Structure and Physiological Regulation of AMPK


1
Center for Cancer and Cell Biology, Van Andel Research Institute, 333 Bostwick Ave. N.E., Grand Rapids, MI 49503, USA
2
VARI/SIMM Center, Center for Structure and Function of Drug Targets, CAS-Key Laboratory of Receptor Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(11), 3534; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113534
Submission received: 17 October 2018
/
Revised: 5 November 2018
/
Accepted: 6 November 2018
/
Published: 9 November 2018
(This article belongs to the Special Issue AMP-Activated Protein Kinase Signalling)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Adenosine monophosphate (AMP)-activated protein kinase (AMPK) is a heterotrimeric αβγ complex that functions as a central regulator of energy homeostasis. Energy stress manifests as a drop in the ratio of adenosine triphosphate (ATP) to AMP/ADP, which activates AMPK’s kinase activity, allowing it to upregulate ATP-generating catabolic pathways and to reduce energy-consuming catabolic pathways and cellular programs. AMPK senses the cellular energy state by competitive binding of the three adenine nucleotides AMP, ADP, and ATP to three sites in its γ subunit, each, which in turn modulates the activity of AMPK’s kinase domain in its α subunit. Our current understanding of adenine nucleotide binding and the mechanisms by which differential adenine nucleotide occupancies activate or inhibit AMPK activity has been largely informed by crystal structures of AMPK in different activity states. Here we provide an overview of AMPK structures, and how these structures, in combination with biochemical, biophysical, and mutational analyses provide insights into the mechanisms of adenine nucleotide binding and AMPK activity modulation.
Keywords:
energy metabolism; AMPK; activation loop; AID; α-linker; β-linker; CBS; LKB1; CaMKK2; αRIM1. AMPK Is a Master Regulator of Energy Homeostasis That Is Dysregulated in Disease
AMPK is the primary energy sensor and regulator of energy homeostasis in eukaryotes. It is activated by energy stress in response to increased ATP consumption (e.g., exercise, cell proliferation, anabolism) or decreased ATP production (e.g., low glucose levels, oxidative stress, hypoxia), which are sensed as low ratios of ATP to AMP and ADP. Upon activation, AMPK phosphorylates downstream targets to directly or indirectly modulate the activities of rate-limiting metabolic enzymes, transcription and translation factors, proliferation and growth pathways, and epigenetic regulators. Collectively, this increases oxidative phosphorylation, autophagy, and uptake and metabolism of glucose and fatty acids, and decreases the synthesis of fatty acids, cholesterol, proteins, and ribosomal RNAs (rRNAs), as well as decreasing cell growth and proliferation [1,2,3,4,5,6]. Due to its central roles in metabolism, AMPK is dysregulated in diabetes, obesity, cardiometabolic disease, and cancer, and it is a promising pharmacological target [1,2,5,7,8,9,10], especially for the treatment of type 2 diabetes [11,12,13].
2. AMPK Consists of a Stable Core Attached to Moveable Domains
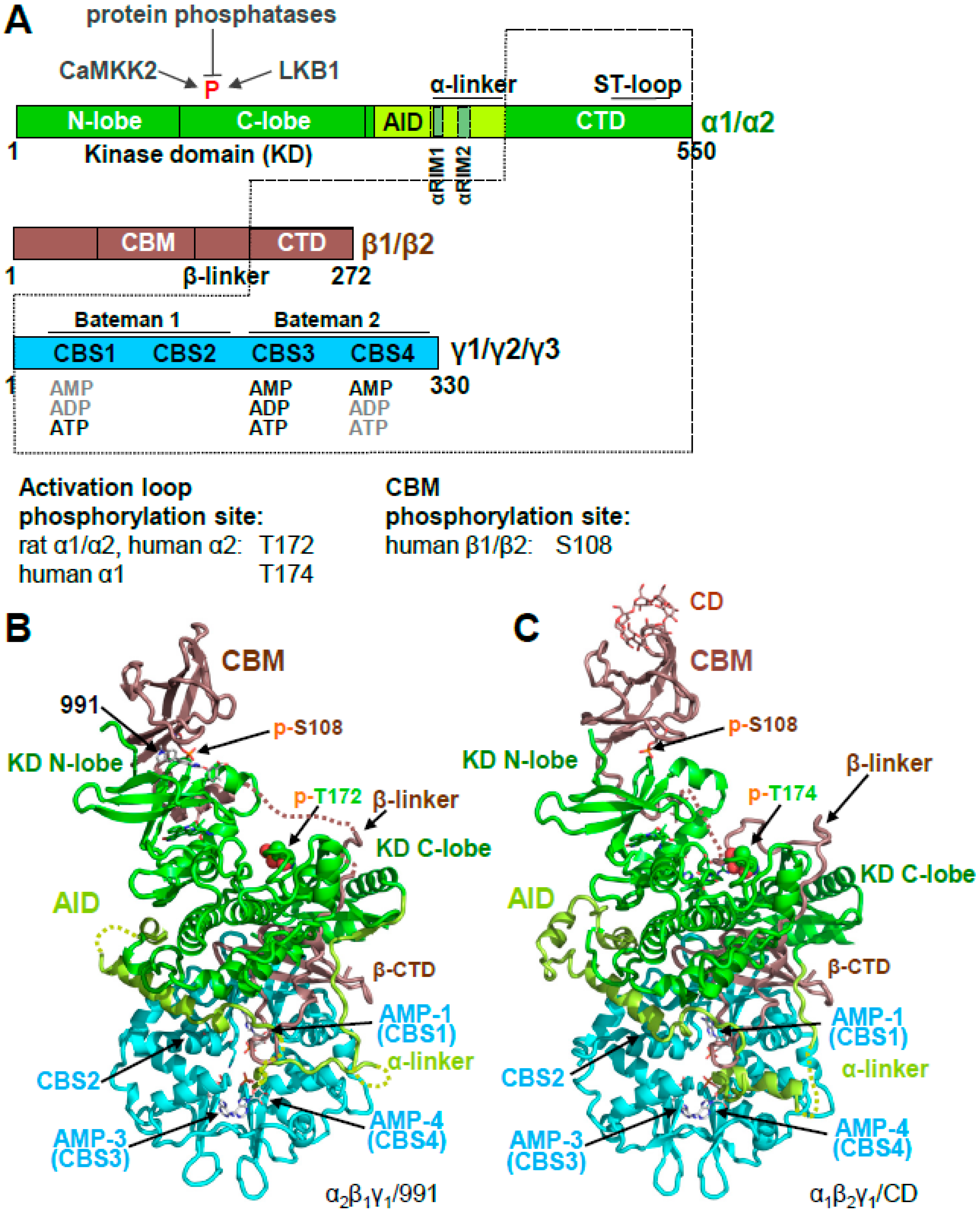
AMPK is a heterotrimeric αβγ protein kinase. In mammals, it is encoded by two alternative α subunits (α1 and α2), two alternative β subunits (β1 and β2), and three alternative γ subunits (γ1, γ2, and γ3) that can form up to 12 different αβγ isoforms [14]. The α subunits contain a canonical Ser/Thr kinase domain (KD), an autoinhibitory domain (AID), an adenine nucleotide sensor segment termed an α-linker, and a β subunit-interacting C-terminal domain (α-CTD), the latter of which contains the ST loop, which harbors proposed phosphorylation sites for AKT [15], PKA [16], and GSK [17]. The β subunits are composed of a myristoylated, unstructured N-terminus, a glycogen-binding carbohydrate-binding module (CBM), a scaffolding C-terminal domain (β-CTD) that interacts with both the γ subunit, and the α-CTD, and the extended β-linker loop that connects the CBM with the β-CTD (Figure 1A,B). The three alternative γ subunits consist of N-termini of different lengths and unknown function, followed by a conserved adenine nucleotide-binding domain that contains four cystathione β-synthetase (CBS) AMP/ADP/ATP binding sites (Figure 1). CBS1, 3, and 4 are functional, whereas in CBS2, the ribose-binding Asp residue is replaced by an Arg, and no nucleotide binding has been observed for CBS2 in heterotrimer structures.
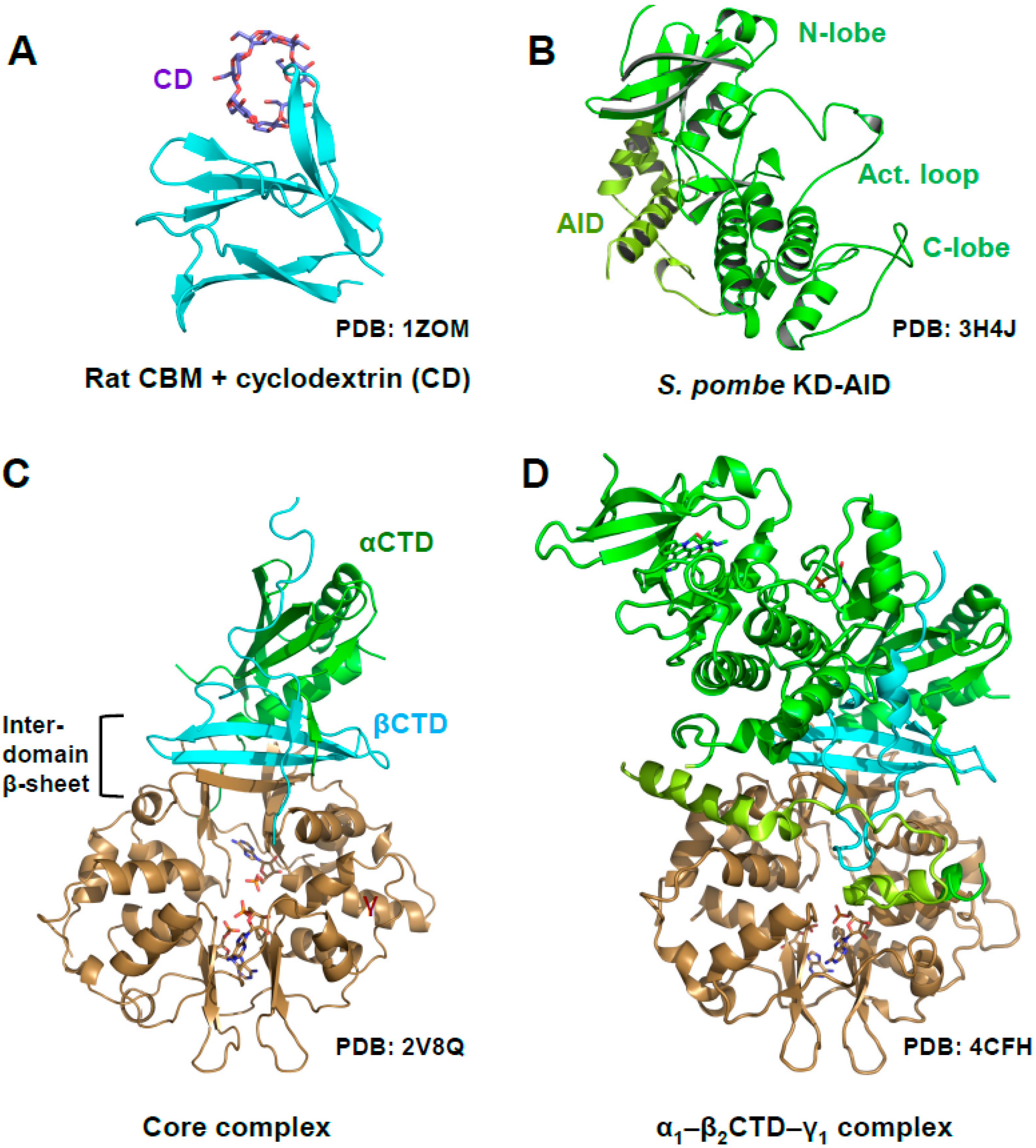
AMPK is a highly dynamic complex with a stable core formed by the γ subunit and the α- and β-CTDs, in which the β-CTD is sandwiched between the α and γ subunits (Figure 1A, core highlighted by dotted lines). Attached to the core are moveable domains whose position is determined by ligand binding and posttranslational modifications. As such, the holo-complex cannot be crystallized in the absence of multiple stabilizing ligands and/or protein engineering. Consequently, the first structures of AMPK consisted of isolated domains, e.g., the KD [18,19,20,21], the CBM bound to the glycogen mimic cyclodextrin [22], the yeast and mammalian nucleotide-bound scaffolding cores [23,24,25,26], the AID [27], and the yeast KD–AID complex [21] (Figure 2).
Activation Loop Phosphorylation Orchestrates the Catalytic Center for Phosphoryl Transfer
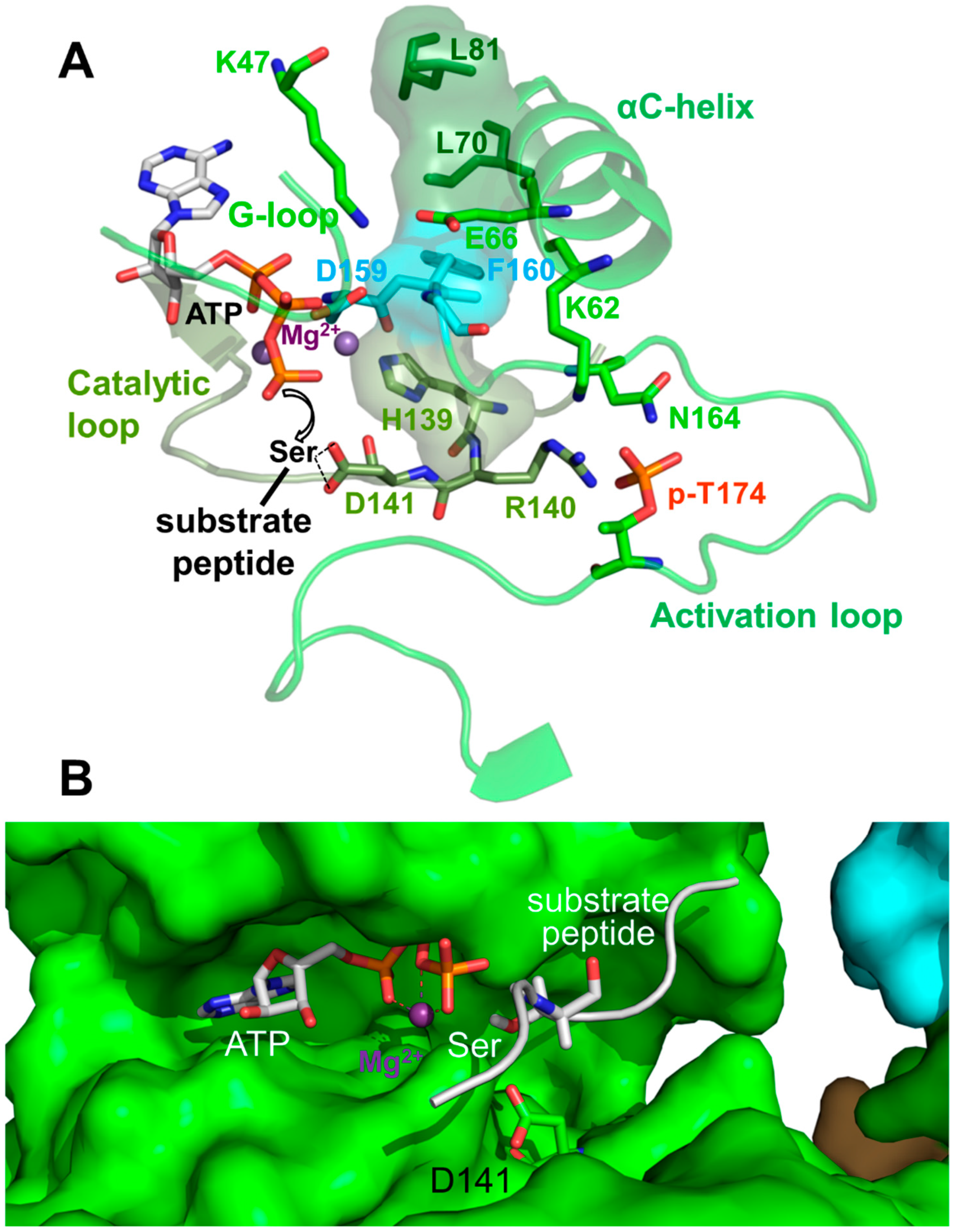
Kinase domains have a highly conserved structure consisting of a smaller N-terminal lobe (N-lobe), composed of a β-sheet and the αB and αC helices, and a larger α-helical C-terminal lobe (C-lobe; see Figure 1B and Figure 2B). The cleft between the lobes is the binding site for substrate peptides and Mg2+–ATP. The two lobes are separated by a flexible hinge at the back that allows them to move towards each other to cycle through substrate-accessible open and catalytically-competent closed conformations as part of the kinase catalytic cycle. Key regulatory elements of the KD are: (i) the activation loop at the entrance of the catalytic cleft; (ii) the αC helix in the N-lobe, which positions the ATP-binding lysine (K47 in human α1) and the Mg2+-binding DFG (Asp-Phe-Gly) loop; and (iii) the peptide substrate-binding catalytic loop in the C-lobe (Figure 3) [28,29,30].
AMPK belongs to the RD (Arg-Asp) kinases, in many of which phosphorylation stabilizes the activation loop through a charge interaction between the negatively charged activation loop phosphate, and the positively charged residues from the αC helix (K62 in AMPK α1), the activation loop (N164), and the catalytic loop (R140). This conformation in turn stabilizes the αC helix and positions the arginine (R) and adjacent aspartate (D) of the catalytic loop for substrate binding (Figure 3). The hallmark of active protein kinases is therefore a precisely positioned set of motifs for substrate- and ATP-binding, in which four residues from the catalytic loop (H139), the Mg2+-binding DFG loop (F160), the αC helix (L70) and the αC-αD loop (L81) are stacked against each other [28,29,30], as found in structures of active AMPK (Figure 3).
3. AMPK Is Activated Both by Direct Allosteric Activation and by Increasing Net Activation Loop Phosphorylation
AMPK activity is regulated at three different levels: at the level of (i) activation loop phosphorylation by upstream kinases, (ii) protection against activation loop dephosphorylation by protein phosphatases, and (iii) at the level of phosphorylation-independent, allosteric kinase activation (Figure 1A). Activation loop phosphorylation increases the AMPK activity by about 100-fold, while allosteric regulation changes AMPK activity up to ten-fold in mammalian cells and about two-fold in recombinant, bacterially produced AMPK [24,31,32,33]. AMP activates, and ATP inhibits, AMPK through all three mechanisms. ADP more weakly protects against activation loop dephosphorylation, does not allosterically activate AMPK [33,34,35,36], and it may not stimulate activation loop phosphorylation [33,37], although the latter is controversial [36].
The two main mammalian AMPK activation loop-phosphorylating kinases are the tumor suppressor LKB1 in complex with STRAD and MO25, and Ca2+/calmodulin-dependent protein kinase kinase β (CaMKK2) [38,39,40,41,42]. While CaMKK2 mediates Ca2+-dependent AMPK phosphorylation, AMP binding to the γ subunit increases activation loop phosphorylation through LKB1 by inducing a conformation that stabilizes formation of a complex between myristoylated AMPK, Axin, and LKB1/STRAD/MO25 [37,43]. However, the structural details of this interaction remain unknown. In addition, activation loop phosphorylation is also modulated by phosphorylation of the ST loop [15,16,17] and by ubiquitination of AMPK [44] and LKB1 [45].
In addition to adenine nucleotides, glucose, glycogen, and nicotinamide adenine dinucleotides are also important energy metabolites. Glucose has recently been identified as an important AMPK activity regulator, but it does so without direct AMPK binding [43,46]. In contrast, both glycogen and NADPH and NADH can directly bind AMPK: glycogen at the CBM [22,47], and in a reconstituted system, NADPH and NADH at the adenine nucleotide sensor site CBS3 [34,48]. However, the physiological relevance of the glycogen [47,49,50] and NADPH/NADH [34,48] interactions for AMPK activity regulation remains unclear.
Finally, a number of pharmacological activators bind AMPK at a unique site at the interface between CBM and KD (so called allosteric drug and metabolite [ADaM] site), as first shown for Merck compound 991 [51], and derivatives of the Abbot compound A769662 [52]. Binding greatly stabilizes the association of the highly dynamic CBM with the KD [53], an interaction that is also modulated by CBM phosphorylation and carbohydrate binding [49,53]. ADaM site agonists activate AMPK both directly and through increased protection against activation loop dephosphorylation, whose structural details will be covered in detail in a separate article in this issue.
Besides activity regulation, the level of AMPK is regulated by ubiquitination and proteasomal degradation in brown adipose tissue [54], testis [55], certain cancers [55,56], and in the presence of high levels of glucose [57].
3.1. The γ Subunit Contains Three Functional Adenine Nucleotide Binding Sites
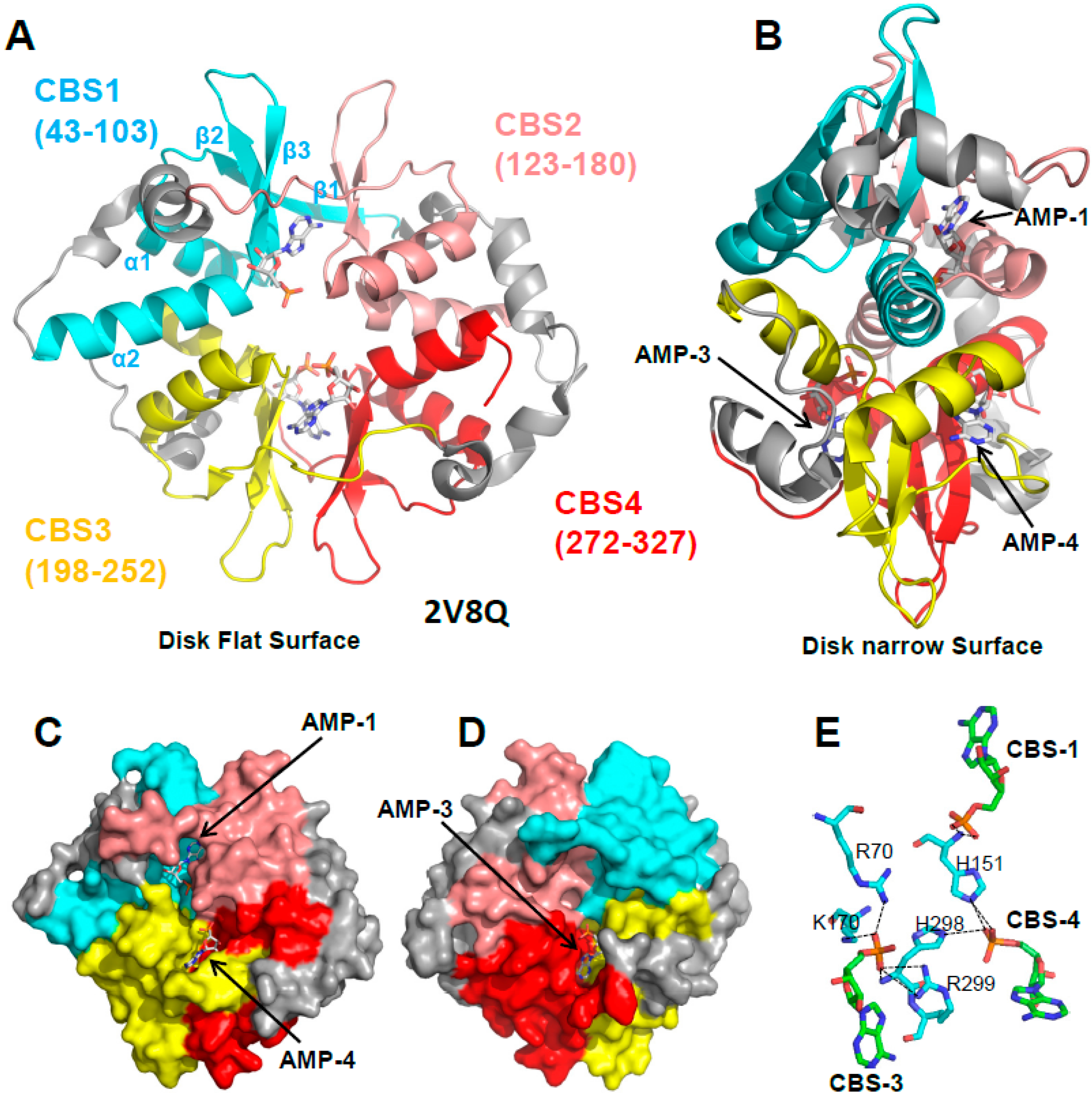
The structure of the yeast and mammalian AMPK core scaffolds revealed a disk-shaped γ subunit composed of four CBS sites. Each CBS consists of a strand-helix-strand-strand-helix fold (β1-α1-β2-β3-α2) with long intervening loops (Figure 4A). β1 is often incomplete, but where present, it forms a three-stranded sheet with the two central β-strands (β2 and β3). The β-sheet of one CBS packs parallel with the sheet of a neighboring CBS. The interface between the two sheets forms two clefts, one on the top flat side and one on the bottom flat side of the disk, which are the binding sites for adenine nucleotides (Figure 4C,D). Therefore, each binding site requires a tandem CBS pair to form a functional unit termed the Bateman domain (CBS1 + CBS2 = Bateman domain 1, CBS3 + CBS4 = Bateman domain 2). The structures of the core complexes in the presence of AMP [24,58], ADP [34], or ATP [24,58] revealed adenine nucleotide binding at three sites in mammalian AMPK: CBS1, CBS3, and CBS4.
3.2. CBS3 Is the Adenine Nucleotide Sensor Site
While the structure of the core complexes revealed how adenine nucleotides bind the γ subunit, they did not provide information on how the binding signal is transduced to the KD in the α subunit. In 2011, the Gamblin and Carling groups crystallized an AMPK complex containing rat α1, human β2 CTD, and rat γ1 [34]. While this complex is not regulated by protection against activation loop dephosphorylation [51], it retained direct AMPK activation by AMP and ADP. The structure revealed that the α-linker that connects AID and α-CTD directly bound the γ subunit [34], which has been validated in all subsequent AMP-bound AMPK complex structures with a resolved α-linker. A segment of the linker, termed regulatory subunit-interacting motif 2 (αRIM2) [27,59], interacts with AMP at CBS3, suggesting that αRIM2 functions as an adenine nucleotide sensor, and that it mediates the transduction of the adenine-binding signal to the KD [27,34,59]. This function has been validated by several experimental approaches. First, the mutation of either of the two key αRIM2 residues (E362 and R363 in rat α1 and human α2; E364 and R365 in human α1) abolished or largely reduced both AMP-dependent direct AMPK activation [27,49,51] and AMP-dependent protection against activation loop dephosphorylation [49]. Second, AMP increases, and ATP decreases the interaction between isolated α-linker and core AMPK in a reconstituted system, and the AMP increase requires intact E364 and R365 [49]. Third, the AMP-mimetic synthetic AMPK activator C2 activates AMPK α-isotype-selectively (it fully activates α1-containing complexes, but only partially α2 complexes), and this selectivity can be fully reversed by a swap of the αRIM2 regions [60,61].
4. If CBS3 Is the Sensor Site, What Are the Roles of CBS1 and CBS4?
Of the three functional CBS sites, only CBS3 interacts with the α subunit, an interaction that is directly modulated by AMP and ATP. In contrast, CBS1 and CBS4 do not interact with any part of the α- or β-subunit. Moreover, CBS4 binds AMP very tightly [24,58] and it is unlikely to exchange AMP under physiological conditions, yet mutations in CBS4 abolish regulation by AMP [36,58], while mutations in CBS1 have either no [58] or only a small [36] effect on AMPK regulation. Important insight came from a mutational study. When CBS1, CBS4, and the ATP-binding site in the KD are mutated, so that CBS3 remains the only functional adenine nucleotide binding site, it binds AMP only very weakly and with 10–100 times lower affinity than ATP [48]. Since the cellular ATP concentrations are much higher than AMP and ADP concentrations, CBS3 by itself would remain almost completely ATP-bound under both normal and energy stress conditions. However, the phosphates of adenine nucleotides bound to CBS1, 3, and 4 coordinately bind a set of charged and polar amino acids (Figure 4E), so that binding to one site affects binding to the other two sites. Through these coordinated interactions, AMP bound at CBS4, together with additional interactions from αRIM2, stabilizes AMP at CBS3. This increases CBS3’s affinity for AMP by two orders of magnitude, and its AMP/ATP binding preference by two to three orders of magnitude [48], allowing CBS3 to sensitively detect physiological energy stress versus non-stress adenine nucleotide levels. Conversely, both CBS3 and CBS1 strongly stabilize AMP-binding at CBS4, so that under physiological conditions CBS4 remains essentially non-exchangeably AMP-bound and CBS1 largely ATP-bound [48].
5. AMP-Binding at CBS3 Destabilizes an Inhibitory AID–KD Interaction
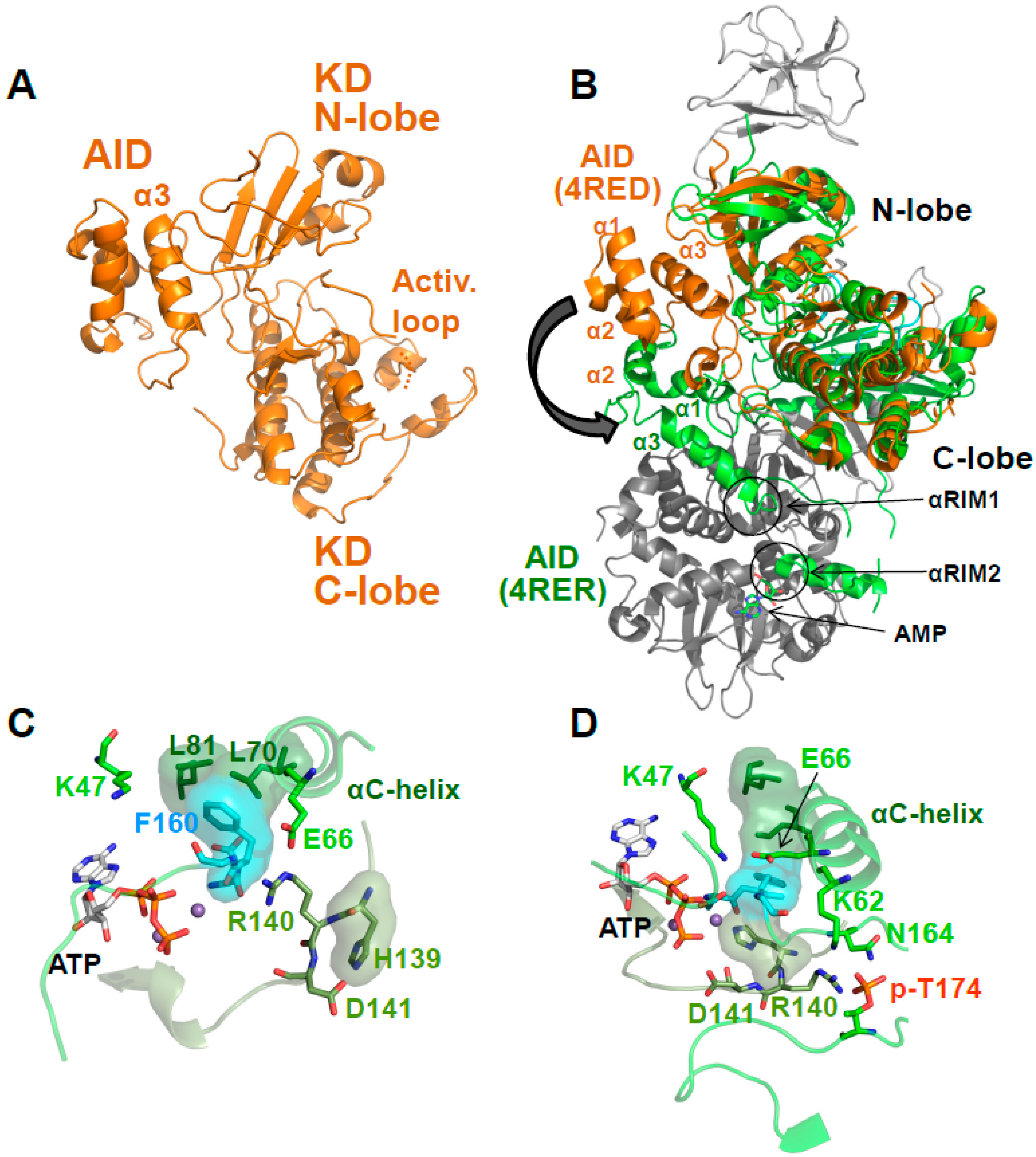
The KD is followed by the AID, a small 48 amino acid domain that inhibits kinase activity about tenfold in the context of a KD–AID fragment [62,63]. Crystal structures of the fission yeast [21] and human [49] AMPK KD–AID fragments revealed a three-helical AID, whose C-terminal helix (α3) directly binds the hinge between the KD N- and C-lobes at the backside of the KD (Figure 2B and Figure 5A). In contrast, in structures of active, AMP-bound AMPK [34,49], the AID is rotated away from the KD and bound to the γ subunit (see structure overlay in Figure 5B). The AID–KD interaction arrests the KD in a unique inactive conformation, in which the ATP binding K47, the Mg2+-binding DFG loop, and the substrate-binding catalytic loop are misaligned, and H139 of the regulatory spine is out of register [21,49] (so called “HRD-out” conformation [30]; Figure 5C). The inhibitory function of the KD–AID interaction was further validated by the mutation of interface residues in either the KD or the AID, all of which made AMPK constitutively active [21,49]. Conversely, binding of the AID to the γ subunit, as seen in structures of AMP-bound AMPK [34,49], allows the KD to adopt the active conformation [27,34,51,59] (Figure 5D). Consistently, mutations in AID-interacting γ subunit residues make AMPK constitutively inactive [59].
A Highly Conserved Interaction Network Links αRIM2/CBS3 and AID-αRIM1/CBS2 Binding
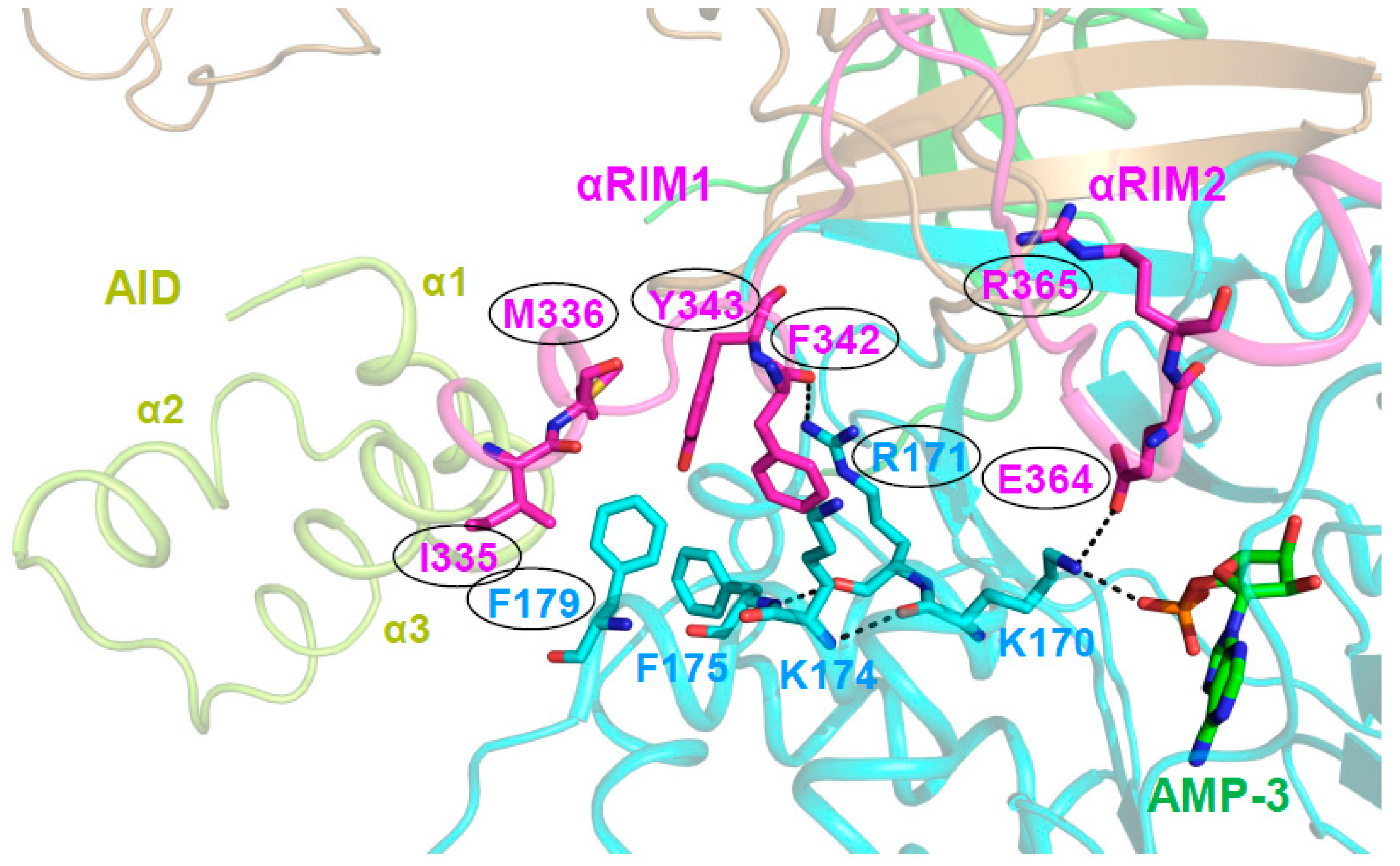
The structure of AMP-bound AMPK α1‒β2CTD‒γ1 [34] first revealed the AID conformation in active AMPK, in which the border of the AID and the N-terminus of the α-linker, termed αRIM1, binds the γ-subunit at the unoccupied CBS2 [27,34,51,59]. Mutational analysis by Ja-Wei Wu’s group provided a molecular pathway to link αRIM2 binding of AMP-occupied CBS3 to direct AMPK kinase activation. They first showed that αRIM1/CBS2 interface amino acids corresponding to human α1 I335/M3364 and F342/Y343, and human γ1 R171 and F179 are required for AMP-mediated relief of AMPK autoinhibition [59]. In active, AMP-bound holo-AMPK, the direct interaction of γ1 K170 with both AMP/CBS3 and αRIM2 α1 E364 positions three key residues at the αRIM1 interface. First, the residue following K170, R171, forms Van der Waals interactions and a backbone hydrogen bond with αRIM1 α1 F342. Second, the K170-interacting residues K174 and F175 form Van der Waals bonds with F342 and both Van der Waals and π-stacking interactions with γ1 F179. The latter is the linchpin of the interface and directly interacts with all four αRIM1 residues that are required for the relief of AMPK autoinhibition (I335, M336, Y343, and F342; Figure 6). Similarly, E364, R171, and F179 are also all required for the relief of AMPK autoinhibition [59]. The mutational analysis thus provides strong support that this AMP-stabilized interaction network that is seen in all active structures of holo-AMPK is responsible for shifting the AID equilibrium from the inactive, KD-bound conformation to the active, γ/CBS2-bound conformation.
ATP binding is thought to disrupt this network. In the structure of the core AMPK complex co-crystallized with ATP [58], ATP was bound to CBS4 and CBS1, which sterically interfered with nucleotide binding at CBS3, and caused rearrangement and disruption of the interaction network [58,59]. However, the physiological relevance of this structure remains unclear, since under physiological conditions, CBS4 does not seem to exchange AMP (see above; [24,48,58]). Therefore, a final understanding of how ATP disrupts the CBS3–α-linker–AID network will require the structure of the holo-AMPK complex, including the α-linker, in ATP-bound conformation.
ADaM site ligands, while not focus of this review, directly activate AMPK by a completely different mechanism. Through binding of both the CBM and the KD [51,52] and stabilization of the CBM–KD interaction [53], the N-terminus of the β-linker at the CBM border adopts a helix that packs parallel to the αC-helix, and it has therefore been named C-interacting helix [51]. This suggested that ADaM site ligands may activate AMPK by stabilizing αC through induced formation of the C-interacting helix, reminiscent of the regulatable αC stabilization of several other protein kinases [64]. Support for this model came from the mutation of H233 in the C-interacting helix, which reduced activation by the ADaM site ligand 991 [51], and by direct demonstration through hydrogen/deuterium exchange mass spectrometry (HDX-MS) that 991 binding strongly and selectively stabilizes αC [48].
6. Regulation of Activation Loop Accessibility
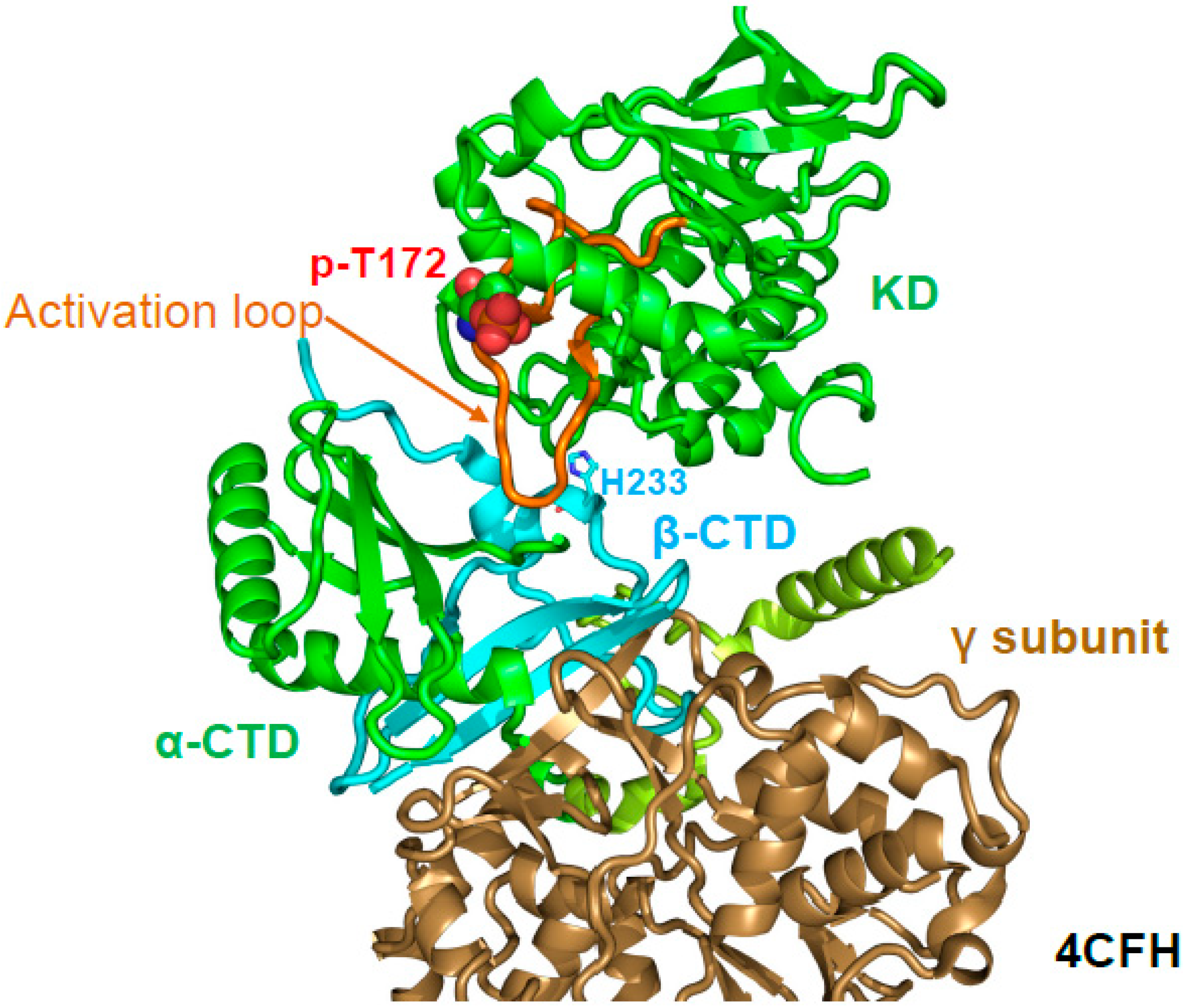
A major regulatory mechanism for AMPK activation by AMP and ADP is the protection of activation loop p-T172 (human α1 T174) against dephosphorylation. p-T172 protection can be demonstrated in a cell-free, reconstituted system independent of the phosphatase used (e.g., PP2C, PP2A, λ-phosphatase), and AMP does not, or only slightly inhibit the dephosphorylation of a different substrate, casein, by PP2Cα [32]. Therefore, reduced dephosphorylation is not due to phosphatase inhibition, but to an AMP/ADP-induced change in the activation loop accessibility. The crystal structure of AMP-bound, phosphorylated AMPK α1–β2CTD–γ1 (PDB: 4CFH) first demonstrated that the activation loop directly interacts with the core of AMPK [34]. Specifically, the stable β-CTD directly bound and stabilized the activation loop (Figure 7). The authors therefore proposed that the core shields the activation loop from phosphatase access. In agreement, mutation of the activation loop-interacting β2 H235 increased p-T172 dephosphorylation in the context of holo-AMPK [34]. However, the construct used in the structure was not regulated by protection against activation loop dephosphorylation [51], indicating that additional parts of AMPK, likely either the β-linker and/or the CBM, were also required for AMP-mediated, and probably ADaM site ligand-mediated protection against activation loop dephosphorylation. Consistently, in structures in which the β-linker is largely resolved (e.g., β2-linker in 4RER [49], β1-linker in 5ISO [65]), p-T172 is clearly protected by the β-linker, especially in the case of the β2-linker. Finally, how can the activation loop in AMP-bound conformation be largely inaccessible to protein phosphatases without affecting accessibility to the T172-phosphorylating upstream protein kinases? Answers to these fundamental questions will likely require the structure of holo-AMPK in the alternative, ATP-bound state and analysis of AMPK’s conformational landscape and dynamics in solution.
7. Conclusions and Future Directions
AMPK is a molecular machine consisting of the adenine nucleotide-binding core (γ subunit plus α- and β-CTDs), the catalytic KD, and at least four dynamic domains (AID, CBM, and the α- and β-linkers). We propose that adenine nucleotides, ADaM site ligands, and CBM phosphorylation affect the conformation of the KD through induced movements of the dynamic domains, while phosphorylation of activation loop and S/T loop modulate the KD conformation directly. Through concerted efforts, the mechanism of direct, allosteric AMPK activation through AID movement and αC stabilization is relatively well understood. However, the structural basis of direct inhibition by ATP, of activation loop accessibility regulation through ligands and possibly phosphorylation, and of the AMP-induced interaction with Axin and the LKB1 complex all remain poorly understood. The most important future challenges in AMPK structural biology will therefore be the determination of the structures of holo-AMPK in its inhibited, ATP-bound conformation, and in complex with Axin and LKB/STRAD/MO25.
Author Contributions
The manuscript was written by K.M. with input from all authors.
Funding
This research was funded by the Van Andel Research Institute (H.E.X. and K.M.) and the National Institutes of Health (R01 GM129436 to K.M.).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| ADaM site | Allosteric drug and metabolite-binding site |
| AID | Autoinhibitory domain |
| AMPK | AMP-activated protein kinase |
| αRIM | α-regulatory subunit interaction motif |
| CaMKK2 | Ca2+/calmodulin-dependent protein kinase kinase β |
| CBM | Carbohydrate-binding module |
| CBS | Cystathionine β-synthetase |
| CTD | C-terminal domain |
| HDX-MS | Hydrogen deuterium exchange mass spectrometry |
| KD | Kinase domain |
| LKB1 | Liver kinase B1 |
| MO25 | Mouse protein-25 |
| PP2A | Protein phosphatase 2A |
| PP2C | Protein phosphatase 2C |
| STRAD | STE20-related kinase adaptor |
References
- Yuan, H.X.; Xiong, Y.; Guan, K.L. Nutrient sensing, metabolism, and cell growth control. Mol. Cell 2013, 49, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Keeping the home fires burning: AMP-activated protein kinase. J. R. Soc. Interface 2018, 15, 20170774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G. AMPK: A target for drugs and natural products with effects on both diabetes and cancer. Diabetes 2013, 62, 2164–2172. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Targeting an energy sensor to treat diabetes. Science 2017, 357, 455–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMP-activated protein kinase: A target for drugs both ancient and modern. Chem. Biol. 2012, 19, 1222–1236. [Google Scholar] [CrossRef] [PubMed]
- Guigas, B.; Viollet, B. Targeting AMPK: From Ancient Drugs to New Small-Molecule Activators. EXS 2016, 107, 327–350. [Google Scholar] [PubMed]
- Cokorinos, E.C.; Delmore, J.; Reyes, A.R.; Albuquerque, B.; Kjobsted, R.; Jorgensen, N.O.; Tran, J.L.; Jatkar, A.; Cialdea, K.; Esquejo, R.M.; et al. Activation of Skeletal Muscle AMPK Promotes Glucose Disposal and Glucose Lowering in Non-human Primates and Mice. Cell Metab. 2017, 25, 1147–1159.e10. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.W.; Guan, H.P.; Ehrhart, J.; Petrov, A.; Prahalada, S.; Tozzo, E.; Yang, X.; Kurtz, M.M.; Trujillo, M.; Gonzalez Trotter, D.; et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 2017, 357, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Steneberg, P.; Lindahl, E.; Dahl, U.; Lidh, E.; Straseviciene, J.; Backlund, F.; Kjellkvist, E.; Berggren, E.; Lundberg, I.; Bergqvist, I.; et al. PAN-AMPK activator O304 improves glucose homeostasis and microvascular perfusion in mice and type 2 diabetes patients. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Ross, F.A.; MacKintosh, C.; Hardie, D.G. AMP-activated protein kinase: A cellular energy sensor that comes in 12 flavours. FEBS J. 2016, 283, 2987–3001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, S.A.; Ross, F.A.; Gowans, G.J.; Tibarewal, P.; Leslie, N.R.; Hardie, D.G. Phosphorylation by Akt within the ST loop of AMPK-alpha1 down-regulates its activation in tumour cells. Biochem. J. 2014, 459, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Hurley, R.L.; Barre, L.K.; Wood, S.D.; Anderson, K.A.; Kemp, B.E.; Means, A.R.; Witters, L.A. Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J. Biol. Chem. 2006, 281, 36662–36672. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Bridges, D.; Nakada, D.; Skiniotis, G.; Morrison, S.J.; Lin, J.D.; Saltiel, A.R.; Inoki, K. Inhibition of AMPK catabolic action by GSK3. Mol. Cell 2013, 50, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Littler, D.R.; Walker, J.R.; Davis, T.; Wybenga-Groot, L.E.; Finerty, P.J., Jr.; Newman, E.; Mackenzie, F.; Dhe-Paganon, S. A conserved mechanism of autoinhibition for the AMPK kinase domain: ATP-binding site and catalytic loop refolding as a means of regulation. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2010, 66, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, V.; Zhao, K.; Wyce, A.; Schwartz, M.F.; Lo, W.S.; Berger, S.L.; Marmorstein, R. Structure and dimerization of the kinase domain from yeast Snf1, a member of the Snf1/AMPK protein family. Structure 2006, 14, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Handa, N.; Takagi, T.; Saijo, S.; Kishishita, S.; Takaya, D.; Toyama, M.; Terada, T.; Shirouzu, M.; Suzuki, A.; Lee, S.; et al. Structural basis for compound C inhibition of the human AMP-activated protein kinase alpha2 subunit kinase domain. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Jiao, Z.H.; Zheng, L.S.; Zhang, Y.Y.; Xie, S.T.; Wang, Z.X.; Wu, J.W. Structural insight into the autoinhibition mechanism of AMP-activated protein kinase. Nature 2009, 459, 1146–1149. [Google Scholar] [CrossRef] [PubMed]
- Polekhina, G.; Gupta, A.; van Denderen, B.J.; Feil, S.C.; Kemp, B.E.; Stapleton, D.; Parker, M.W. Structural basis for glycogen recognition by AMP-activated protein kinase. Structure 2005, 13, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Amodeo, G.A.; Rudolph, M.J.; Tong, L. Crystal structure of the heterotrimer core of Saccharomyces cerevisiae AMPK homologue SNF1. Nature 2007, 449, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Heath, R.; Saiu, P.; Leiper, F.C.; Leone, P.; Jing, C.; Walker, P.A.; Haire, L.; Eccleston, J.F.; Davis, C.T.; et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 2007, 449, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Townley, R.; Shapiro, L. Crystal structures of the adenylate sensor from fission yeast AMP-activated protein kinase. Science 2007, 315, 1726–1729. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Townley, R.; Shapiro, L. Structural insight into AMPK regulation: ADP comes into play. Structure 2007, 15, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xin, F.J.; Wang, J.; Hu, J.; Zhang, Y.Y.; Wan, S.; Cao, L.S.; Lu, C.; Li, P.; Yan, S.F.; et al. Conserved regulatory elements in AMPK. Nature 2013, 498, E8–E10. [Google Scholar] [CrossRef] [PubMed]
- Kornev, A.P.; Haste, N.M.; Taylor, S.S.; Eyck, L.F. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 17783–17788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornev, A.P.; Taylor, S.S. Dynamics-Driven Allostery in Protein Kinases. Trends Biochem. Sci. 2015, 40, 628–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meharena, H.S.; Chang, P.; Keshwani, M.M.; Oruganty, K.; Nene, A.K.; Kannan, N.; Taylor, S.S.; Kornev, A.P. Deciphering the structural basis of eukaryotic protein kinase regulation. PLoS Biol. 2013, 11, e1001680. [Google Scholar] [CrossRef] [PubMed]
- Sanders, M.J.; Ali, Z.S.; Hegarty, B.D.; Heath, R.; Snowden, M.A.; Carling, D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J. Biol. Chem. 2007, 282, 32539–32548. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.P.; Helps, N.R.; Cohen, P.T.; Hardie, D.G. 5’-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995, 377, 421–425. [Google Scholar] [PubMed]
- Gowans, G.J.; Hawley, S.A.; Ross, F.A.; Hardie, D.G. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013, 18, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carling, D.; Clarke, P.R.; Zammit, V.A.; Hardie, D.G. Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities. Eur. J. Biochem. 1989, 186, 129–136. [Google Scholar] [PubMed]
- Oakhill, J.S.; Steel, R.; Chen, Z.P.; Scott, J.W.; Ling, N.; Tam, S.; Kemp, B.E. AMPK is a direct adenylate charge-regulated protein kinase. Science 2011, 332, 1433–1435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Guo, H.; Zhang, C.S.; Lin, S.Y.; Yin, Z.; Peng, Y.; Luo, H.; Shi, Y.; Lian, G.; Zhang, C.; et al. AMP as a low-energy charge signal autonomously initiates assembly of AXIN-AMPK-LKB1 complex for AMPK activation. Cell Metab. 2013, 18, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Makela, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, R.L.; Anderson, K.A.; Franzone, J.M.; Kemp, B.E.; Means, A.R.; Witters, L.A. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 2005, 280, 29060–29066. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.L.; Wu, Y.Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Yang, X.; Qin, B.; Liu, T.; Zhang, H.; Guo, W.; Lee, S.B.; Kim, J.J.; Yuan, J.; Pei, H.; et al. Deubiquitination and Activation of AMPK by USP10. Mol. Cell 2016, 61, 614–624. [Google Scholar] [PubMed]
- Lee, S.W.; Li, C.F.; Jin, G.; Cai, Z.; Han, F.; Chan, C.H.; Yang, W.L.; Li, B.K.; Rezaeian, A.H.; Li, H.Y.; et al. Skp2-dependent ubiquitination and activation of LKB1 is essential for cancer cell survival under energy stress. Mol. Cell 2015, 57, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.W.; Zhu, M.; et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polekhina, G.; Gupta, A.; Michell, B.J.; van Denderen, B.; Murthy, S.; Feil, S.C.; Jennings, I.G.; Campbell, D.J.; Witters, L.A.; Parker, M.W.; et al. AMPK beta subunit targets metabolic stress sensing to glycogen. Curr. Biol. 2003, 13, 867–871. [Google Scholar] [CrossRef]
- Gu, X.; Yan, Y.; Novick, S.J.; Kovich, A.; Goswami, D.; Ke, J.; Tan, M.H.E.; Wang, L.; Li, X.; de Waal, P.; et al. Deconvoluting AMP-dependent kinase (AMPK) adenine nucleotide binding and sensing. J. Biol. Chem. 2017, 292, 12653–12666. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, L.; Zhou, X.E.; Ke, J.; de Waal, P.W.; Gu, X.; Tan, M.H.; Wang, D.; Wu, D.; Xu, H.E.; et al. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res. 2015, 25, 50–66. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.; Ghilagaber, S.; Nikolaev, A.; Hardie, D.G. The glycogen-binding domain on the AMPK beta subunit allows the kinase to act as a glycogen sensor. Cell Metab. 2009, 9, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Sanders, M.J.; Carmena, D.; Bright, N.J.; Haire, L.F.; Underwood, E.; Patel, B.R.; Heath, R.B.; Walker, P.A.; Hallen, S.; et al. Structural basis of AMPK regulation by small molecule activators. Nat. Commun. 2013, 4, 3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, M.F.; Rajamohan, F.; Harris, M.S.; Caspers, N.L.; Magyar, R.; Withka, J.M.; Wang, H.; Borzilleri, K.A.; Sahasrabudhe, P.V.; Hoth, L.R.; et al. Structural Basis for AMPK Activation: Natural and Synthetic Ligands Regulate Kinase Activity from Opposite Poles by Different Molecular Mechanisms. Structure 2014, 22, 1161–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Bridges, M.D.; Yan, Y.; de Waal, P.; Zhou, X.E.; Suino-Powell, K.M.; Xu, H.E.; Hubbell, W.L.; Melcher, K. Conformational heterogeneity of the allosteric drug and metabolite (ADaM) site in AMP-activated protein kinase (AMPK). J. Biol. Chem. 2018, 239, 16994–17007. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Gong, J.; Zhao, T.; Zhao, J.; Lam, P.; Ye, J.; Li, J.Z.; Wu, J.; Zhou, H.M.; Li, P. Downregulation of AMP-activated protein kinase by Cidea-mediated ubiquitination and degradation in brown adipose tissue. EMBO J. 2008, 27, 1537–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pineda, C.T.; Ramanathan, S.; Fon Tacer, K.; Weon, J.L.; Potts, M.B.; Ou, Y.H.; White, M.A.; Potts, P.R. Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 2015, 160, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Vila, I.K.; Yao, Y.; Kim, G.; Xia, W.; Kim, H.; Kim, S.J.; Park, M.K.; Hwang, J.P.; Gonzalez-Billalabeitia, E.; Hung, M.C.; et al. A UBE2O-AMPKalpha2 Axis that Promotes Tumor Initiation and Progression Offers Opportunities for Therapy. Cancer Cell 2017, 31, 208–224. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.O.; Lee, S.K.; Kim, N.; Kim, J.H.; You, G.Y.; Moon, J.W.; Jie, S.; Kim, S.J.; Lee, Y.W.; Kang, H.J.; et al. E3 ubiquitin ligase, WWP1, interacts with AMPKalpha2 and down-regulates its expression in skeletal muscle C2C12 cells. J. Biol. Chem. 2013, 288, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, J.; Zhang, Y.Y.; Yan, S.F.; Neumann, D.; Schlattner, U.; Wang, Z.X.; Wu, J.W. AMP-activated protein kinase undergoes nucleotide-dependent conformational changes. Nat. Struct. Mol. Biol. 2012, 19, 716–718. [Google Scholar] [CrossRef] [PubMed]
- Xin, F.J.; Wang, J.; Zhao, R.Q.; Wang, Z.X.; Wu, J.W. Coordinated regulation of AMPK activity by multiple elements in the alpha-subunit. Cell Res. 2013, 23, 1237–1240. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.W.; Foretz, M.; Bultot, L.; Fullerton, M.D.; Deak, M.; Ross, F.A.; Hawley, S.A.; Shpiro, N.; Viollet, B.; Barron, D.; et al. Mechanism of action of compound-13: An alpha1-selective small molecule activator of AMPK. Chem. Biol. 2014, 21, 866–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langendorf, C.G.; Ngoei, K.R.; Scott, J.W.; Ling, N.X.; Issa, S.M.; Gorman, M.A.; Parker, M.W.; Sakamoto, K.; Oakhill, J.S.; Kemp, B.E. Structural basis of allosteric and synergistic activation of AMPK by furan-2-phosphonic derivative C2 binding. Nat. Commun. 2016, 7, 10912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crute, B.E.; Seefeld, K.; Gamble, J.; Kemp, B.E.; Witters, L.A. Functional domains of the alpha1 catalytic subunit of the AMP-activated protein kinase. J. Biol. Chem. 1998, 273, 35347–35354. [Google Scholar] [CrossRef] [PubMed]
- Pang, T.; Xiong, B.; Li, J.Y.; Qiu, B.Y.; Jin, G.Z.; Shen, J.K.; Li, J. Conserved alpha-helix acts as autoinhibitory sequence in AMP-activated protein kinase alpha subunits. J. Biol. Chem. 2007, 282, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, L.; Rastelli, G. alphaC helix displacement as a general approach for allosteric modulation of protein kinases. Drug Discov. Today 2013, 18, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Willows, R.; Sanders, M.J.; Xiao, B.; Patel, B.R.; Martin, S.R.; Read, J.; Wilson, J.R.; Hubbard, J.; Gamblin, S.J.; Carling, D. Phosphorylation of AMPK by upstream kinases is required for activity in mammalian cells. Biochem. J. 2017, 474, 3059–3073. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Overall structure of human adenosine monophosphate (AMP)-activated protein kinase (AMPK). (A). Domain structure and AMPK isoforms. Activation loop and carbohydrate-binding module (CBM) phosphorylation sites of different isoforms are indicated below the domain map (B,C). Crystal structures of phosphorylated, AMP-bound AMPK α2β1γ1/991 ((B); PDB: 4CFE) and α1β2γ1/cyclodextrin (CD) ((C); PDB: 4RER).
Figure 1.
Overall structure of human adenosine monophosphate (AMP)-activated protein kinase (AMPK). (A). Domain structure and AMPK isoforms. Activation loop and carbohydrate-binding module (CBM) phosphorylation sites of different isoforms are indicated below the domain map (B,C). Crystal structures of phosphorylated, AMP-bound AMPK α2β1γ1/991 ((B); PDB: 4CFE) and α1β2γ1/cyclodextrin (CD) ((C); PDB: 4RER).

Figure 2.
Structure of AMPK domains and subcomplexes. (A) Rat CBM bound to cyclodextrin; (B) Fission yeast kinase domain–autoinhibitory domain (KD‒AID) complex; (C) AMP-bound, phosphorylated mammalian AMPK core complex (rat α1-human β2-rat γ1); (D) AMP-bound, phosphorylated rat α1—human β2CTD—rat γ1 complex.
Figure 2.
Structure of AMPK domains and subcomplexes. (A) Rat CBM bound to cyclodextrin; (B) Fission yeast kinase domain–autoinhibitory domain (KD‒AID) complex; (C) AMP-bound, phosphorylated mammalian AMPK core complex (rat α1-human β2-rat γ1); (D) AMP-bound, phosphorylated rat α1—human β2CTD—rat γ1 complex.

Figure 3.
Active protein kinase catalytic cleft. (A) Key residues and structural elements of phosphorylated AMP-bound α1β2γ1 AMPK (4RER). Active kinase structures are characterized by a precisely positioned set of motifs for substrate- and adenosine triphosphate (ATP)-binding, in which four residues (L70, L81, H139, F160; shown in stick plus translucent surface presentation) are stacked against each other to form a regulatory spine. In this conformation, the activation loop p-T174 (p-T172 in human α2) positions R140 and D141 from the catalytic loop for peptide substrate binding, and K62 from the αC-helix for aligning the ATP-binding K47 and the Mg2+-binding DFG loop. The AMPK active protein kinase cleft resembles the canonical protein kinase A (PKA) site. To better visualize the active structure, we modeled the serine residue of a substrate peptide and the co-substrate ATP from the structure of PKA (PDB: 1ATP) in the catalytic cleft. Spheres: Mg2+ ions. (B) Surface presentation of the AMPK catalytic cleft (4RER) overlaid with a stick model of the aligned substrate peptide and ATP from the structure of substrate-bound CDK2 (PDB: 1QMZ). The Ser hydroxyl-positioning AMPK D141 is shown in green stick representation.
Figure 3.
Active protein kinase catalytic cleft. (A) Key residues and structural elements of phosphorylated AMP-bound α1β2γ1 AMPK (4RER). Active kinase structures are characterized by a precisely positioned set of motifs for substrate- and adenosine triphosphate (ATP)-binding, in which four residues (L70, L81, H139, F160; shown in stick plus translucent surface presentation) are stacked against each other to form a regulatory spine. In this conformation, the activation loop p-T174 (p-T172 in human α2) positions R140 and D141 from the catalytic loop for peptide substrate binding, and K62 from the αC-helix for aligning the ATP-binding K47 and the Mg2+-binding DFG loop. The AMPK active protein kinase cleft resembles the canonical protein kinase A (PKA) site. To better visualize the active structure, we modeled the serine residue of a substrate peptide and the co-substrate ATP from the structure of PKA (PDB: 1ATP) in the catalytic cleft. Spheres: Mg2+ ions. (B) Surface presentation of the AMPK catalytic cleft (4RER) overlaid with a stick model of the aligned substrate peptide and ATP from the structure of substrate-bound CDK2 (PDB: 1QMZ). The Ser hydroxyl-positioning AMPK D141 is shown in green stick representation.

Figure 4.
AMP binds three of the four CBS sites of the γ subunit. (A,B) Cartoon representation of the γ subunit in two different orientations. AMP molecules are shown in stick representation. The four CBS sites are shown in different colors with the secondary structure elements of CBS1 labeled. (C,D) Surface representation of the front and back sides of the disk flat surfaces illustrating the AMP-occupied binding pockets 1, 3, and 4, and the empty CBS2 pocket. (E) The phosphate groups (orange) of the three AMP molecules (cyan C atoms) coordinately interact with a set of polar γ subunit residues (green C atoms); O: red, N: blue.
Figure 4.
AMP binds three of the four CBS sites of the γ subunit. (A,B) Cartoon representation of the γ subunit in two different orientations. AMP molecules are shown in stick representation. The four CBS sites are shown in different colors with the secondary structure elements of CBS1 labeled. (C,D) Surface representation of the front and back sides of the disk flat surfaces illustrating the AMP-occupied binding pockets 1, 3, and 4, and the empty CBS2 pocket. (E) The phosphate groups (orange) of the three AMP molecules (cyan C atoms) coordinately interact with a set of polar γ subunit residues (green C atoms); O: red, N: blue.

Figure 5.
The AID is in equilibrium between KD- and γ-bound conformations. (A) Cartoon structure of the human α1 KD-AID complex. (B) Overlay of the inactive KD-AID structure with the structure of active holo-AMPK (α subunit: green; β- and γ-subunits: grey). The arrow indicates the repositioning of the AID in the active structure. (C,D) Catalytic center of the inactive (C) and active (D) AMPK conformation. Stick plus translucent surface presentations indicate the regulatory spine residues L70, L81, H139, and F160. Mg2+-ATP was modeled into both structures for orientation, even though it cannot bind to the inactive structure shown in panel C. Spheres: Mg2+ ions.
Figure 5.
The AID is in equilibrium between KD- and γ-bound conformations. (A) Cartoon structure of the human α1 KD-AID complex. (B) Overlay of the inactive KD-AID structure with the structure of active holo-AMPK (α subunit: green; β- and γ-subunits: grey). The arrow indicates the repositioning of the AID in the active structure. (C,D) Catalytic center of the inactive (C) and active (D) AMPK conformation. Stick plus translucent surface presentations indicate the regulatory spine residues L70, L81, H139, and F160. Mg2+-ATP was modeled into both structures for orientation, even though it cannot bind to the inactive structure shown in panel C. Spheres: Mg2+ ions.

Figure 6.
αRIM2/CBS3 and AID-αRIM1/CBS2 interactions are linked. Structure of human AMP-bound AMPK α1β2γ1 (4RER) with key residues shown in a stick presentation; the α-linker is shown in magenta, the γ subunit in cyan, and the AID in light green. AMP bound at CBS3 and αRIM2 E364 directly interact with γ1 K170, which positions the αRIM1-binding residues R171, and indirectly through K174 and F175, F179, thus stabilizing the AID‒γ subunit interaction. Consistently, mutations of the αRIM1/γ subunit (and αRIM2/CBS3) interface residues highlighted by oval outlines (human α1: F342D/Y343D, I335D/M336D, E364, R365; γ1: R171A, F179D) are constitutively AMP-non-responsive. Dashed lines indicate hydrogen bonds.
Figure 6.
αRIM2/CBS3 and AID-αRIM1/CBS2 interactions are linked. Structure of human AMP-bound AMPK α1β2γ1 (4RER) with key residues shown in a stick presentation; the α-linker is shown in magenta, the γ subunit in cyan, and the AID in light green. AMP bound at CBS3 and αRIM2 E364 directly interact with γ1 K170, which positions the αRIM1-binding residues R171, and indirectly through K174 and F175, F179, thus stabilizing the AID‒γ subunit interaction. Consistently, mutations of the αRIM1/γ subunit (and αRIM2/CBS3) interface residues highlighted by oval outlines (human α1: F342D/Y343D, I335D/M336D, E364, R365; γ1: R171A, F179D) are constitutively AMP-non-responsive. Dashed lines indicate hydrogen bonds.

Figure 7.
The β-CTD binds and stabilizes the activation loop. Structure of AMP-bound, phosphorylated AMPK α1–β2CTD–γ1 (PDB: 4CFH). The activation loop is highlighted in orange, and p-T172 is shown in sphere presentation.
Figure 7.
The β-CTD binds and stabilizes the activation loop. Structure of AMP-bound, phosphorylated AMPK α1–β2CTD–γ1 (PDB: 4CFH). The activation loop is highlighted in orange, and p-T172 is shown in sphere presentation.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yan, Y.; Zhou, X.E.; Xu, H.E.; Melcher, K. Structure and Physiological Regulation of AMPK. Int. J. Mol. Sci. 2018, 19, 3534. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113534
AMA Style
Yan Y, Zhou XE, Xu HE, Melcher K. Structure and Physiological Regulation of AMPK. International Journal of Molecular Sciences. 2018; 19(11):3534. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113534
Chicago/Turabian StyleYan, Yan, X. Edward Zhou, H. Eric Xu, and Karsten Melcher. 2018. "Structure and Physiological Regulation of AMPK" International Journal of Molecular Sciences 19, no. 11: 3534. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113534
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.