Involvement of 5′AMP-Activated Protein Kinase (AMPK) in the Effects of Resveratrol on Liver Steatosis

 , , and
, , and
Abstract
:
1. Introduction
2. In Vitro Studies
3. In Vivo Studies on Hepatic Steatosis Prevention
4. In Vivo Studies on Hepatic Steatosis Treatment
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | acetyl-CoA-carboxylase |
| ACOX1 | Acyl-Coenzyme A oxidase 1 |
| AICAR | 5-Aminoimidazole-4-carboxamide ribonucleotide |
| AKT | Protein kinase B |
| ALP | Alkaline phosphatase, |
| ALT | Alanine aminotransferase |
| AMP | Adenosine monophosphate |
| AMPK | 5′AMP-activated protein kinase |
| AST | Aspartate aminotransferase |
| ATGL | Adipose triglyceride lipase |
| ATP | Adenosine triphosphate |
| BW | Body weight |
| CaMKKβ | Ca2+/calmodulin-dependent protein kinase kinase β |
| cAMP | Cyclic adenosine monophosphate |
| CHREBP | Carbohydrate response element binding protein |
| CPT | Carnitine palmitoyl transferase |
| CS | Citrate synthase |
| FAS | Fatty acid synthase |
| FATP5 | Fatty acid transport protein 5 |
| FFA | Free fatty acid |
| FOXO 1 | Forkhead box protein O1 |
| GGT | Gamma glutamil transpeptidase |
| GPAT1 | Glycerol-3-phosphate acyltransferase 1 |
| G6PDH | Glucose-6P-dehydrogenase |
| GSH | Glutathione |
| GPx | Glutathione peroxidase |
| HBV | Hepatitis B virus |
| HFD | High-fat diet |
| HFHS | High-fat high-sucrose diet |
| HSL | Hormone sensitive lipase |
| IkBα | Inhibitor of nuclear factor kappa subunit α |
| IL | Interleukin |
| LDH | Lactate dehydrogenase |
| LFD | Low fat diet |
| LKB1 | Liver kinase B1 |
| LXR | Liver X receptor |
| MCAD | Mitochondrial medium-chain acyl-CoA dehydrogenase |
| MDA | Malonaldehyde |
| ME | Malic enzyme |
| MTP | Microsomal triglyceride transfer protein |
| NAD+ | Oxidized nicotinamide adenine dinucleotide |
| NADH | Reduced nicotinamide adenine dinucleotide |
| NAFLD | Non-alcoholic fatty liver disease |
| NF-kB p65 | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| O+A | Oleic acid plus alcohol |
| PDE | Phosphodiesterase |
| pIkBα | Phospho-inhibitory subunit of NF-KBα |
| PPAR | Peroxisome proliferator-activated receptor |
| PRKA | Protein kinase A |
| ROS | Reactive oxygen species |
| RSV | Resveratrol |
| si RNA | Small interfering RNA |
| SIRT1 | Silent information regulator 1; Surtuin-1 |
| SCD | Stearoyl-Coenzyme A desaturase 1 |
| SQSTM1 | Sequestosome 1 |
| SOD | Superoxide dismutase |
| SREBF | Sterol regulatory element-binding transcription factor |
| SREBP | Sterol regulatory element-binding protein |
| STD | Standard diet |
| TG | Triglyceride |
| TNF-α | Tumor necrosis factor- α |
| UCP | Uncoupling protein |
References
- Milton-Laskibar, I.; Aguirre, L.; Fernández-Quintela, A.; Rolo, A.P.; Soeiro Teodoro, J.; Palmeira, C.M.; Portillo, M.P. Lack of additive effects of resveratrol and energy restriction in the treatment of hepatic steatosis in rats. Nutrients 2017, 9, 737. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, L.; Portillo, M.P.; Hijona, E.; Bujanda, L. Effects of resveratrol and other polyphenols in hepatic steatosis. World J. Gastroenterol. 2014, 20, 7366–7380. [Google Scholar] [CrossRef] [PubMed]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. Ampk as a therapeutic target for treating metabolic diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Carling, D. Ampk signalling in health and disease. Curr. Opin. Cell Biol. 2017, 45, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. Ampk: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Carling, D. The amp-activated protein kinase cascade—A unifying system for energy control. Trends Biochem. Sci. 2004, 29, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Minireview: The amp-activated protein kinase cascade: The key sensor of cellular energy status. Endocrinology 2003, 144, 5179–5183. [Google Scholar] [CrossRef] [PubMed]
- Cool, B.; Zinker, B.; Chiou, W.; Kifle, L.; Cao, N.; Perham, M.; Dickinson, R.; Adler, A.; Gagne, G.; Iyengar, R.; et al. Identification and characterization of a small molecule ampk activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006, 3, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Williams, J.R.; Muckett, P.J.; Mayer, F.V.; Liljevald, M.; Bohlooly, Y.M.; Carling, D. Liver-specific activation of ampk prevents steatosis on a high-fructose diet. Cell Rep. 2017, 18, 3043–3051. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Ampk: A target for drugs and natural products with effects on both diabetes and cancer. Diabetes 2013, 62, 2164–2172. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Quintela, A.; Milton-Laskibar, I.; González, M.; Portillo, M.P. Antiobesity effects of resveratrol: Which tissues are involved? Ann. N. Y. Acad. Sci. 2017, 1403, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Heebøll, S.; Thomsen, K.L.; Pedersen, S.B.; Vilstrup, H.; George, J.; Grønbæk, H. Effects of resveratrol in experimental and clinical non-alcoholic fatty liver disease. World J. Hepatol. 2014, 6, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Heebøll, S.; Thomsen, K.L.; Clouston, A.; Sundelin, E.I.; Radko, Y.; Christensen, L.P.; Ramezani-Moghadam, M.; Kreutzfeldt, M.; Pedersen, S.B.; Jessen, N.; et al. Effect of resveratrol on experimental non-alcoholic steatohepatitis. Pharmacol. Res. 2015, 95–96, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Heebøll, S.; El-Houri, R.B.; Hellberg, Y.E.; Haldrup, D.; Pedersen, S.B.; Jessen, N.; Christensen, L.P.; Grønbaek, H. Effect of resveratrol on experimental non-alcoholic fatty liver disease depends on severity of pathology and timing of treatment. J. Gastroenterol. Hepatol. 2016, 31, 668–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Bruno, J.; Easlon, E.; Lin, S.J.; Cheng, H.L.; Alt, F.W.; Guarente, L. Tissue-specific regulation of sirt1 by calorie restriction. Genes Dev. 2008, 22, 1753–1757. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Auwerx, J. Pgc-1alpha, sirt1 and ampk, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, N.B.; Xu, X.J.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. Ampk and sirt1: A long-standing partnership? Am. J. Physiol. Endocrinol. Metab. 2010, 298, E751–E760. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. Sirt1 regulates hepatocyte lipid metabolism through activating amp-activated protein kinase. J. Biol. Chem. 2008, 283, 20015–20026. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. Sirt1 modulation of the acetylation status, cytosolic localization, and activity of lkb1. Possible role in amp-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef] [PubMed]
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 2008, 14, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P.; et al. Amp-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting camp phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Zang, M.; Xu, S.; Maitland-Toolan, K.A.; Zuccollo, A.; Hou, X.; Jiang, B.; Wierzbicki, M.; Verbeuren, T.J.; Cohen, R.A. Polyphenols stimulate amp-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes 2006, 55, 2180–2191. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Chen, L.L.; Xiao, F.X.; Sun, H.; Ding, H.C.; Xiao, H. Resveratrol improves non-alcoholic fatty liver disease by activating amp-activated protein kinase. Acta Pharmacol. Sin. 2008, 29, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Liu, D. Resveratrol suppresses t0901317-induced hepatic fat accumulation in mice. AAPS J. 2013, 15, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Suh, H.R.; Yoon, Y.; Lee, K.J.; Kim, D.G.; Kim, S.; Lee, B.H. Protective effect of resveratrol derivatives on high-fat diet induced fatty liver by activating amp-activated protein kinase. Arch. Pharm. Res. 2014, 37, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.Y.; Chen, Y.; Rui, B.B.; Hu, C.M. Resveratrol ameliorates lipid accumulation in HepG2 cells, associated with down-regulation of lipin1 expression. Can. J. Physiol. Pharmacol. 2016, 94, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, M.L.; Zhou, Y.; Yi, L.; Gao, Y.X.; Ran, L.; Chen, S.H.; Zhang, T.; Zhou, X.; Zou, D.; et al. Resveratrol improves hepatic steatosis by inducing autophagy through the camp signaling pathway. Mol. Nutr. Food Res. 2015, 59, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Ma, J.; Wang, W.; Zhang, L.; Xu, J.; Wang, K.; Li, D. Resveratrol supplement inhibited the NF-κB inflammation pathway through activating AMPKα-sirt1 pathway in mice with fatty liver. Mol. Cell. Biochem. 2016, 422, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Zang, M.; Zuccollo, A.; Hou, X.; Nagata, D.; Walsh, K.; Herscovitz, H.; Brecher, P.; Ruderman, N.B.; Cohen, R.A. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J. Biol. Chem. 2004, 279, 47898–47905. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.Y.; Repa, J.J. The liver x receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J. Biol. Chem. 2007, 282, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Liang, G.; Ou, J.; Bashmakov, Y.; Lobaccaro, J.M.; Shimomura, I.; Shan, B.; Brown, M.S.; Goldstein, J.L.; Mangelsdorf, D.J. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000, 14, 2819–2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajmo, J.M.; Liang, X.; Rogers, C.Q.; Pennock, B.; You, M. Resveratrol alleviates alcoholic fatty liver in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G833–G842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberdi, G.; Rodríguez, V.M.; Macarulla, M.T.; Miranda, J.; Churruca, I.; Portillo, M.P. Hepatic lipid metabolic pathways modified by resveratrol in rats fed an obesogenic diet. Nutrition 2013, 29, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Tung, Y.C.; Lin, Y.H.; Chen, H.J.; Chou, S.C.; Cheng, A.C.; Kalyanam, N.; Ho, C.T.; Pan, M.H. Piceatannol exerts anti-obesity effects in c57bl/6 mice through modulating adipogenic proteins and gut microbiota. Molecules 2016, 21, 1419. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Kita, T.; Yamasaki, S.; Kawahara, T.; Ueno, Y.; Yamada, M.; Mukai, Y.; Sato, S.; Kurasaki, M.; Saito, T. Maternal resveratrol intake during lactation attenuates hepatic triglyceride and fatty acid synthesis in adult male rat offspring. Biochem. Biophys. Rep. 2017, 9, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Rivera, L.; Morón, R.; Zarzuelo, A.; Galisteo, M. Long-term resveratrol administration reduces metabolic disturbances and lowers blood pressure in obese Zucker rats. Biochem. Pharmacol. 2009, 77, 1053–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, G.M.; Jung, U.J.; Park, H.J.; Kwon, E.Y.; Jeon, S.M.; McGregor, R.A.; Choi, M.S. Resveratrol ameliorates diabetes-related metabolic changes via activation of amp-activated protein kinase and its downstream targets in db/db mice. Mol. Nutr. Food Res. 2012, 56, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Hong, H.J.; Guan, J.; Kim, D.G.; Yang, E.J.; Koh, G.; Park, D.; Han, C.H.; Lee, Y.J.; Lee, D.H. Resveratrol improves insulin signaling in a tissue-specific manner under insulin-resistant conditions only: In vitro and in vivo experiments in rodents. Metabolism 2012, 61, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.C.; Chen, Y.F.; Hsu, W.H.; Yang, C.W.; Kao, C.H.; Tsai, T.F. Resveratrol helps recovery from fatty liver and protects against hepatocellular carcinoma induced by hepatitis b virus × protein in a mouse model. Cancer Prev. Res. 2012, 5, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Chen, S.; Li, Z.; Zhao, X.; Li, W.; Sun, Y.; Zhang, Z.; Ling, W.; Feng, X. Effects and mechanisms of resveratrol on the amelioration of oxidative stress and hepatic steatosis in KKAy mice. Nutr. Metab. (Lond.) 2014, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Lim, Y.; Yang, S.J. Involvement of resveratrol in crosstalk between adipokine adiponectin and hepatokine fetuin-A in vivo and in vitro. J. Nutr. Biochem. 2015, 26, 1254–1260. [Google Scholar] [CrossRef] [PubMed]

 : regulation direction;
: regulation direction;  : regulation inhibition).
: regulation direction; : regulation inhibition).
: regulation inhibition).
: regulation direction; : regulation inhibition).

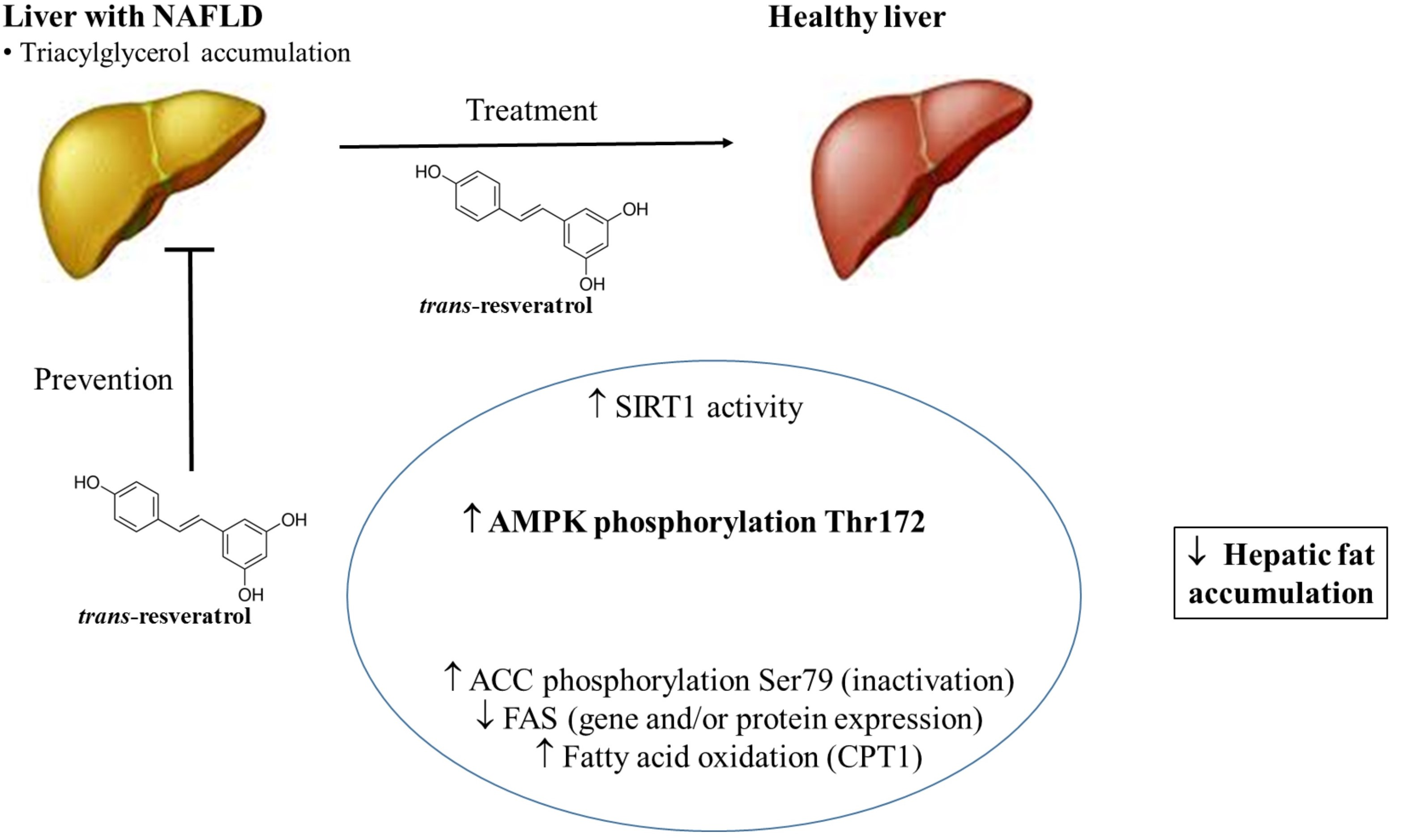{kind=link}
{kind=link}
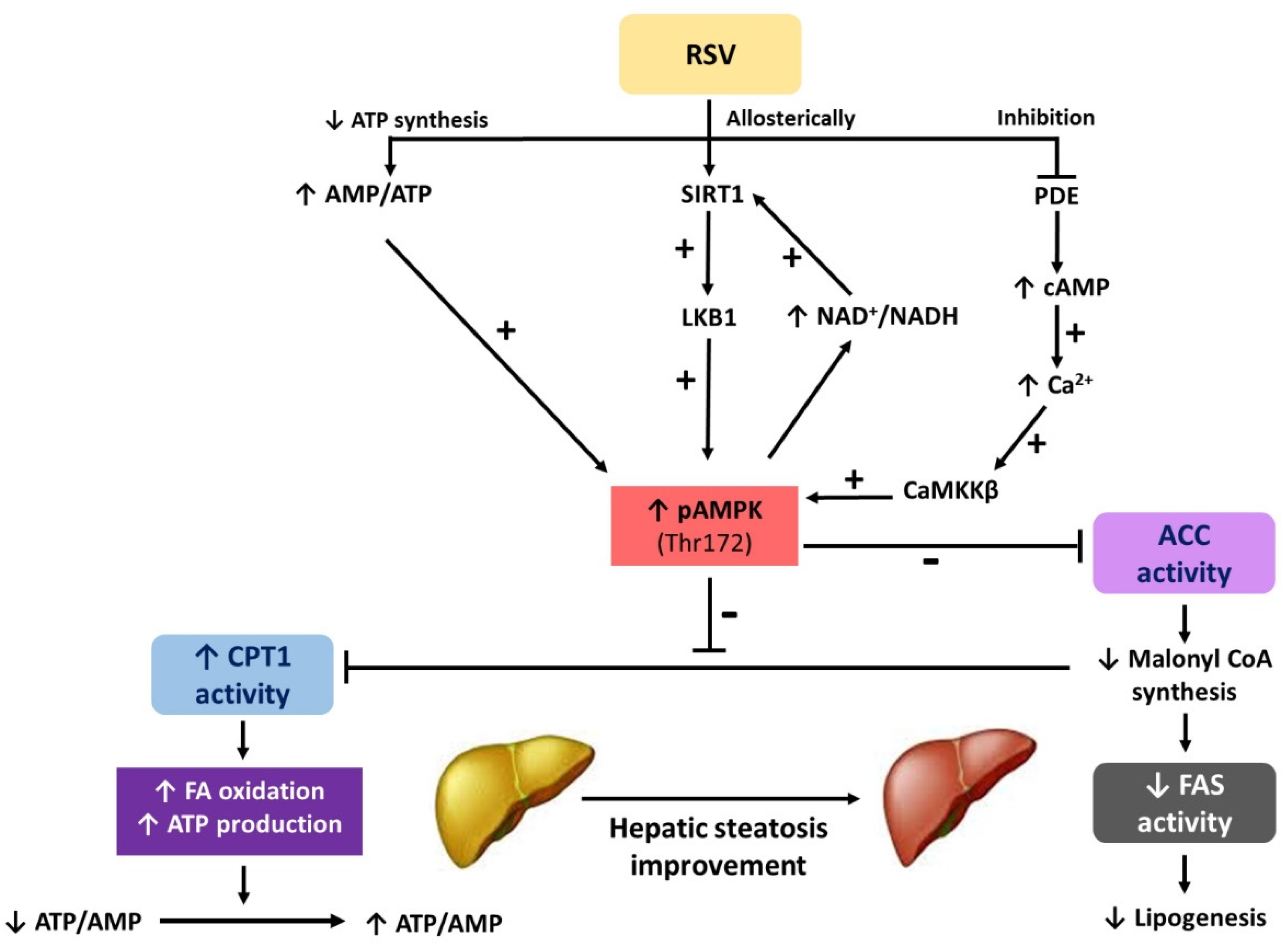{kind=link}
| Reference | Cell Line | Experimental Design | Effect of Resveratrol | Mechanism of Action |
|---|---|---|---|---|
| [23] | HepG2 | 24 h culture with RSV (10 µmol/L) 1 h culture with RSV (50 µmol/L) (AMPKα1 activity determination) | Prevention of high-glucose-induced lipid accumulation (in HepG2 cells) | ↑ AMPK (Thr-172) and ACC (Ser-79) phosphorylation (10 µmol/L RSV) ↑ AMPKα1 activity |
| [18] | HepG2 HEK293 | HepG2 24 h culture with RSV (1–100 μM) HEK293 cells pretreated with splitomicin (100 μM) for 24 h and incubated with RSV (50 μM) for an additional 1 h | ↓ High glucose-induced TG accumulation: RSV (10–50 μM) | ↑ SIRT1 activity (dose dependent (10–100 μM RSV) ↓ FAS protein expression ↑ AMPKα phosphorylation (Thr172) ↑ ACC phosphorylation (Ser79) |
| [24] | HepG2 | 6 or 24 h culture with 10, 25, and 50 μM of RSV | ↓ TG accumulation | ↑ AMPKα phosphorylation (Thr172) ↓ srebf1 and fasn gene expressions |
| [25] | Hepa 1–6 cell line (murine hepatocytes) | 24 h culture with T0901317 (1 μM, LXR activator: ↑ liver fat accumulation) + RSV (40 μM), with or without compound C (10 μM, AMPK inhibitor) | ↓ T0901317-induced fat accumulation | ↓ T0901317-induced fat accumulation (via AMPK activation) |
| [26] | H4IIEC3 rat hepatoma cells | 24 h culture with FFA [oleic acid and palmitic acid 2:1, 0.5 mM] or T0901317 (10 μM, LXR activator) + RSV or SY-102 (RSV derivative, 3.3–50 μM) | ↓ FFA-induced lipid accumulation by RSV (50 mΜ) and SY-102 (30 μM) ↓ T0901317-induced SREBP-1 maturation by RSV (30 μM) and SY-102 (10 μM) | ↓ srebf1 and fasn gene expression by SY-102 (50 μM) via AMPK/LXR pathway |
| [27] | HepG2 | 48 h treatment Control, oleic acid + alcohol (O + A), O + A-RSV (5, 15, 45, 13 5 µM), O + A-AICAR-RSV 45 µM, O + A-Compound C-RSV 45 µM | ↓ Lipid accumulation (15, 45, 135 μM) ↓ Hepatocyte TG content (45 and 135 μM) Attenuated hepatic steatosis | ↑ AMPKα phosphorylation * ↑ ACC phosphorylation * ↓ SREBP1c and lipin protein expression |
| [28] | HepG2 | 24 h culture with 0.2 mM palmitate Additional 24 h treated with RSV (20–80 μM) Also exposed to 3-MA autophagy inhibitor for 1 h or siRNAs before the addition of RSV | ↓ Lipid content | ↑ LC3-II protein expression and SQSTM1 protein degradation (3-MA pre-treatment inhibited this effect) ↑ SIRT1 protein expression/activity and cyclic AMP levels ↑ AMPK (Thr-172) and PRKA (Ser-96) phosphorylation |
| [29] | Primary hepatocytes from C57BL/6 mice | Treatment with NEFA, NEFA + RSV (50 and 100 μM), NEFA + Nicotinamide, NEFA + Compound C, NEFA + RSV + Nicotinamide, NEFA + RSV + Compound C Treatment length not specified | ↓ NEFA increased expression of several inflammatory markers | ↑ AMPK phosphorylation * ↑ sirt1 gene and SIRT1 protein expressions ↓ phosphorylation IκBα and NF-κB p65 ↓ il-1β, il-6, and tnf-α gene expression |
| Reference | Animal Model | Experimental Design | Effect of Resveratrol | Mechanism of Action |
|---|---|---|---|---|
| [33] | One-year-old male C57BL/6NIA mice | HFD (60% of calories as fat) RSV dose: 22.4 mg/kg bw/day Length: 6 months | Fatty liver development prevention (organ size) Prevention of cellular integrity loss and large lipid droplet accumulation | ↑AMPK phosphorylation (Thr172) ↑ACC phosphorylation (Ser79) |
| [35] | 6–8 week-old male C57BL/6J mice | LFD (10% of calories as fat) RSV dose: 200 and 400 mg/kg bw/day 3 groups: LF diet+ethanol, LF diet + ethanol + RSV200, LF diet + ethanol + RSV400 Length: 2 weeks | Prevention of liver weight, liver lipid droplets, hepatic TG content and serum ALT level increase | ↑sirt1 gene and SIRT1 protein expressions ↑ AMPKα and β phosphorylation * ↑ total AMPK levels ↑ ACC phosphorylation * ↓ SREBP1c protein expression ↓ fasn, gpat1, scd1, accα, me ↑acox1, mcad and cpt1a gene expression ↓ pparγ gene expression |
| [36] | Male Sprague Dawley rats | Obesogenic diet (45% of calories as fat) RSV dose: 30 mg/kg bw/day Length: 6 weeks | ↓ Hepatic fat content | ↑ AMPK phosphorylation (Thr172) ↑ ACC phosphorylation (Ser79) |
| [25] | Male C57BL/6 mice induced by LXR receptor | Groups: Control, T0901317 (LXR activator) and T0901317+ RSV RSV dose: 200 mg/kg bw/day Length: 5 days | Prevention of the increase in liver size, fat accumulation and TG content (induced by LXR activator) | ↑ AMPK phosphorylation (Thr172) ↑ACC phosphorylation (Ser79) ↓ srebf1, chrebp and acc expression (RSV alone) |
| [29] | 4 week-old C57BL/6 mice | HFD (60% of calories as fat) RSV dose: 30 mg/kg bw/day Length: 60 day | ↓ Liver weight ↓ GGT, AST, ALT, ALP, LDH plasma levels ↓ IL-1β, IL-6, and TNF-α plasma levels | ↑ AMPK phosphorylation (Thr172) ↑ SIRT1 protein expression ↓ IkBα and NF-kB p65 phosphorylation * ↓ il-1β, il-6, and tnf-α gene expression |
| [37] | 5 week-old male C57BL/6 mice | HFD (45% of energy as fat) RSV dose: 0.1% resveratrol (w/w) Length: 18 weeks | No changes in liver weight and serum AST and ALT levels | ↑ AMPK phosphorylation (Thr172) ↑ACC phosphorylation (Ser79) ↓ FAS protein expression ↓ Hepatic adipogenic protein expression |
| [38] | Pups from female Wistar rats | Control diet RSV dose: 20 mg/kg bw/day Length: 3 weeks (lactation period) | ↓ Hepatic lipid accumulation | ↑ AMPK phosphorylation (Ser403) ↑ SIRT1 protein expression ↓ Active/precursor SREBP-1c protein ratio ↓ ACC protein expression ↓ FAS protein expression ↓Hepatic adipogenic protein expression |
| Reference | Animal Model | Experimental Design | Effect of Resveratrol | Mechanism of Action |
|---|---|---|---|---|
| [24] | Male Wistar rats (180–200 g) | Acute treatment: Fed stated rats RSV dose: 100 mg/kg bw/day Length: 4 h | ↓ Hepatic fat content | ↑ AMPK phosphorylation (Thr172) ↓ srebf1 and fasn gene expressions |
| Chronic treatment: High-fat diet (59% of calories as fat) RSV dose: 100 mg/kg bw/day Length: 10 weeks | ||||
| [39] | Obese male Zucker rats and lean heterozygous littermates | STD RSV dose: 10 mg/kg bw/day Length: 8 weeks | No change in liver weight ↓ Liver TG and cholesterol content | ↑ AMPK phosphorylation (Thr172) ↑ ACC phosphorylation * |
| [40] | 4 week-old male C57BL/KsJ-db/db mice | RSV dose: 0.005% and 0.02% (w/w) Length: 6 weeks | ↓ Hepatic fat content (only in 0.02% RSV group) | ↓ ACC phosphorylation * ↑ srebf1 gene expression (0.02% RSV) ↑ PPARα protein expression (0.02% RSV) ↑ UCP2 protein expression ↑ AMPK phosphorylation * |
| [41] | 5 week-old male C57BL/6N mice | HFD RSV dose: 30 mg/kg bw/day Length: 2 weeks | ↓ Hepatic fat content | ↑ AKT phosphorylation (Ser473 and Thr308) ↓ AMPKα phosphorylation (Thr172) |
| [42] | 4 week-old male C57BL/6 mice expressing HBV X protein | RSV dose: 30 mg/kg bw/day Length: 2, 3, 7, and 14 days | Histopatology alteration reversion ↓ Serum ALT levels | ↓ srebf1 and lxrα gene expressions (from day 2 in advance) ↑ AMPK phosphorylation (Thr172) (from day 3 in advance) ↓ pparγ and acc gene expressions. ↑ SIRT1 protein expression and activity (from day 7 in advance) ↓ fasn gene expression |
| [26] | Male ICR mice (20–25 g) | HFD. RSV dose: 15 or 45 mg/kg bw/day. (same doses of SY-102, a RSV derivative). Length: 2 days | ↓ Hepatic TG levels (by SY-102 and RSV) | ↑ AMPK phosphorylation (Thr172) (by SY-102) ↓ srebf1 and fasn mRNA levels (by SY-102 and RSV) |
| [43] | 8 week-old male KKAy mice (genetic model of obesity) 8 week-old male C57BL/6J mice (control) | Chow diet (AIN93G) RSV dose: 2 or 4 g/kg diet Length: 12 weeks | ↓ Hepatic fat content (Oil Red) and TG levels Hepatic steatosis attenuation (histological study) ↓ MDA levels | ↑ AMPK phosphorylation (Thr172) ↑ SIRT1 protein expression ↑ FOXO1 phosphorylation (Thr24) ↓ ROS levels ↑ GSH levels, GPx and SOD activities ↑ hsl gene expression and HSL phosphorylation (Ser660) ↑ atgl gene expression and ATGL protein expression |
| [44] | 6 week-old male C57BL/6J mice | HFD RSV dose: 8 mg/kg bw/day Length: 4 weeks | ↓ Liver weight ↓ Plasma levels of Fetuin-A and ALT ↓ Hepatic index | ↑ ampk gene expression ↓ fetuin-A gene expression ↓ Fetuin-A protein expression ↓ nfκβ gene expression |
| [28] | 8 week-old 129/SvJ mice (male) | HFD (60% of energy as fat) RSV dose: 0.4% (w/w) Length: 8 weeks | ↓ Hepatic fat content | ↑ cyclic AMP levels ↑ PRKA phosphorylation (Ser96) ↑ AMPK phosphorylation (Thr172) ↑ SIRT1 protein expression |
| [1] | 6 week-old male Wistar rats | HFHS RSV dose: 30 mg/kg bw/day Length: 6 weeks | ↓ Hepatic TG content ↑ Plasma TG release (from liver) ↓ Liver fatty acid uptake | ↑ AMPK phosphorylation (Thr172) ↑ CPT1a activity ↑ CS activity ↑ MTP activity ↓ FATP5 protein expression |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trepiana, J.; Milton-Laskibar, I.; Gómez-Zorita, S.; Eseberri, I.; González, M.; Fernández-Quintela, A.; Portillo, M.P. Involvement of 5′AMP-Activated Protein Kinase (AMPK) in the Effects of Resveratrol on Liver Steatosis. Int. J. Mol. Sci. 2018, 19, 3473. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113473
Trepiana J, Milton-Laskibar I, Gómez-Zorita S, Eseberri I, González M, Fernández-Quintela A, Portillo MP. Involvement of 5′AMP-Activated Protein Kinase (AMPK) in the Effects of Resveratrol on Liver Steatosis. International Journal of Molecular Sciences. 2018; 19(11):3473. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113473
Chicago/Turabian StyleTrepiana, Jenifer, Iñaki Milton-Laskibar, Saioa Gómez-Zorita, Itziar Eseberri, Marcela González, Alfredo Fernández-Quintela, and María P. Portillo. 2018. "Involvement of 5′AMP-Activated Protein Kinase (AMPK) in the Effects of Resveratrol on Liver Steatosis" International Journal of Molecular Sciences 19, no. 11: 3473. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113473