Discovery of Novel Druggable Sites on Zika Virus NS3 Helicase Using X-ray Crystallography-Based Fragment Screening

 and
and
Abstract
:
1. Introduction
2. Results
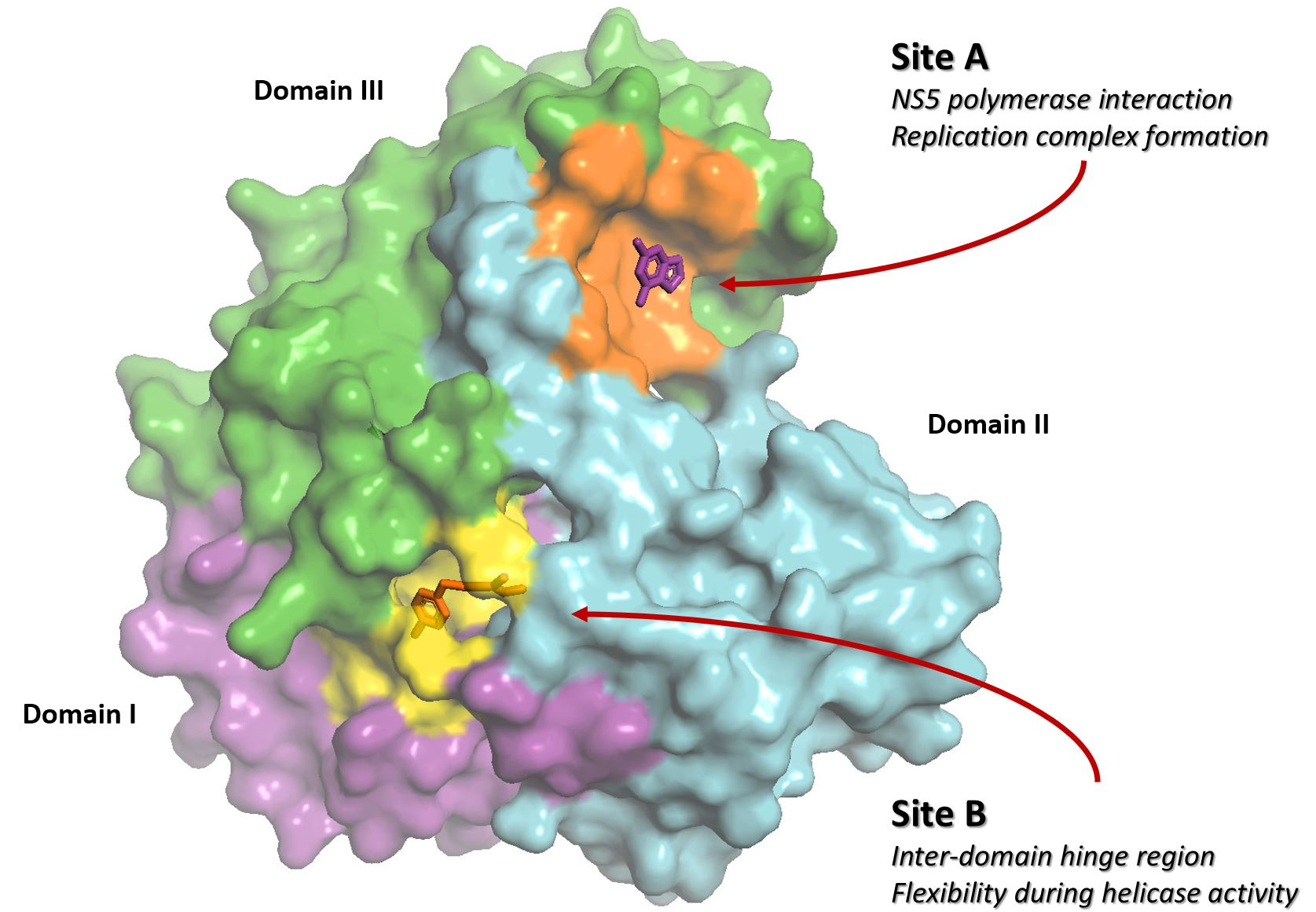
2.1. FBS-X Reveals Two Novel Drug Binding Sites on ZIKV NS3-Hel
2.2. Site A Is at a Critical Interface of the NS3-NS5 Flaviviral Replication Complex (RC)
2.3. Binding of Fragments 1 and 2 to Site A Offers Multiple Drug Design Prospects
2.3.1. Fragment 1
2.3.2. Fragment 2
2.4. Site B Is at a Flexible, Interdomain Hinge That is Highly Conserved across Flaviviruses
2.5. Fragments 3 and 4 Collectively Reveal Features Enabling Efficient Binding at Site B
2.5.1. Fragment 3
2.5.2. Fragment 4
3. Discussion
4. Materials and Methods
4.1. Fragment Libraries
4.2. Protein Purification and Crystallization
4.3. Fragment Soaking and X-ray Data Collection
4.4. NMR STD Experiments
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | ZIKV | DENV | Kunjin | JEV | YFV | MVEV | TBEV |
|---|---|---|---|---|---|---|---|
| PDB Code | This Work | 2JLQ | 2QEQ | 2Z83 | 1YKS | 2V8O | - |
| Site A | R439 | R | R | R | K | R | R |
| I441 | I | I | I | A | I | E | |
| N568 | N | T | T | E | S | A | |
| N569 | N | N | N | H | N | N | |
| T570 | Q | T | A | E | I | A | |
| I571 | I | I | I | I | I | V | |
| M572 | L | L | L | L | L | D | |
| M595 | L | I | L | C | L | K | |
| A597 | A | A | A | E | A | A | |
| C600 | Y | Y | Y | S | Y | F | |
| S601 | A | S | A | S | S | K | |
| Site B | V191 | V | V | V | V | V | V |
| A287 | A | A | A | A | A | A | |
| H288 | H | H | H | H | H | H | |
| F314 | F | F | F | A | F | L | |
| T316 | T | T | T | T | T | T | |
| P320 | P | P | P | P | P | P | |
| E413 | E | I | S | I | E | E | |
| T449 | T | T | T | S | T | T | |
| S452 | S | S | S | S | S | S | |
| Y508 | Y | Y | Y | Y | Y | Y | |
| P510 | P | P | P | V | P | P |
References
- Mansuy, J.M.; Dutertre, M.; Mengelle, C.; Fourcade, C.; Marchou, B.; Delobel, P.; Izopet, J.; Martin-Blondel, G. Zika virus: High infectious viral load in semen, a new sexually transmitted pathogen? Lancet Infect. Dis. 2016, 16, 405. [Google Scholar] [CrossRef]
- De Oliveira, W.K.; de Franca, G.V.A.; Carmo, E.H.; Duncan, B.B.; de Souza Kuchenbecker, R.; Schmidt, M.I. Infection-related microcephaly after the 2015 and 2016 Zika virus outbreaks in Brazil: A surveillance-based analysis. Lancet 2017, 390, 861–870. [Google Scholar] [CrossRef]
- Cumulative Zika Virus Disease Case Counts in the United States 2015–2018. Available online: https://www.cdc.gov/zika/reporting/case-counts.html (accessed on 25 August 2018).
- Baud, D.; Gubler, D.J.; Schaub, B.; Lanteri, M.C.; Musso, D. An update on Zika virus infection. Lancet 2017, 390, 2099–2109. [Google Scholar] [CrossRef]
- Cao-Lormeau, V.M.; Blake, A.; Mons, S.; Lastere, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barre Syndrome outbreak associated with Zika virus infection in French Polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierson, T.C.; Diamond, M.S. Flaviviruses, 6th ed.; Wolter Kluwer: Philadelphia, PN, USA, 2013; Volume 2. [Google Scholar]
- Zhang, Z.; Li, Y.; Loh, Y.R.; Phoo, W.W.; Hung, A.W.; Kang, C.; Luo, D. Crystal structure of unlinked NS2B-NS3 protease from Zika virus. Science 2016, 354, 1597–1600. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Ji, X.; Yang, X.; Xie, W.; Yang, K.; Chen, C.; Wu, C.; Chi, H.; Mu, Z.; Wang, Z.; et al. The crystal structure of Zika virus helicase: Basis for antiviral drug design. Protein Cell 2016, 7, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Singleton, M.R.; Dillingham, M.S.; Wigley, D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007, 76, 23–50. [Google Scholar] [CrossRef] [PubMed]
- Matusan, A.E.; Kelley, P.G.; Pryor, M.J.; Whisstock, J.C.; Davidson, A.D.; Wright, P.J. Mutagenesis of the dengue virus type 2 NS3 proteinase and the production of growth-restricted virus. J. Gen. Virol. 2001, 82, 1647–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, A.M.; Frick, D.N. Hepatitis C virus subgenomic replicon requires an active NS3 RNA helicase. J. Virol. 2006, 80, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Matusan, A.E.; Pryor, M.J.; Davidson, A.D.; Wright, P.J. Mutagenesis of the Dengue virus type 2 NS3 protein within and outside helicase motifs: Effects on enzyme activity and virus replication. J. Virol. 2001, 75, 9633–9643. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Coloma, J.; Garcia-Sastre, A.; Aggarwal, A.K. Structure of the NS3 helicase from Zika virus. Nat. Struct. Mol. Biol. 2016, 23, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Kwong, A.D.; Rao, B.G.; Jeang, K.T. Viral and cellular RNA helicases as antiviral targets. Nat. Rev. Drug Discov. 2005, 4, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Li, H.; Kong, D.; Cao, S.; Peng, G.; Zhou, R.; Chen, H.; Song, Y. Structure-based discovery of two antiviral inhibitors targeting the NS3 helicase of Japanese encephalitis virus. Sci. Rep. 2016, 6, 34550. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Y.F.; Vasudevan, S.G. The Transactions of NS3 and NS5 in Flaviviral RNA Replication. Adv. Exp. Med. Biol. 2018, 1062, 147–163. [Google Scholar] [PubMed]
- Lee, H.; Ren, J.; Nocadello, S.; Rice, A.J.; Ojeda, I.; Light, S.; Minasov, G.; Vargas, J.; Nagarathnam, D.; Anderson, W.F.; et al. Identification of novel small molecule inhibitors against NS2B/NS3 serine protease from Zika virus. Antiviral Res. 2017, 139, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, J.; Hansen, G.; Nitsche, C.; Klein, C.D.; Zhang, L.; Hilgenfeld, R. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science 2016, 353, 503–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.C.; Hsieh, Y.C.; Lee, S.J.; Wu, S.H.; Liao, C.L.; Tsao, C.H.; Chao, Y.S.; Chern, J.H.; Wu, C.P.; Yueh, A. Novel dengue virus-specific NS2B/NS3 protease inhibitor, BP2109, discovered by a high-throughput screening assay. Antimicrob. Agents Chemother. 2011, 55, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Salam, K.A.; Akimitsu, N. Hepatitis C virus NS3 inhibitors: Current and future perspectives. Biomed. Res. Int. 2013, 2013, 467869. [Google Scholar] [CrossRef] [PubMed]
- Shadrick, W.R.; Ndjomou, J.; Kolli, R.; Mukherjee, S.; Hanson, A.M.; Frick, D.N. Discovering new medicines targeting helicases: Challenges and recent progress. J. Biomol. Screen. 2013, 18, 761–781. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Coutard, B.; Decroly, E.; Li, C.; Sharff, A.; Lescar, J.; Bricogne, G.; Barral, K. Assessment of Dengue virus helicase and methyltransferase as targets for fragment-based drug discovery. Antiviral Res. 2014, 106, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.D.; Patel, D.; Dharia, C.; Fromer, M.W.; Ahmed, S.; Frenkel, Y.; Vijayan, R.S.; Eck, J.T.; Ho, W.C.; Das, K.; et al. Detecting allosteric sites of HIV-1 reverse transcriptase by X-ray crystallographic fragment screening. J. Med. Chem. 2013, 56, 2738–2746. [Google Scholar] [CrossRef] [PubMed]
- Huschmann, F.U.; Linnik, J.; Sparta, K.; Uhlein, M.; Wang, X.; Metz, A.; Schiebel, J.; Heine, A.; Klebe, G.; Weiss, M.S.; et al. Structures of endothiapepsin-fragment complexes from crystallographic fragment screening using a novel, diverse and affordable 96-compound fragment library. Acta Crystallogr. F Struct. Biol. Commun. 2016, 72, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.Y.; Siddaramaiah, L.K.; Ranade, R.M.; Nguyen, J.; Jian, T.; Zhang, Z.; Gillespie, J.R.; Buckner, F.S.; Verlinde, C.L.; Fan, E.; et al. A binding hotspot in Trypanosoma cruzi histidyl-tRNA synthetase revealed by fragment-based crystallographic cocktail screens. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 1684–1698. [Google Scholar] [CrossRef] [PubMed]
- Sondergaard, C.R.; Garrett, A.E.; Carstensen, T.; Pollastri, G.; Nielsen, J.E. Structural artifacts in protein-ligand X-ray structures: Implications for the development of docking scoring functions. J. Med. Chem. 2009, 52, 5673–5684. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Zhang, L.; Ramachandra, M.; Kusukawa, J.; Ebner, K.E.; Padmanabhan, R. Association between NS3 and NS5 proteins of dengue virus type 2 in the putative RNA replicase is linked to differential phosphorylation of NS5. J. Biol. Chem. 1995, 270, 19100–19106. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Y.; Saw, W.G.; Zhao, Y.; Chan, K.W.; Singh, D.; Chong, Y.; Forwood, J.K.; Ooi, E.E.; Gruber, G.; Lescar, J.; et al. The C-terminal 50 amino acid residues of dengue NS3 protein are important for NS3-NS5 interaction and viral replication. J. Biol. Chem. 2015, 290, 2379–2394. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.K.; Zhang, S. Computational prediction of protein hot spot residues. Curr. Pharm. Des. 2012, 18, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Rajamani, D.; Thiel, S.; Vajda, S.; Camacho, C.J. Anchor residues in protein-protein interactions. Proc. Natl. Acad. Sci. USA 2004, 101, 11287–11292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincetti, P.; Caporuscio, F.; Kaptein, S.; Gioiello, A.; Mancino, V.; Suzuki, Y.; Yamamoto, N.; Crespan, E.; Lossani, A.; Maga, G.; et al. Discovery of Multitarget Antivirals Acting on Both the Dengue Virus NS5-NS3 Interaction and the Host Src/Fyn Kinases. J. Med. Chem. 2015, 58, 4964–4975. [Google Scholar] [CrossRef] [PubMed]
- Webber, S.E.; Tikhe, J.; Worland, S.T.; Fuhrman, S.A.; Hendrickson, T.F.; Matthews, D.A.; Love, R.A.; Patick, A.K.; Meador, J.W.; Ferre, R.A.; et al. Design, synthesis, and evaluation of nonpeptidic inhibitors of human rhinovirus 3C protease. J. Med. Chem. 1996, 39, 5072–5082. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Pei, J.; Lai, L. Statistical Analysis and Prediction of Covalent Ligand Targeted Cysteine Residues. J. Chem. Inf. Model. 2017, 57, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Weik, M.; Ravelli, R.B.; Kryger, G.; McSweeney, S.; Raves, M.L.; Harel, M.; Gros, P.; Silman, I.; Kroon, J.; Sussman, J.L. Specific chemical and structural damage to proteins produced by synchrotron radiation. Proc. Natl. Acad. Sci. USA 2000, 97, 623–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbuzov, A.E.; Bastanova, M.S. The question of the tautomerism of Isatin. Russ. Chem. Bull. 1952, 1, 443–451. [Google Scholar] [CrossRef]
- Cerchiaro, G.; Ferreira, A.M. Oxindoles and copper complexes with oxindole-derivatives as potential pharmacological agents. J. Braz. Chem. Soc. 2006, 17, 1473–1485. [Google Scholar] [CrossRef] [Green Version]
- Pal, D.; Chakrabarti, P. Different types of interactions involving cysteine sulfhydryl group in proteins. J. Biomol. Struct. Dyn. 1998, 15, 1059–1072. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, S.; Caaveiro, J.M.M.; Nakakido, M.; Tanabe, A.; Nagatoishi, S.; Tamura, Y.; Matsuda, N.; Liu, D.; Hoang, Q.Q.; Tsumoto, K. Discovery and Optimization of Inhibitors of the Parkinson’s Disease Associated Protein DJ-1. ACS Chem. Biol. 2018, 13, 2783–2793. [Google Scholar] [CrossRef] [PubMed]
- Tonge, P.J. Drug-Target Kinetics in Drug Discovery. ACS Chem. Neurosci. 2018, 9, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Rice, C.M. Three conformational snapshots of the hepatitis C virus NS3 helicase reveal a ratchet translocation mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Valdar, W.S. Scoring residue conservation. Proteins 2002, 48, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Zondlo, N.J. Aromatic-proline interactions: Electronically tunable CH/pi interactions. Acc. Chem. Res. 2013, 46, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing toward the reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Studier, F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Weeks, S.D.; Drinker, M.; Loll, P.J. Ligation independent cloning vectors for expression of SUMO fusions. Protein Expr. Purif. 2007, 53, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battye, T.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Smart, O.S.; Womack, T.O.; Flensburg, C.; Keller, P.; Paciorek, W.; Sharff, A.; Vonrhein, C.; Bricogne, G. Exploiting structure similarity in refinement: Automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Liebschner, D.; Afonine, P.V.; Moriarty, N.W.; Poon, B.K.; Sobolev, O.V.; Terwilliger, T.C.; Adams, P.D. Polder maps: Improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D Biol. Crystallogr. 2017, 73, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.L.; Shaka, A.J. Water Suppression That Works. Excitation Sculpting Using Arbitrary Wave-Forms and Pulsed-Field Gradients. J. Magn. Reson. A 1995, 112, 275–279. [Google Scholar] [CrossRef]
- Mayer, M.; Meyer, B. Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew. Chem. Int. Ed. Engl. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]



| Parameters | Results |
|---|---|
| Total number of compounds selected for screening | 200 |
| Number of compounds successfully soaked into individual crystals for data collection 1 | 154 |
| Number of datasets that diffracted to better than 3 Å | 88 |
| Median resolution of collected datasets | 2.2 Å |
| Number of hits 2 | 4 |
| Binding Site | A (NS3-NS5 interface) | B (Interdomain hinge region) | ||
|---|---|---|---|---|
| Fragment Hits | 1 | 2 | 3 | 4 |
| Chemical name | 2,6-di-aminopurine | 5-methylisatin | 3-amino-1-[2-(4-chlorophenyl)ethyl] thiourea | 2,4-dicholorobenzamide |
| Structural formula |  |  |  |  |
| Molecular weight (Da) | 150 | 160 | 230 | 190 |
| Data collection 1 | ||||
| Cell parameters (space group P21): | ||||
| a, b, c (Å) | 49.92, 73.22, 58.45 | 50.12, 73.35, 59.06 | 49.89, 73.46, 60.72 | 49.95, 74.17, 58.58 |
| β (°) | 90.85 | 91.66 | 99.51 | 90.7 |
| Resolution range (Å) | 46–1.9 | 46.6–2.2 | 49.21–2.35 | 49.8–1.80 |
| Unique reflections | 33305 (2063) | 21559 (1783) | 17920 (1742) | 39559 (2239) |
| Rmerge | 0.10 (1.21) | 0.187 (1.63) | 0.178 (1.62) | 0.12 (1.74) |
| Rmeas | 0.12(1.44) | 0.22 (1.93) | 0.21 (1.95) | 0.14 (2.05) |
| I/σ(I) | 10.3 (1.3) | 6.8 (1.1) | 7.4 (1.1) | 11.1 (1.2) |
| CC1/2 | 0.99 (0.99) | 0.97 (0.99) | 0.99 (0.36) | 0.99 (0.51) |
| Completeness (%) | 99.6 (94.4) | 98.7 (95.1) | 98.6 (96.8) | 99.6 (95.2) |
| Redundancy | 6.8 (6.5) | 6.9 (6.9) | 7.0(6.5) | 6.8 (6.6) |
| Refinement | ||||
| Rwork | 0.194 | 0.219 | 0.202 | 0.185 |
| Rfree | 0.224 | 0.242 | 0.264 | 0.217 |
| No. of atoms: | ||||
| Protein | 3544 | 3544 | 3544 | 3434 |
| Ligand/ions | 81 | 69 | 107 | 43 |
| Water molecules | 354 | 276 | 149 | 491 |
| Mean B factor (Å2) | 36.6 | 44.5 | 52.0 | 28.1 |
| R.m.s. deviations: | ||||
| Bond lengths (Å) | 0.008 | 0.008 | 0.011 | 0.008 |
| Bond angles (°) | 0.96 | 0.96 | 0.91 | 0.94 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munawar, A.; Beelen, S.; Munawar, A.; Lescrinier, E.; Strelkov, S.V. Discovery of Novel Druggable Sites on Zika Virus NS3 Helicase Using X-ray Crystallography-Based Fragment Screening. Int. J. Mol. Sci. 2018, 19, 3664. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113664
Munawar A, Beelen S, Munawar A, Lescrinier E, Strelkov SV. Discovery of Novel Druggable Sites on Zika Virus NS3 Helicase Using X-ray Crystallography-Based Fragment Screening. International Journal of Molecular Sciences. 2018; 19(11):3664. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113664
Chicago/Turabian StyleMunawar, Ali, Steven Beelen, Ahmad Munawar, Eveline Lescrinier, and Sergei V. Strelkov. 2018. "Discovery of Novel Druggable Sites on Zika Virus NS3 Helicase Using X-ray Crystallography-Based Fragment Screening" International Journal of Molecular Sciences 19, no. 11: 3664. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113664