Proteomic Analysis of Plasma Membrane Proteins of Antler Stem Cells Using Label-Free LC–MS/MS


Abstract
:1. Introduction
2. Results
2.1. Summary of Label-Free Liquid Chromatography–Mass Spetrometry (LC–MS/MS) Results
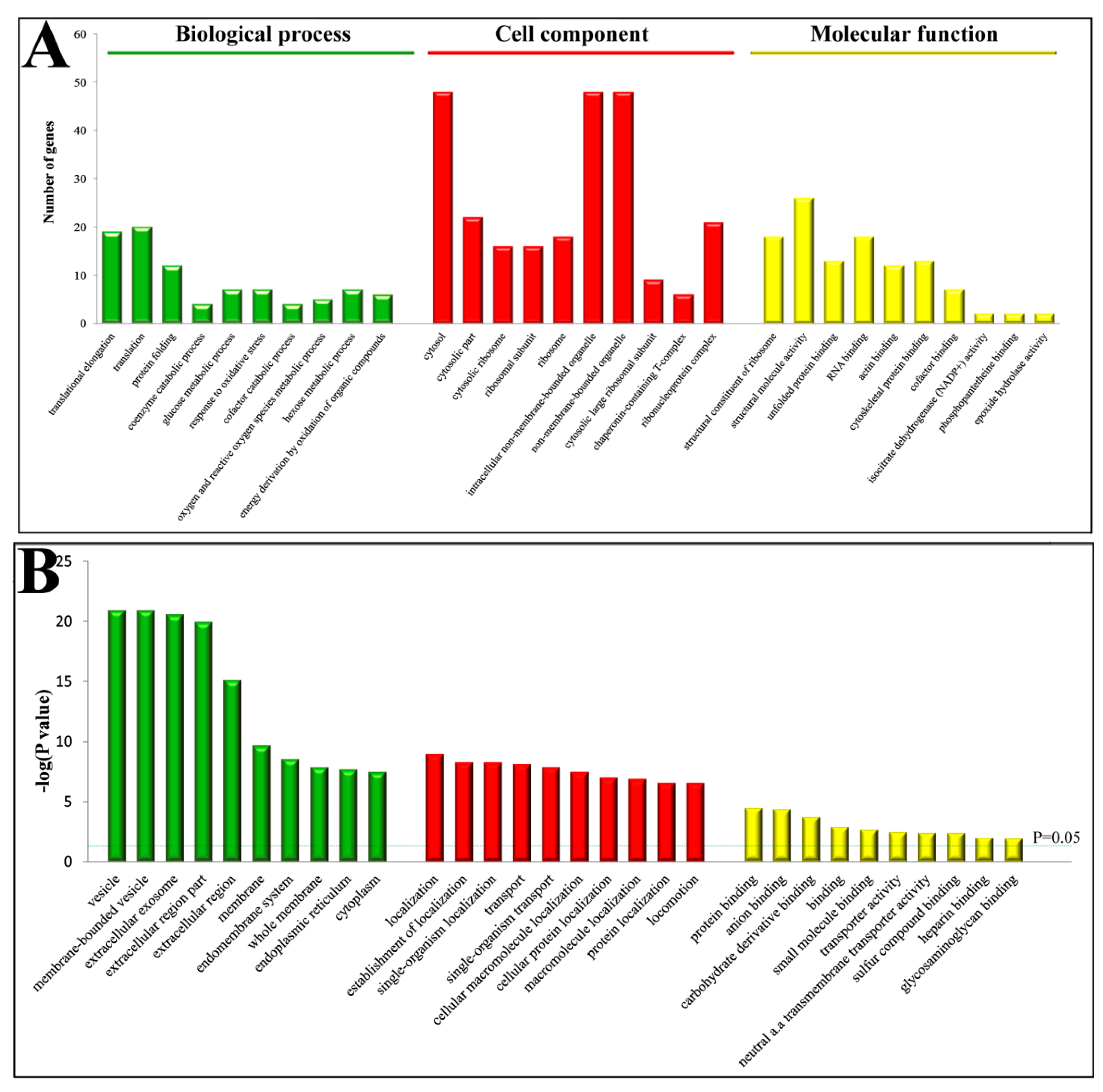
2.2. Functional Classification of Differentially Expressed Proteins (DEPs)
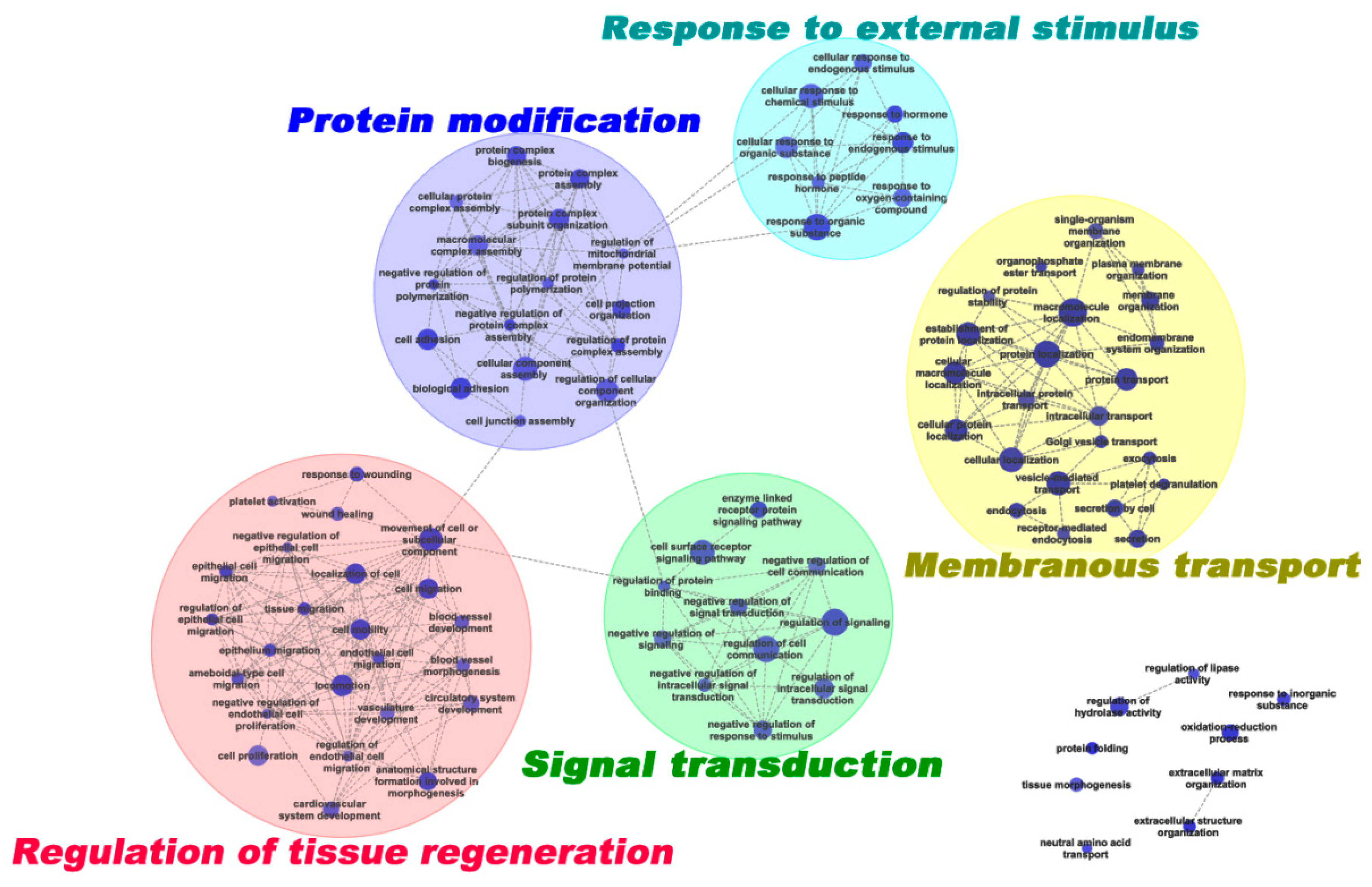
2.3. Interactive Network DEPs
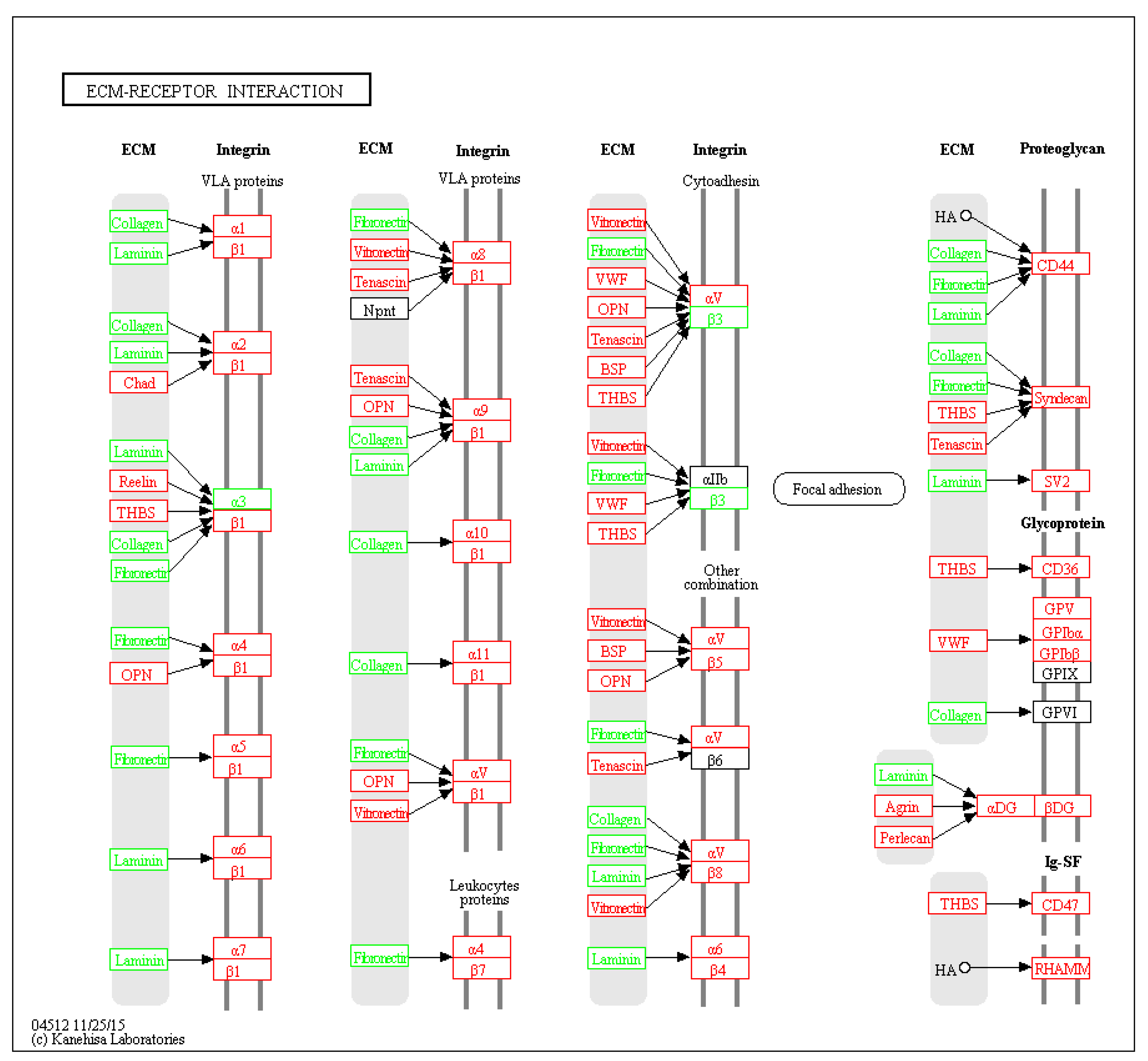
2.4. Enriched Signaling Pathways
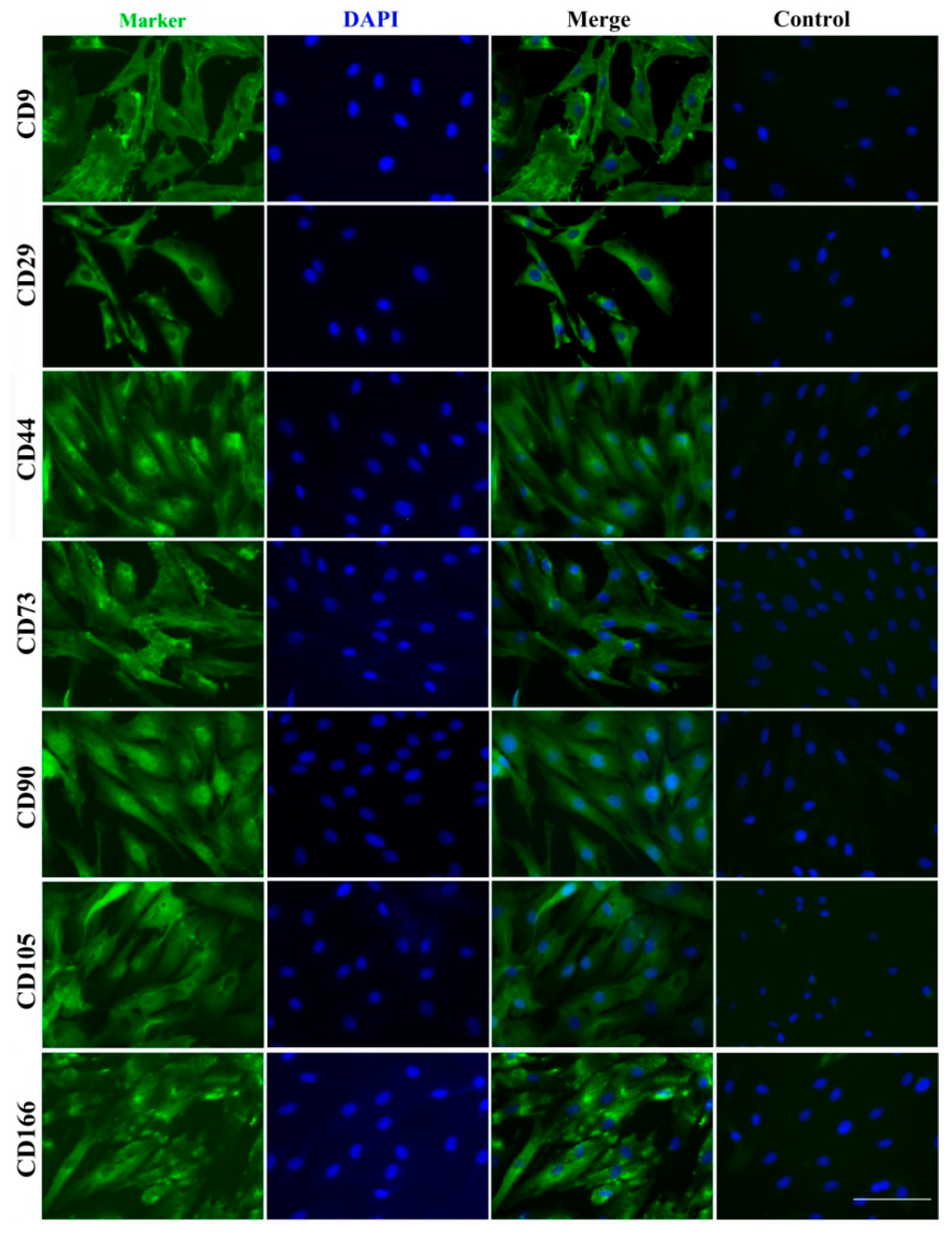
2.5. Expressed Stem Cell Surface Markers
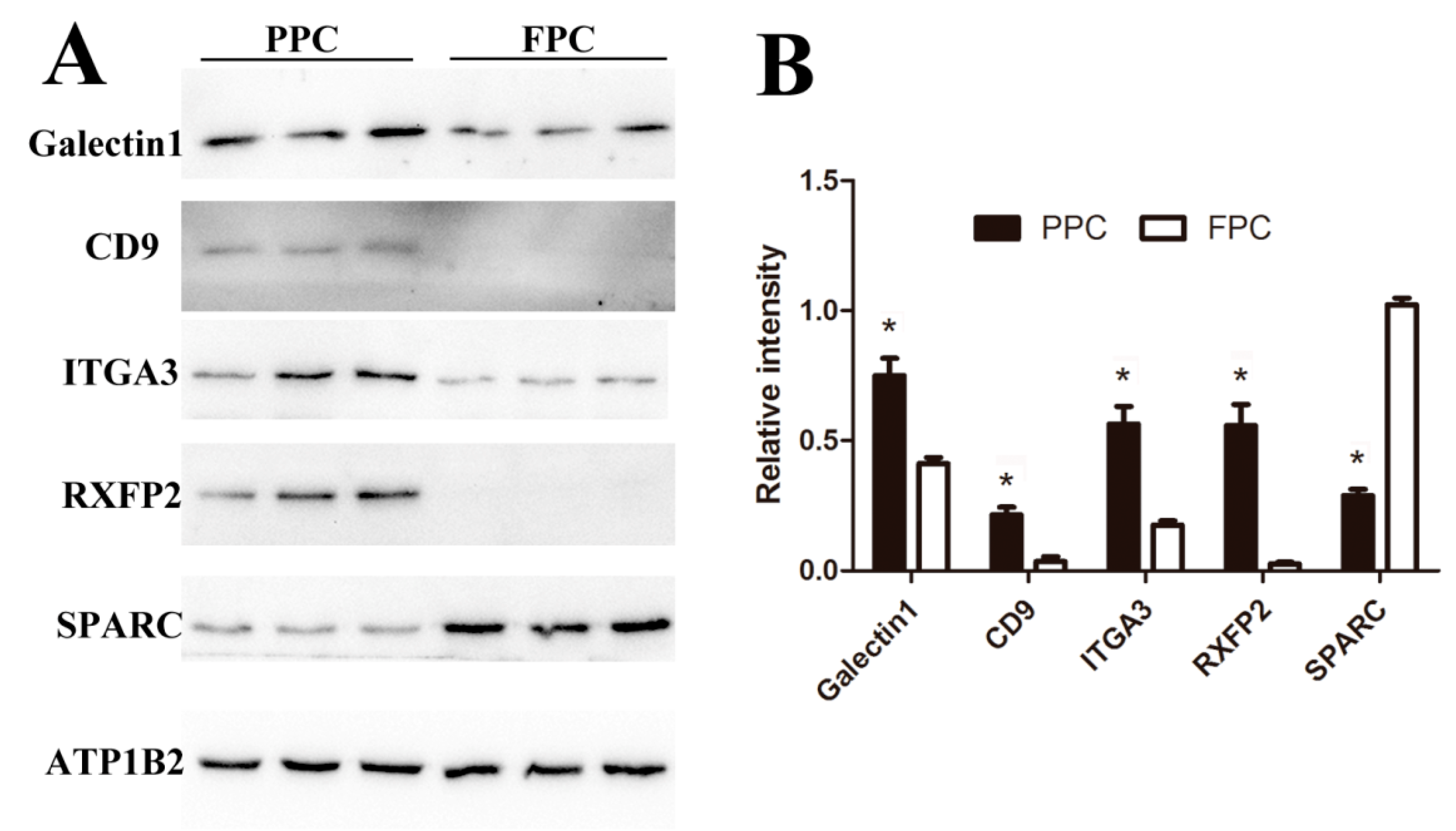
2.6. Validated DEPs
3. Discussion
4. Materials and Methods
4.1. Tissue Sampling and Cell Culture
4.2. Plasma Membrane Protein Isolation
4.3. Protein Identification and Quantification Using LC–MS/MS
4.4. Bioinformatics Analysis of DEPs
4.5. Western Blotting
4.6. Immunofluorescent Staining
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stocum, D.L. Regenerative Biology & Medicine. In Tissue Restoration through Regenerative Biology and Medicine; Elsevier Academic Press: Boston, MA, USA, 2006; Volume 176, pp. 1–20. [Google Scholar]
- Li, C.; Yang, F.; Sheppard, A. Adult stem cells and mammalian epimorphic regeneration-insights from studying annual renewal of deer antlers. Curr. Stem Cell Res. Ther. 2009, 4, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Goss, R.J. Future directions in antler research. Anat. Rec. 1995, 241, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Suttie, J.M.; Clark, D.E. Morphological observation of antler regeneration in red deer (Cervus elaphus). J. Morphol. 2004, 262, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Suttie, J.M.; Clark, D.E. Histological examination of antler regeneration in red deer (Cervus elaphus). Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. 2005, 282, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, U.; Stoffels, E.; Stoffels, D.; Kierdorf, H.; Szuwart, T.; Clemen, G. Histological studies of bone formation during pedicle restoration and early antler regeneration in roe deer and fallow deer. Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. 2003, 273, 741–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Mackintosh, C.G.; Martin, S.K.; Clark, D.E. Identification of key tissue type for antler regeneration through pedicle periosteum deletion. Cell Tissue Res. 2007, 328, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Rolf, H.J.; Kierdorf, U.; Kierdorf, H.; Schulz, J.; Seymour, N.; Schliephake, H.; Napp, J.; Niebert, S.; Wolfel, H.; Wiese, K.G. Localization and characterization of STRO-1 cells in the deer pedicle and regenerating antler. PLoS ONE 2008, 3, e2064. [Google Scholar] [CrossRef] [PubMed]
- Wallin, E.; Heijne, G.V.; Wallin, E.; von Heijne, G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 2008, 7, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Josic, D.; Clifton, J.G. Mammalian plasma membrane proteomics. Proteomics 2010, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.R.; Liu, H.M.; Shao, Y.C.; Zhou, P.Y.; Ying, S.U.; Wang, L.; Xing, X.M. Comparative Proteomic Analysis in Different Growth Stages of Sika Deer Velvet Antler. Chin. J. Anim. Vet. Sci. 2016, 47, 493–501. [Google Scholar]
- Dong, Z.; Ba, H.; Zhang, W.; Coates, D.; Li, C. iTRAQ-Based Quantitative Proteomic Analysis of the Potentiated and Dormant Antler Stem Cells. Int. J. Mol. Sci. 2016, 17, 1778. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Harper, A.; Puddick, J.; Wang, W.; McMahon, C. Proteomes and signalling pathways of antler stem cells. PLoS ONE 2012, 7, e30026. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Suttie, J.M. Tissue collection methods for antler research. Eur. J. Morphol. 2003, 41, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment Map: A Network-Based Method for Gene-Set Enrichment Visualization and Interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef] [PubMed]
- Boginski, V.; Commander, C.W. Identifying Critical Nodes in Protein-Protein Interaction Networks. Clust. Chall. Biol. Netw. 2009, 153–167. [Google Scholar] [CrossRef]
- Seo, M.S.; Park, S.B.; Choi, S.W.; Kim, J.J.; Kim, H.S.; Kang, K.S. Isolation and characterization of antler-derived multipotent stem cells. Cell Transplant. 2014, 23, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Keating, A. Mesenchymal Stromal Cells: New Directions. Cell Stem Cell 2012, 10, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Wiedemar, N.; Drögemüller, C. A 1.8-kb insertion in the 3′-UTR of RXFP2 is associated with polledness in sheep. Anim. Genet. 2015, 46, 457–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correani, V.; Francesco, L.D.; Mignogna, G.; Fabrizi, C.; Leone, S.; Giorgi, A.; Passeri, A.; Casata, R.; Fumagalli, L.; Maras, B. Plasma Membrane Protein Profiling in Beta-Amyloid-Treated Microglia Cell Line. Proteomics 2017, 17, 1600439. [Google Scholar] [CrossRef] [PubMed]
- Goñi, F.M. Non-permanent proteins in membranes: When proteins come as visitors (Review). Mol. Membr. Biol. 2002, 19, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Burn, P. Amphitropic proteins: A new class of membrane proteins. Trends Biochem. Sci. 1988, 13, 79–83. [Google Scholar] [CrossRef]
- Cho, W.; Stahelin, R.V. Membrane-Protein Interactions in Cell Signaling and Membrane Trafficking. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 119–151. [Google Scholar] [CrossRef] [PubMed]
- Milner, R.; Campbell, I.L. The integrin family of cell adhesion molecules has multiple functions within the CNS. J. Neurosci. Res. 2002, 69, 286–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuttfeld, E.; Ballmerhofer, K. Structure and function of VEGF receptors. Iubmb Life 2010, 61, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lowry, P.R.; Zhou, X.; Depry, C.; Wei, Z.; Wong, G.W.; Zhang, J. PI3K/Akt signaling requires spatial compartmentalization in plasma membrane microdomains. Proc. Natl. Acad. Sci. USA 2011, 108, 14509–14514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Oosterom, A.T.V.; Bruijn, E.A.D. Vascular Endothelial Growth Factor and Angiogenesis. Pharmacol. Rev. 2004, 56, 549. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Ji, L.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Morgunova, E.; Tuuttila, A.; Bergmann, U.; Tryggvason, K. Structural insight into the complex formation of latent matrix metalloproteinase 2 with tissue inhibitor of metalloproteinase 2. Proc. Natl. Acad. Sci. USA 2002, 99, 7414–7419. [Google Scholar] [CrossRef] [PubMed]
- Mccawley, L.J.; Matrisian, L.M. Matrix metalloproteinases: Multifunctional contributors to tumor progression. Mol. Med. Today 2000, 6, 149–156. [Google Scholar] [CrossRef]
- Rojiani, M.V.; Alidina, J.; Esposito, N.; Rojiani, A.M. Expression of MMP-2 correlates with increased angiogenesis in CNS metastasis of lung carcinoma. Int. J. Clin. Exp. Pathol. 2010, 3, 775–781. [Google Scholar] [PubMed]
- Lunter, P.C.; van Kilsdonk, J.W.J.; van Beek, H.; Cornelissen, I.M.H.A.; Bergers, M.; Willems, P.H.G.M.; van Muijen, G.N.P.; Swart, G.W.M. Activated Leukocyte Cell Adhesion Molecule (ALCAM/CD166/MEMD), a Novel Actor in Invasive Growth, Controls Matrix Metalloproteinase Activity. Cancer Res. 2005, 65, 8801–8808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldbrunner, R.H.; Bernstein, J.J.; Tonn, J.C. ECM-mediated glioma cell invasion. Microsc. Res. Tech. 1998, 43, 250–257. [Google Scholar] [CrossRef]
- Mecham, R.P. The Extracellular Matrix: An Overview; Springer: Heidelberg, Germany, 2010; pp. 1–17. [Google Scholar]
- Price, J.; Faucheux, C.; Allen, S. Deer antlers as a model of Mammalian regeneration. Curr. Top. Dev. Biol. 2005, 67, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Li, C. Deer antler regeneration: A stem cell-based epimorphic process. Birth Defects Res. Part C Embryo Today Rev. 2012, 96, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, N.; Kielbowicz, Z.; Nowacki, W.; Bajzert, J.; Reichert, P.; Biezynski, J.; Zebrowski, J.; Haczkiewicz, K.; Cegielski, M. Antlerogenic stem cells: Molecular features and potential in rabbit bone regeneration. Connect. Tissue Res. 2015, 57, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Pazhanisamy, S. Adult Stem Cell and Embryonic Stem Cell Markers. Mater. Methods 2013, 3, 200. [Google Scholar] [CrossRef]
- Suttie, J.M.; Lincoln, G.A.; Kay, R.N. Endocrine control of antler growth in red deer stags. J. Reprod. Fertil. 1984, 71, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.L.; Oldham, J.M.; Ambler, G.R.; Bass, J.J.; Spencer, G.S.; Hodgkinson, S.C.; Breier, B.H.; Gluckman, P.D.; Suttie, J.M. Presence of insulin-like growth factor-I receptors and absence of growth hormone receptors in the antler tip. Endocrinology 1992, 130, 2513–2520. [Google Scholar] [CrossRef] [PubMed]
- Sadighi, M.; Haines, S.R.; Skottner, A.; Harris, A.J.; Suttie, J.M. Effects of insulin-like growth factor-I (IGF-I) and IGF-II on the growth of antler cells in vitro. J. Endocrinol. 1994, 143, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, W.; Manley, T.; Suttie, J.M. No direct mitogenic effect of sex hormones on antlerogenic cells detected in vitro. Gen. Comp. Endocrinol. 2001, 124, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Taub, R. Liver regeneration 4: Transcriptional control of liver regeneration. FASEB J. 1996, 10, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Yokosuka, O.; Chiba, T.; Imazeki, F.; Kato, M.; Hashida, J.; Ueda, Y.; Sugano, S.; Hashimoto, K.; Saisho, H. Gene expression profiling reveals the mechanism and pathophysiology of mouse liver regeneration. J. Biol. Chem. 2003, 278, 29813–29818. [Google Scholar] [CrossRef] [PubMed]
- Nelson, F.; Campbell, J.S.; Riehle, K.J. Liver regeneration. Hepatology 2006, 43, S45–S53. [Google Scholar] [Green Version]
- Brockes, J.P. Amphibian limb regeneration: Rebuilding a complex structure. Science 1997, 276, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Kragl, M.; Knapp, D.; Nacu, E.; Khattak, S.; Maden, M.; Epperlein, H.H.; Tanaka, E.M. Cells keep a memory of their tissue origin during axolotl limb regeneration. Nature 2009, 460, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Li, C. Development of deer antler model for biomedical research. Recent Adv. Res. Updates 2003, 2, 19. [Google Scholar]
- Cholewa, B.; Pellitterihahn, M.C.; Scarlett, C.O.; Ahmad, N. Large-Scale Label-Free ComparativeProteomics Analysisof Polo-Like Kinase 1 Inhibition via the Small-Molecule InhibitorBI 6727 (Volasertib) in BRAFV600E Mutant Melanoma Cells. J. Proteome Res. 2014, 13, 5041–5050. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367. [Google Scholar] [CrossRef] [PubMed]
- Javed, A.; Zaidi, S.K.; Gutierrez, S.E.; Lengner, C.J.; Harrington, K.S.; Hovhannisyan, H.; Cho, B.C.; Pratap, J.; Pockwinse, S.M.; Montecino, M.; et al. In situ immunofluorescence analysis: Immunofluorescence microscopy. Methods Mol. Biol. 2004, 285, 23–28. [Google Scholar] [CrossRef] [PubMed]
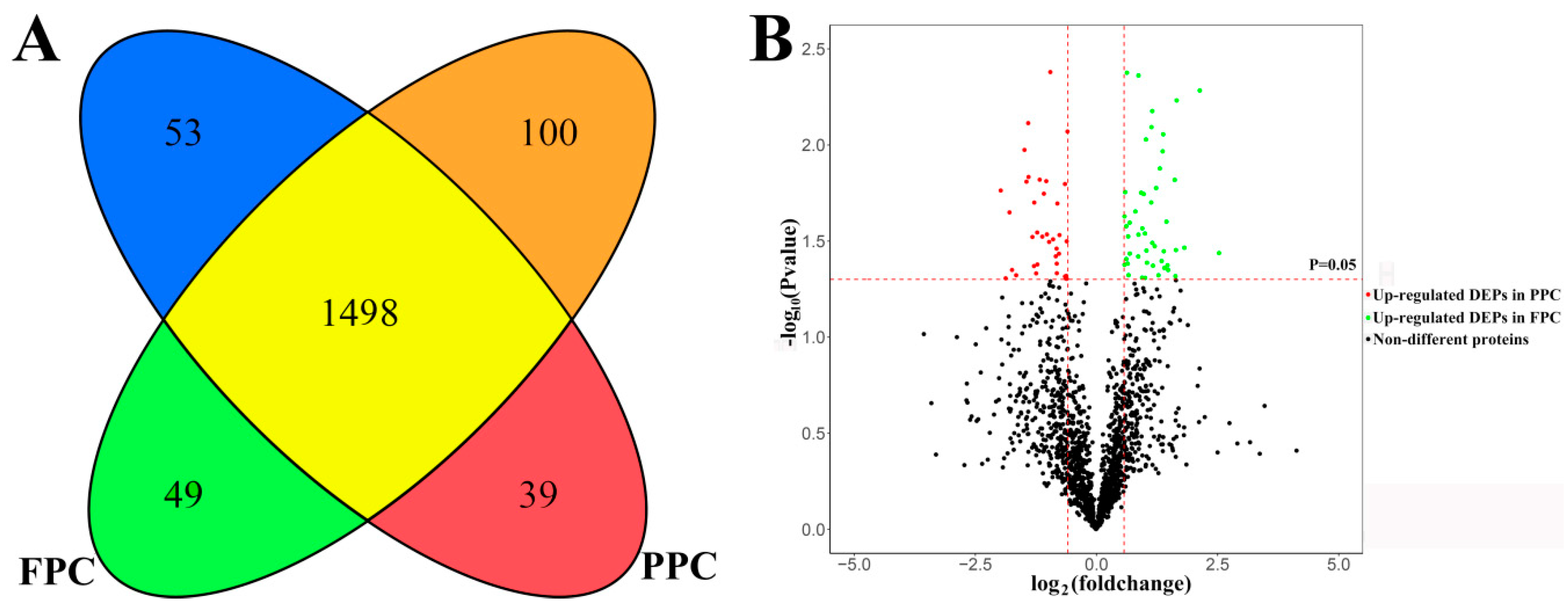
 ) and 100 solely in the FPCs (
) and 100 solely in the FPCs (  ). 1498 co-existed in both the PPCs and FPCs without significant difference in expression level (
). 1498 co-existed in both the PPCs and FPCs without significant difference in expression level (  ), 39 were significantly up-regulated in the PPCs (
), 39 were significantly up-regulated in the PPCs (  ) and 49 up-regulated in the FPCs (
) and 49 up-regulated in the FPCs (  ). (B) A volcano plot displays fold changes and p-values of starting MaxQuant data. Fold change >1.5 and p < 0.05 were defined as significant difference.
) and 100 solely in the FPCs ( ). 1498 co-existed in both the PPCs and FPCs without significant difference in expression level ( ), 39 were significantly up-regulated in the PPCs ( ) and 49 up-regulated in the FPCs ( ). (B) A volcano plot displays fold changes and p-values of starting MaxQuant data. Fold change >1.5 and p < 0.05 were defined as significant difference.
). (B) A volcano plot displays fold changes and p-values of starting MaxQuant data. Fold change >1.5 and p < 0.05 were defined as significant difference.
) and 100 solely in the FPCs ( ). 1498 co-existed in both the PPCs and FPCs without significant difference in expression level ( ), 39 were significantly up-regulated in the PPCs ( ) and 49 up-regulated in the FPCs ( ). (B) A volcano plot displays fold changes and p-values of starting MaxQuant data. Fold change >1.5 and p < 0.05 were defined as significant difference.


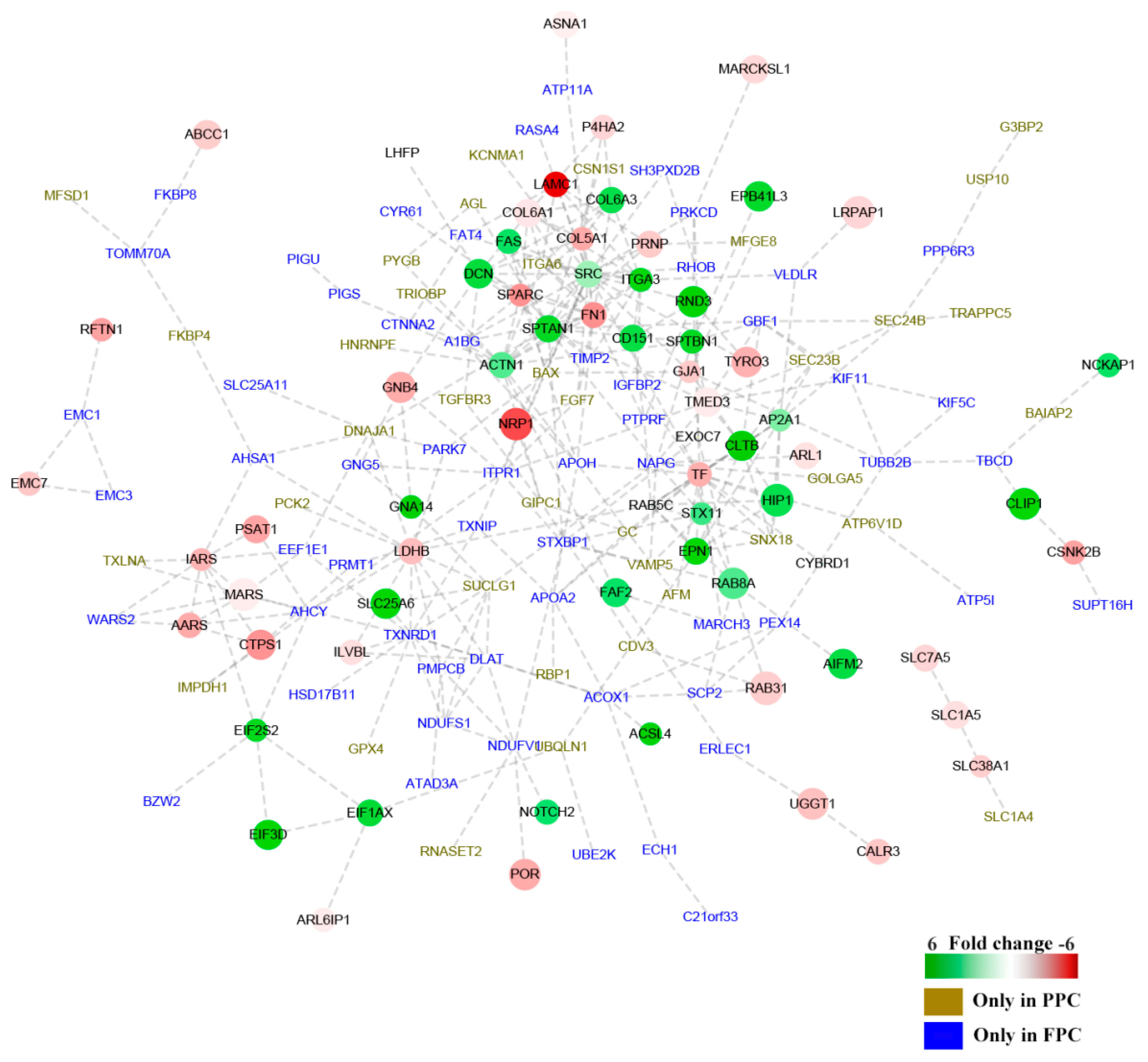
 ) and 58 solely detected in the FPCs (
) and 58 solely detected in the FPCs (  ). “
). “  ” Denotes up-regulated proteins, and “
” Denotes up-regulated proteins, and “  ” denotes down-regulated proteins in the PPCs. Bar color was considered as a logarithmic scale from −6.00 to 6.00.
) and 58 solely detected in the FPCs ( ). “ ” Denotes up-regulated proteins, and “ ” denotes down-regulated proteins in the PPCs. Bar color was considered as a logarithmic scale from −6.00 to 6.00.
” denotes down-regulated proteins in the PPCs. Bar color was considered as a logarithmic scale from −6.00 to 6.00.
) and 58 solely detected in the FPCs ( ). “ ” Denotes up-regulated proteins, and “ ” denotes down-regulated proteins in the PPCs. Bar color was considered as a logarithmic scale from −6.00 to 6.00.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway Description | Protein Count | p Value | Matching Proteins (Labels), “↑” Up-Regulation, “↓” Down-Regulation |
|---|---|---|---|
| Extracellular matrix (ECM)-receptor interaction | 7 | 0.00882 | COL5A1↓, COL6A1↓, COL6A3↑, FN1↓, ITGA3↑, ITGA6↑, LAMC1↓ |
| Adherens junction | 6 | 0.0111 | ACTN1↑, BAIAP2↑, CTNNA2↓, PTPRF↓, PTPRM↓, SRC↑ |
| Arrhythmogenic right ventricular cardiomyopathy | 6 | 0.0111 | ACTN1↑, CTNNA2↓, DSG2↑, GJA1↓, ITGA3↑, ITGA6↑ |
| Protein processing in endoplasmic reticulum | 8 | 0.018 | BAX↑, DNAJA1↑, ERLEC1↑, ERP29↓, SEC23B↑, SEC24B↑, UBQLN1↑, UGGT1↓ |
| Focal adhesion | 9 | 0.018 | ACTN1↑, COL5A1↓, COL6A1↓, COL6A3↑, FN1↓, ITGA3↑, ITGA6↑, LAMC1↓, SRC↑ |
| Aminoacyl-tRNA biosynthesis | 4 | 0.0499 | AARS↓, IARS↓, MARS↓, WARS2↓ |
| Gene/Protein | Protein IDs | Average LFQ (Label-Free Quantitation) Intensity |
|---|---|---|
| CD9 (MRP1) | P30932 | 3.24 × 108 |
| CD29 (ITGB1) | P53712 | 3.36 × 109 |
| CD44 (HCAM) | L8ITJ7 | 1.45 × 108 |
| CD73 (NT5E) | Q05927 | 6.20 × 108 |
| CD90 (THY1) | L8IGG9 | 9.42 × 108 |
| CD105 (ENG) | E1B7I8 | 5.164 × 107 |
| CD166 (ALCAM) | F1MHN8 | 4.75 × 108 |
| Item | Value |
|---|---|
| Enzyme | Trypsin |
| Max Missed Cleavages | 2 |
| Main search | 6 ppm |
| First search | 20 ppm |
| Mass spectrometry (MS/MS) Tolerance | 20 ppm |
| Fixed modifications | Carbamidomethyl (C) |
| Variable modifications | Oxidation (M), Acetyl (Protein N-term) |
| Database | uniprot-Pecora_94642_20170405 |
| Database pattern | Reverse |
| Peptide FDR (False Discovery Rate) | ≤0.01 |
| Protein FDR | ≤0.01 |
| Time window (match between runs) | 2min |
| Protein Quantification | Razor and unique peptides were used for protein quantification. |
| LFQ | True |
| LFQ min. ratio count | 1 |
| Terms | Manufacturer and Product Code | Isotype | Application |
|---|---|---|---|
| CD9 | LSBio; LS-C46004 | Mouse, IgG2 | WB 1 1:500; IF 2 1:100 |
| CD29 | Proteintech;12594-1 | Rabbit, IgG | IF 1:100 |
| CD44 | Proteintech; 15675-1 | Rabbit, IgG | IF 1:200 |
| CD73 | Santa; sc-25603 | Rabbit, IgG | IF 1:500 |
| CD90 | Bioss; bs-0778R | Rabbit, IgG | IF 1:100 |
| CD105 | Elabscience; ESH135 | Mouse, IgG1 | IF 1:500 |
| CD166 | Bioss; bs-1251R | Rabbit, IgG | IF 1:100 |
| Galectin-1 | Self-produced | Rabbit, serum | WB 1:100 |
| SPARC | Proteintech; 15274-1 | Rabbit, IgG | WB 1:500 |
| ITGA3 | Bioss; bs-6328R | Rabbit, IgG | WB 1:200 |
| ATP1B2 | Bioss; bs-23414R | Rabbit, IgG | WB 1:500 |
| RXFP2 | Self-produced | Rabbit, serum | WB 1:200 |
| Rabbit IgG-Isotype control | Abcam, ab172730 | IF 1:200 | |
| Mouse IgG-Isotype control | Abcam, ab37355 | IF 1:200 | |
| HRP-conjugated goat anti-rabbit IgG (H + L) | Beyotime, A0208 | WB 1:2000 | |
| HRP-conjugated goat anti-mouse IgG | Beyotime, A0216 | WB: 1:2000 | |
| Goat anti-rabbit IgG H&L (Alexa Fluor 488) | Abcam, ab150077 | IF 1:1000 | |
| Goat Anti-mouse IgG H&L (Alexa Fluor 488) | Abcam, 150113 | IF 1:1000 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Ba, H.; Li, C.; Zhao, Q.; Li, C. Proteomic Analysis of Plasma Membrane Proteins of Antler Stem Cells Using Label-Free LC–MS/MS. Int. J. Mol. Sci. 2018, 19, 3477. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113477
Wang D, Ba H, Li C, Zhao Q, Li C. Proteomic Analysis of Plasma Membrane Proteins of Antler Stem Cells Using Label-Free LC–MS/MS. International Journal of Molecular Sciences. 2018; 19(11):3477. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113477
Chicago/Turabian StyleWang, Datao, Hengxing Ba, Chenguang Li, Quanmin Zhao, and Chunyi Li. 2018. "Proteomic Analysis of Plasma Membrane Proteins of Antler Stem Cells Using Label-Free LC–MS/MS" International Journal of Molecular Sciences 19, no. 11: 3477. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113477