Soares, P.; Gadd, M.S.; Grzes, K.M.; Ellis, L.; Epemolu, O.; Shimamura, S.; Bantscheff, M.; Grandi, P.; et al. Potent and selective chemical probe of hypoxic signalling downstream of HIF-alpha hydroxylation via VHL inhibition. Nat. Commun. 2016, 7, 13312. [Google Scholar] [CrossRef] [PubMed]
- Soares, P.; Gadd, M.S.; Frost, J.; Galdeano, C.; Ellis, L.; Epemolu, O.; Rocha, S.; Read, K.D.; Ciulli, A. Group-Based Optimization of Potent and Cell-Active Inhibitors of the von Hippel-Lindau (VHL) E3 Ubiquitin Ligase: Structure-Activity Relationships Leading to the Chemical Probe (2S,4R)-1-((S)-2-(1-Cyanocyclopropanecarboxamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (VH298). J. Med. Chem. 2018, 61, 599–618. [Google Scholar] [PubMed]
- Courtney, K.D.; Infante, J.R.; Lam, E.T.; Figlin, R.A.; Rini, B.I.; Brugarolas, J.; Zojwalla, N.J.; Lowe, A.M.; Wang, K.; Wallace, E.M.; et al. Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2alpha Antagonist in Patients with Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J. Clin. Oncol. 2018, 36, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, E.; Jung, J.; Lee, J.W.; Kim, H.J.; Kim, J.; Yoo, H.J.; Lee, H.J.; Chae, S.Y.; Jeon, S.M.; et al. microRNA-155 positively regulates glucose metabolism via PIK3R1-FOXO3a-cMYC axis in breast cancer. Oncogene 2018, 37, 2982–2991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Ma, X.; Yao, Y.; Li, H.; Fan, Y.; Zhang, Y.; Zhao, C.; Wang, L.; Ma, M.; Lei, Z.; et al. miR-155 regulates the proliferation and invasion of clear cell renal cell carcinoma cells by targeting E2F2. Oncotarget 2016, 7, 20324–20337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, R.; Ma, Y.L.; Liang, W.; Li, H.H.; Ma, Z.J.; Yu, X.; Liao, Y.H. MicroRNA-155 modulates Treg and Th17 cells differentiation and Th17 cell function by targeting SOCS1. PLoS ONE 2012, 7, e46082. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Harris, A.L.; Martelli, F.; Kulshreshtha, R. Hypoxia response and microRNAs: No longer two separate worlds. J. Cell. Mol. Med. 2008, 12, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.D.; Chang, S. Development of Novel Therapeutic Agents by Inhibition of Oncogenic MicroRNAs. Int. J. Mol. Sci. 2017, 19, 65. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.F. Drug target miRNAs: Chances and challenges. Trends Biotechnol. 2014, 32, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Wigerup, C.; Pahlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jedeszko, C.; Paez-Ribes, M.; Di Desidero, T.; Man, S.; Lee, C.R.; Xu, P.; Bjarnason, G.A.; Bocci, G.; Kerbel, R.S. Postsurgical adjuvant or metastatic renal cell carcinoma therapy models reveal potent antitumor activity of metronomic oral topotecan with pazopanib. Sci. Transl. Med. 2015, 7, 282ra250. [Google Scholar] [CrossRef] [PubMed]
- Oevermann, K.; Buer, J.; Hoffmann, R.; Franzke, A.; Schrader, A.; Patzelt, T.; Kirchner, H.; Atzpodien, J. Capecitabine in the treatment of metastatic renal cell carcinoma. Br. J. Cancer 2000, 83, 583–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannir, N.M.; Thall, P.F.; Ng, C.S.; Wang, X.; Wooten, L.; Siefker-Radtke, A.; Mathew, P.; Pagliaro, L.; Wood, C.; Jonasch, E. A phase II trial of gemcitabine plus capecitabine for metastatic renal cell cancer previously treated with immunotherapy and targeted agents. J. Urol. 2008, 180, 867–872; discussion 872. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Durrani, F.A.; Rustum, Y.M. Selective modulation of the therapeutic efficacy of anticancer drugs by selenium containing compounds against human tumor xenografts. Clin. Cancer Res. 2004, 10, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Durrani, F.A.; Toth, K.; Rustum, Y.M. Se-methylselenocysteine offers selective protection against toxicity and potentiates the antitumour activity of anticancer drugs in preclinical animal models. Br. J. Cancer 2014, 110, 1733–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakharia, Y.; Garje, R.; Brown, J.A.; Nepple, K.G.; Dahmoush, L.; Gibson-Corley, K. Phase1 clinical trial of high doses of Seleno-L-methionine (SLM), in sequential combination with axitinib in previously treated and relapsed clear cell renal cell carcinoma (ccRCC) patients. J. Clin. Oncol. 2018, 36 (Suppl. 6), 630. [Google Scholar] [CrossRef]
- Zakharia, Y.; Bhattacharya, A.; Rustum, Y.M. Selenium targets resistance biomarkers enhancing efficacy while reducing toxicity of anti-cancer drugs: Preclinical and clinical development. Oncotarget 2018, 9, 10765–10783. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Incidence of HIF-α and PHDs Protein Expression in Primary Human ccRCC, Head & neck (H/N) and Colorectal Cancer (CRC) Tumor Biopsies: | |||||
| Tumors | HIF-1α | HIF-2α | HIF-1α and/or HIF-2α | PHD2 | PHD3 |
| ccRCC | 45% (40/88) | 78% (69/88) | 92% (81/88) | 35% (31/88) | 0% (0/88) |
| H/N | 23% (40/173) | 16% (23/146) | 38% (46/122) | 86% (180/210) | 21% (32/153) |
| CRC | 13% (8/62) | 15% (10/65) | 26% (17/64) | 90% (55/61) | 50% (31/62) |
| VEGF(A) | |||||
| Tumors | Incidence of Positions | Average Immunoscope | |||
| ccRCC | 54% (48/88) | 2.3 (weak) | |||
| H/N | 79% (136/173) | 4.24 (moderate) | |||
| CRC | 97% (60/62) | 5.68 (strong) | |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rustum, Y.M.; Chintala, S.; Durrani, F.A.; Bhattacharya, A. Non-Coding Micro RNAs and Hypoxia-Inducible Factors Are Selenium Targets for Development of a Mechanism-Based Combination Strategy in Clear-Cell Renal Cell Carcinoma—Bench-to-Bedside Therapy. Int. J. Mol. Sci. 2018, 19, 3378. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113378
Rustum YM, Chintala S, Durrani FA, Bhattacharya A. Non-Coding Micro RNAs and Hypoxia-Inducible Factors Are Selenium Targets for Development of a Mechanism-Based Combination Strategy in Clear-Cell Renal Cell Carcinoma—Bench-to-Bedside Therapy. International Journal of Molecular Sciences. 2018; 19(11):3378. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113378
Chicago/Turabian StyleRustum, Youcef M., Sreenivasulu Chintala, Farukh A. Durrani, and Arup Bhattacharya. 2018. "Non-Coding Micro RNAs and Hypoxia-Inducible Factors Are Selenium Targets for Development of a Mechanism-Based Combination Strategy in Clear-Cell Renal Cell Carcinoma—Bench-to-Bedside Therapy" International Journal of Molecular Sciences 19, no. 11: 3378. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113378