TGFβ Superfamily Members as Regulators of B Cell Development and Function—Implications for Autoimmunity

Abstract
:1. Introduction
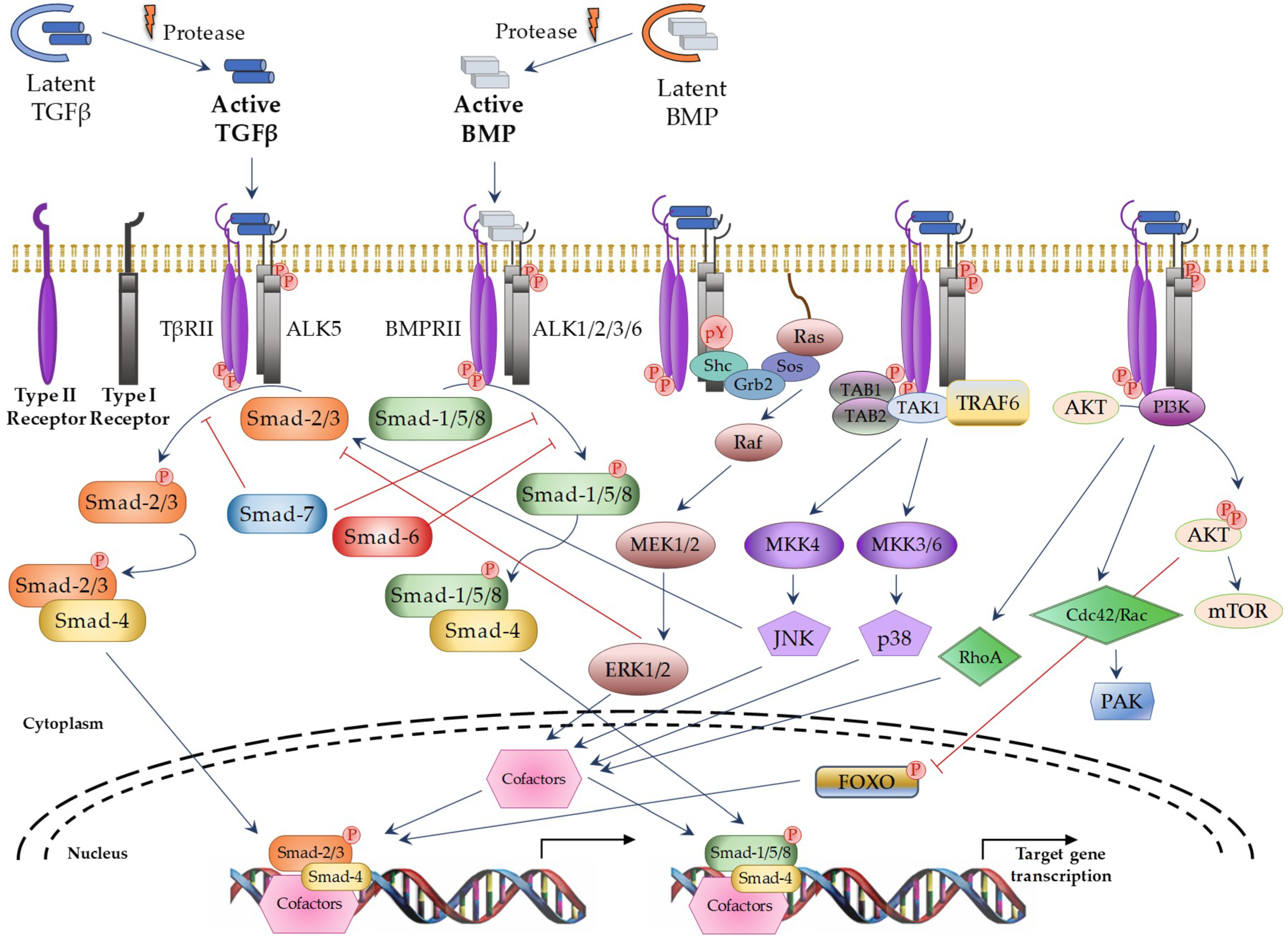
TGFβ Superfamily Members and Signaling
2. TGFβ Superfamily in B Cells
2.1. B Cell Development
2.2. B Cell Proliferation and Survival
2.3. Activation, Immunoglobulin (Ig) Production and Differentiation
3. B Cell Tolerance and Autoimmunity
3.1. Mechanisms for B Cell-Mediated Autoimmunity and Tolerance
3.2. TGFβ Induced IgA and Mucosal Homeostasis
4. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaswen, L.; Kulkarni, A.B.; Fredrickson, T.; Mittleman, B.; Schiffman, R.; Payne, S.; Longenecker, G.; Mozes, E.; Karlsson, S. Autoimmune manifestations in the transforming growth factor-β1 knockout mouse. Blood 1996, 87, 1439–1445. [Google Scholar] [PubMed]
- Gorelik, L.; Flavell, R.A. Abrogation of TGFβ signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity 2000, 12, 171–181. [Google Scholar] [CrossRef]
- Travis, M.A.; Sheppard, D. TGF-β Activation and Function in Immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, A.; Houston, S.A.; Sherwood, E.; Casulli, J.; Travis, M.A. Regulation of Innate and Adaptive Immunity by TGFβ. Adv. Immunol. 2017, 134, 137–233. [Google Scholar]
- Honda, K.; Littman, D.R. The microbiota in adaptive immune homeostasis and disease. Nature 2016, 535, 75–84. [Google Scholar] [CrossRef]
- Kim, D.; Yoo, S.-A.; Kim, W.-U. Gut microbiota in autoimmunity: potential for clinical applications. Arch. Pharm. Res. 2016, 39, 1565–1576. [Google Scholar] [CrossRef]
- Weiss, A.; Attisano, L. The TGFbeta Superfamily Signaling Pathway. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 47–63. [Google Scholar] [CrossRef]
- Hata, A.; Chen, Y.-G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudnall, A.M.; Arthur, J.W.; Lowery, J.W. Clinical Relevance and Mechanisms of Antagonism Between the BMP and Activin/TGF-β Signaling Pathways. J. Am. Osteopath. Assoc. 2016, 116, 452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Revy, P.; Regnault, A.; Lepelletier, Y.; Dy, M.; Brousse, N.; Amigorena, S.; Hermine, O.; Durandy, A. TGF-β1 prevents the noncognate maturation of human dendritic Langerhans cells. J. Immunol. 1999, 162, 4567–4575. [Google Scholar] [PubMed]
- Ramalingam, R.; Larmonier, C.B.; Thurston, R.D.; Midura-Kiela, M.T.; Zheng, S.G.; Ghishan, F.K.; Kiela, P.R. Dendritic Cell-Specific Disruption of TGF-Receptor II Leads to Altered Regulatory T Cell Phenotype and Spontaneous Multiorgan Autoimmunity. J. Immunol. 2012, 189, 3878–3893. [Google Scholar] [CrossRef] [PubMed]
- Yasmin, N.; Bauer, T.; Modak, M.; Wagner, K.; Schuster, C.; Köffel, R.; Seyerl, M.; Stöckl, J.; Elbe-Bürger, A.; Graf, D.; et al. Identification of bone morphogenetic protein 7 (BMP7) as an instructive factor for human epidermal Langerhans cell differentiation. J. Exp. Med. 2013, 210, 2597–2610. [Google Scholar] [CrossRef] [Green Version]
- Scutera, S.; Riboldi, E.; Daniele, R.; Elia, A.R.; Fraone, T.; Castagnoli, C.; Giovarelli, M.; Musso, T.; Sozzani, S. Production and function of activin A in human dendritic cells. Eur. Cytokine Netw. 2008, 19, 60–68. [Google Scholar] [CrossRef]
- Robson, N.C.; Wei, H.; McAlpine, T.; Kirkpatrick, N.; Cebon, J.; Maraskovsky, E. Activin-A attenuates several human natural killer cell functions. Blood 2009, 113, 3218–3225. [Google Scholar] [CrossRef] [Green Version]
- Gruber, B.L.; Marchese, M.J.; Kew, R.R. Transforming growth factor-β1 mediates mast cell chemotaxis. J. Immunol. 1994, 152, 5860–5867. [Google Scholar]
- Parekh, T.; Saxena, B.; Reibman, J.; Cronstein, B.N.; Gold, L.I. Neutrophil chemotaxis in response to TGF-beta isoforms (TGF-β1, TGF-β2, TGF-β3) is mediated by fibronectin. J. Immunol. 1994, 152, 2456–2466. [Google Scholar] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.H.; Lee, G.T.; Lee, J.H.; Kwon, S.J.; Park, S.H.; Kim, S.J.; Kim, I.Y. Effect of bone morphogenetic protein-6 on macrophages. Immunology 2009, 128, e442–e450. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.J.; Lee, G.T.; Lee, J.-H.; Kim, W.J.; Kim, I.Y. Bone morphogenetic protein-6 induces the expression of inducible nitric oxide synthase in macrophages. Immunology 2009, 128, e758–e765. [Google Scholar] [CrossRef] [PubMed]
- Sierra-Filardi, E.; Puig-Kroger, A.; Blanco, F.J.; Nieto, C.; Bragado, R.; Palomero, M.I.; Bernabeu, C.; Vega, M.A.; Corbi, A.L. Activin A skews macrophage polarization by promoting a proinflammatory phenotype and inhibiting the acquisition of anti-inflammatory macrophage markers. Blood 2011, 117, 5092–5101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Qi, Y.; Wu, N.; Ma, C.; Feng, W.; Cui, X.; Liu, Z. Expression and anti-inflammatory role of activin receptor-interacting protein 2 in lipopolysaccharide-activated macrophages. Sci. Rep. 2017, 7, 10306. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Sanjabi, S.; Flavell, R.A. Transforming Growth Factor-β Controls Development, Homeostasis, and Tolerance of T Cells by Regulatory T Cell-Dependent and -Independent Mechanisms. Immunity 2006, 25, 455–471. [Google Scholar] [CrossRef] [Green Version]
- Marie, J.C.; Liggitt, D.; Rudensky, A.Y. Cellular Mechanisms of Fatal Early-Onset Autoimmunity in Mice with the T Cell-Specific Targeting of Transforming Growth Factor-β Receptor. Immunity 2006, 25, 441–454. [Google Scholar] [CrossRef]
- Kehrl, J.H.; Wakefield, L.M.; Roberts, A.B.; Jakowlew, S.; Alvarez-Mon, M.; Derynck, R.; Sporn, M.B.; Fauci, A.S. Production of transforming growth factor β by human T lymphocytes and its potential role in the regulation of T cell growth. J. Exp. Med. 1986, 163, 1037–1050. [Google Scholar] [CrossRef]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.-K.L.; Flavell, R.A. Transforming growth factor-β regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Sanjabi, S.; Mosaheb, M.M.; Flavell, R.A. Opposing Effects of TGF-β and IL-15 Cytokines Control the Number of Short-Lived Effector CD8+ T Cells. Immunity 2009, 31, 131–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinoco, R.; Alcalde, V.; Yang, Y.; Sauer, K.; Zuniga, E.I. Cell-Intrinsic Transforming Growth Factor-β Signaling Mediates Virus-Specific CD8+ T Cell Deletion and Viral Persistence In Vivo. Immunity 2009, 31, 145–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerwenka, A.; Kovar, H.; Majdic, O.; Holter, W. Fas- and activation-induced apoptosis are reduced in human T cells preactivated in the presence of TGF-β1. J. Immunol. 1996, 156, 459–464. [Google Scholar] [PubMed]
- Liu, Y.; Zhang, P.; Li, J.; Kulkarni, A.B.; Perruche, S.; Chen, W. A critical function for TGF-β signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat. Immunol. 2008, 9, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Beckett, O.; Ma, Q.; Li, M.O. Transforming Growth Factor-β Signaling Curbs Thymic Negative Selection Promoting Regulatory T Cell Development. Immunity 2010, 32, 642–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Mucida, D.; Park, Y.; Kim, G.; Turovskaya, O.; Scott, I.; Kronenberg, M.; Cheroutre, H. Reciprocal TH17 and Regulatory T Cell Differentiation Mediated by Retinoic Acid. Science 2007, 317, 256–260. [Google Scholar] [CrossRef]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFβ in the Context of an Inflammatory Cytokine Milieu Supports De Novo Differentiation of IL-17-Producing T Cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef]
- Kryczek, I.; Wei, S.; Vatan, L.; Escara-Wilke, J.; Szeliga, W.; Keller, E.T.; Zou, W. Cutting edge: opposite effects of IL-1 and IL-2 on the regulation of IL-17+ T cell pool IL-1 subverts IL-2-mediated suppression. J. Immunol. 2007, 179, 1423–1426. [Google Scholar] [CrossRef]
- Gorelik, L.; Constant, S.; Flavell, R.A. Mechanism of Transforming Growth Factor β-induced Inhibition of T Helper Type 1 Differentiation. J. Exp. Med. 2002, 195, 1499–1505. [Google Scholar] [CrossRef]
- Gorelik, L.; Fields, P.E.; Flavell, R.A. Cutting edge: TGF-β inhibits Th type 2 development through inhibition of GATA-3 expression. J. Immunol. 2000, 165, 4773–4777. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.H.; Hufford, M.M.; Olson, M.R. The development and in vivo function of T helper 9 cells. Nat. Rev. Immunol. 2015, 15, 295–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Awasthi, A.; Yosef, N.; Quintana, F.J.; Xiao, S.; Peters, A.; Wu, C.; Kleinewietfeld, M.; Kunder, S.; Hafler, D.A.; et al. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 2012, 13, 991–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshioka, Y.; Ono, M.; Osaki, M.; Konishi, I.; Sakaguchi, S. Differential effects of inhibition of bone morphogenic protein (BMP) signalling on T-cell activation and differentiation. Eur. J. Immunol. 2012, 42, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.P.; Gregory, L.G.; Causton, B.; Campbell, G.A.; Lloyd, C.M. Activin A and TGF-β promote TH9 cell-mediated pulmonary allergic pathology. J. Allergy Clin. Immunol. 2012, 129, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Stahl, F.R.; Schrader, J.; Luth, S.; Presser, K.; Carambia, A.; Flavell, R.A.; Werner, S.; Blessing, M.; Herkel, J.; et al. Activin A Promotes the TGF-β-Induced Conversion of CD4+CD25− T Cells into Foxp3+ Induced Regulatory T Cells. J. Immunol. 2009, 182, 4633–4640. [Google Scholar] [CrossRef] [PubMed]
- Söderberg, S.S.; Karlsson, G.; Karlsson, S. Complex and Context Dependent Regulation of Hematopoiesis by TGF-β Superfamily Signaling. Ann. N. Y. Acad. Sci. 2009, 1176, 55–69. [Google Scholar] [CrossRef] [Green Version]
- Hinge, A.; Filippi, M.-D. Deconstructing the Complexity of TGFβ Signaling in Hematopoietic Stem Cells: Quiescence and Beyond. Curr. Stem Cell Rep. 2016, 2, 388–397. [Google Scholar] [CrossRef] [Green Version]
- Naka, K.; Hirao, A. Regulation of Hematopoiesis and Hematological Disease by TGF-β Family Signaling Molecules. Cold Spring Harb. Perspect. Biol. 2017, 9, a027987. [Google Scholar] [CrossRef]
- Singbrant, S.; Karlsson, G.; Ehinger, M.; Olsson, K.; Jaako, P.; Miharada, K.; Stadtfeld, M.; Graf, T.; Karlsson, S. Canonical BMP signaling is dispensable for hematopoietic stem cell function in both adult and fetal liver hematopoiesis, but essential to preserve colon architecture. Blood 2010, 115, 4689–4698. [Google Scholar] [CrossRef] [Green Version]
- Crisan, M.; Kartalaei, P.S.; Vink, C.S.; Yamada-Inagawa, T.; Bollerot, K.; van IJcken, W.; van der Linden, R.; de Sousa Lopes, S.M.C.; Monteiro, R.; Mummery, C.; et al. BMP signalling differentially regulates distinct haematopoietic stem cell types. Nat. Commun. 2015, 6, 8040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Challen, G.A.; Boles, N.C.; Chambers, S.M.; Goodell, M.A. Distinct Hematopoietic Stem Cell Subtypes Are Differentially Regulated by TGF-β1. Cell Stem Cell 2010, 6, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Ellingsworth, L.R.; Gillis, S.; Wall, R.; Kincade, P.W. Beta transforming growth factors are potential regulators of B lymphopoiesis. J. Exp. Med. 1987, 166, 1290–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehmann, J.A.; LeBien, T.W. Transforming growth factor-beta regulates normal human pre-B cell differentiation. Int. Immunol. 1994, 6, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Nuccie, B.L.; Ritterman, I.; Liesveld, J.L.; Abboud, C.N.; Ryan, D.H. TGF-β down-regulates stromal IL-7 secretion and inhibits proliferation of human B cell precursors. J. Immunol. 1997, 159, 117–125. [Google Scholar] [PubMed]
- Kersten, C.; Dosen, G.; Myklebust, J.H.; Sivertsen, E.A.; Hystad, M.E.; Smeland, E.B.; Rian, E. BMP-6 inhibits human bone marrow B lymphopoiesis—Upregulation of Id1 and Id3. Exp. Hematol. 2006, 34, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Shav-Tal, Y.; Zipori, D. The Role of Activin A in Regulation of Hemopoiesis. Stem Cells 2002, 20, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Zipori, D.; Barda-Saad, M. Role of activin A in negative regulation of normal and tumor B lymphocytes. J. Leukoc. Biol. 2001, 69, 867–873. [Google Scholar] [CrossRef]
- Shoham, T.; Parameswaran, R.; Shav-Tal, Y.; Barda-Saad, M.; Zipori, D. The mesenchymal stroma negatively regulates B cell lymphopoiesis through the expression of activin A. Ann. N. Y. Acad. Sci. 2003, 996, 245–260. [Google Scholar] [CrossRef]
- Massagué, J.; Blain, S.W.; Lo, R.S. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef]
- Kehrl, J.H.; Roberts, A.B.; Wakefield, L.M.; Jakowlew, S.; Sporn, M.B.; Fauci, A.S. Transforming growth factor β is an important immunomodulatory protein for human B lymphocytes. J. Immunol. 1986, 137, 3855–3860. [Google Scholar] [PubMed]
- Kehrl, J.H.; Taylor, A.S.; Delsing, G.A.; Roberts, A.B.; Sporn, M.B.; Fauci, A.S. Further studies of the role of transforming growth factor-beta in human B cell function. J. Immunol. 1989, 143, 1868–1874. [Google Scholar] [PubMed]
- Zhang, Y.; Alexander, P.B.; Wang, X.-F. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef] [PubMed]
- Petit-Koskas, E.; Génot, E.; Lawrence, D.; Kolb, J.-P. Inhibition of the proliferative response of human B lymphocytes to B cell growth factor by transforming growth factor-β. Eur. J. Immunol. 1988, 18, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Kehrl, J.H.; Taylor, A.; Kim, S.J.; Fauci, A.S. Transforming growth factor-β is a potent negative regulator of human lymphocytes. Ann. N. Y. Acad. Sci. 1991, 628, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Warner, G.L.; Ludlow, J.W.; Nelson, D.A.; Gaur, A.; Scott, D.W. Anti-immunoglobulin treatment of murine B-cell lymphomas induces active transforming growth factor beta but pRB hypophosphorylation is transforming growth factor beta independent. Cell Growth Differ. 1992, 3, 175–181. [Google Scholar] [PubMed]
- Smeland, E.B.; Blomhoff, H.K.; Holte, H.; Ruud, E.; Beiske, K.; Funderud, S.; Godal, T.; Ohlsson, R. Transforming growth factor type β (TGF β) inhibits G1 to S transition, but not activation of human B lymphocytes. Exp. Cell Res. 1987, 171, 213–222. [Google Scholar] [CrossRef]
- Bouchard, C.; Fridman, W.H.; Sautès, C. Effect of TGF-β1 on cell cycle regulatory proteins in LPS-stimulated normal mouse B lymphocytes. J. Immunol. 1997, 159, 4155–4164. [Google Scholar]
- Kamesaki, H.; Nishizawa, K.; Michaud, G.Y.; Cossman, J.; Kiyono, T. TGF-β1 Induces the Cyclin-Dependent Kinase Inhibitor p27Kip1 mRNA and Protein in Murine B Cells. J. Immunol. 1998, 160, 770–777. [Google Scholar]
- Kersten, C.; Sivertsen, E.A.; Hystad, M.E.; Forfang, L.; Smeland, E.B.; Myklebust, J.H. BMP-6 inhibits growth of mature human B cells; induction of Smad phosphorylation and upregulation of Id1. BMC Immunol. 2005, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Tsalavos, S.; Segklia, K.; Passa, O.; Petryk, A.; O’Connor, M.B.; Graf, D. Involvement of Twisted Gastrulation in T Cell-Independent Plasma Cell Production. J. Immunol. 2011, 186, 6860–6870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lømo, J.; Blomhoff, H.K.; Beiske, K.; Stokke, T.; Smeland, E.B. TGF-β1 and cyclic AMP promote apoptosis in resting human B lymphocytes. J. Immunol. 1995, 154, 1634–1643. [Google Scholar] [PubMed]
- Wildey, G.M.; Patil, S.; Howe, P.H. Smad3 Potentiates Transforming Growth Factor β (TGFβ)-induced Apoptosis and Expression of the BH3-only Protein Bim in WEHI 231 B Lymphocytes. J. Biol. Chem. 2003, 278, 18069–18077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saltzman, A.; Munro, R.; Searfoss, G.; Franks, C.; Jaye, M.; Ivashchenko, Y. Transforming Growth Factor-β-Mediated Apoptosis in the Ramos B-Lymphoma Cell Line Is Accompanied by Caspase Activation and Bcl-XL Downregulation. Exp. Cell Res. 1998, 242, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Spender, L.C.; O’Brien, D.I.; Simpson, D.; Dutt, D.; Gregory, C.D.; Allday, M.J.; Clark, L.J.; Inman, G.J. TGF-β induces apoptosis in human B cells by transcriptional regulation of BIK and BCL-XL. Cell Death Differ. 2009, 16, 593–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazac, B.B.; Roes, J. TGF-β Receptor Controls B Cell Responsiveness and Induction of IgA In Vivo. Immunity 2000, 13, 443–451. [Google Scholar] [CrossRef]
- Bollum, L.K.; Huse, K.; Oksvold, M.P.; Bai, B.; Hilden, V.I.; Forfang, L.; Yoon, S.O.; Wälchli, S.; Smeland, E.B.; Myklebust, J.H. BMP-7 induces apoptosis in human germinal center B cells and is influenced by TGF-β receptor type I ALK5. PLoS ONE 2017, 12, e0177188. [Google Scholar] [CrossRef]
- Taher, T.E.; Bystrom, J.; Ong, V.H.; Isenberg, D.A.; Renaudineau, Y.; Abraham, D.J.; Mageed, R.A. Intracellular B Lymphocyte Signalling and the Regulation of Humoral Immunity and Autoimmunity. Clin. Rev. Allergy Immunol. 2017, 53, 237–264. [Google Scholar] [CrossRef] [Green Version]
- Kehrl, J.H.; Thevenin, C.; Rieckmann, P.; Fauci, A.S. Transforming growth factor-β suppresses human B lymphocyte Ig production by inhibiting synthesis and the switch from the membrane form to the secreted form of Ig mRNA. J. Immunol. 1991, 146, 4016–4023. [Google Scholar]
- Roes, J.; Choi, B.K.; Cazac, B.B. Redirection of B cell responsiveness by transforming growth factor beta receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 7241–7246. [Google Scholar] [CrossRef]
- Snapper, C.M.; Waegell, W.; Beernink, H.; Dasch, J.R. Transforming growth factor-β1 is required for secretion of IgG of all subclasses by LPS-activated murine B cells in vitro. J. Immunol. 1993, 151, 4625–4636. [Google Scholar]
- Sanjabi, S.; Oh, S.A.; Li, M.O. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb. Perspect. Biol. 2017, 9, a022236. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Rosendahl, A.; Brodin, G.; Cheng, A.M.; Ahgren, A.; Sundquist, C.; Kulkarni, S.; Pawson, T.; Heldin, C.-H.; Heuchel, R.L. Deletion of exon I of SMAD7 in mice results in altered B cell responses. J. Immunol. 2006, 176, 6777–6784. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Ju, W.; Heyer, J.; Wittek, B.; Haneke, T.; Knaus, P.; Kucherlapati, R.; Böttinger, E.P.; Nitschke, L.; Kneitz, B. B Cell-Specific Deficiency for Smad2 In Vivo Leads to Defects in TGF-β-Directed IgA Switching and Changes in B Cell Fate. J. Immunol. 2006, 176, 2389–2396. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef] [PubMed]
- Huse, K.; Bakkebø, M.; Oksvold, M.P.; Forfang, L.; Hilden, V.I.; Stokke, T.; Smeland, E.B.; Myklebust, J.H. Bone morphogenetic proteins inhibit CD40L/IL-21-induced Ig production in human Bcells: Differential effects of BMP-6 and BMP-7. Eur. J. Immunol. 2011, 41, 3135–3145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, K.; Funaba, M.; Tsujimoto, M. A dual role of activin A in regulating immunoglobulin production of B cells. J. Leukoc. Biol. 2008, 83, 1451–1458. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-J.; Kim, P.-H. Further Characterization of Activin A-induced IgA Response in Murine B Lymphocytes. Immune Netw. 2009, 9, 133–137. [Google Scholar] [CrossRef]
- Pone, E.J.; Xu, Z.; White, C.A.; Zan, H.; Casali, P. B cell TLRs and induction of immunoglobulin class-switch DNA recombination. Front. Biosci. Landmark Ed. 2012, 17, 2594–2615. [Google Scholar] [CrossRef]
- Vinuesa, C.G.; Linterman, M.A.; Yu, D.; MacLennan, I.C.M. Follicular Helper T Cells. Annu. Rev. Immunol. 2016, 34, 335–368. [Google Scholar] [CrossRef]
- McCarron, M.J.; Marie, J.C. TGF-β prevents T follicular helper cell accumulation and B cell autoreactivity. J. Clin. Investig. 2014, 124, 4375–4386. [Google Scholar] [CrossRef] [Green Version]
- Marshall, H.D.; Ray, J.P.; Laidlaw, B.J.; Zhang, N.; Gawande, D.; Staron, M.M.; Craft, J.; Kaech, S.M. The transforming growth factor beta signaling pathway is critical for the formation of CD4 T follicular helper cells and isotype-switched antibody responses in the lung mucosa. eLife 2015, 4, e04851. [Google Scholar] [CrossRef]
- Schmitt, N.; Liu, Y.; Bentebibel, S.-E.; Munagala, I.; Bourdery, L.; Venuprasad, K.; Banchereau, J.; Ueno, H. The cytokine TGF-β co-opts signaling via STAT3-STAT4 to promote the differentiation of human TFH cells. Nat. Immunol. 2014, 15, 856–865. [Google Scholar] [CrossRef] [Green Version]
- Locci, M.; Wu, J.E.; Arumemi, F.; Mikulski, Z.; Dahlberg, C.; Miller, A.T.; Crotty, S. Activin A programs the differentiation of human TFH cells. Nat. Immunol. 2016, 17, 976–984. [Google Scholar] [CrossRef] [Green Version]
- Xu, A.; Liu, Y.; Chen, W.; Wang, J.; Xue, Y.; Huang, F.; Rong, L.; Lin, J.; Liu, D.; Yan, M.; et al. TGF-β–Induced Regulatory T Cells Directly Suppress B Cell Responses through a Noncytotoxic Mechanism. J. Immunol. 2016, 196, 3631–3641. [Google Scholar] [CrossRef]
- Wing, J.B.; Tekgüç, M.; Sakaguchi, S. Control of Germinal Center Responses by T-Follicular Regulatory Cells. Front. Immunol. 2018, 9, 1910. [Google Scholar] [CrossRef]
- Li, L.; Yang, S.-H.; Yao, Y.; Xie, Y.-Q.; Yang, Y.-Q.; Wang, Y.-H.; Yin, X.-Y.; Ma, H.-D.; Gershwin, M.; Lian, Z.-X. Block of both TGF-β and IL-2 signaling impedes Neurophilin-1+ regulatory T cell and follicular regulatory T cell development. Cell Death Dis. 2016, 7, e2439. [Google Scholar] [CrossRef]
- Okamura, T.; Sumitomo, S.; Morita, K.; Iwasaki, Y.; Inoue, M.; Nakachi, S.; Komai, T.; Shoda, H.; Miyazaki, J.; Fujio, K.; et al. TGF-β3-expressing CD4+CD25−LAG3+ regulatory T cells control humoral immune responses. Nat. Commun. 2015, 6, 6329. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, Y.; Sumitomo, S.; Ishigaki, K.; Suzuki, A.; Kochi, Y.; Tsuchiya, H.; Ota, M.; Komai, T.; Inoue, M.; Morita, K.; et al. TGF-β3 Inhibits Antibody Production by Human B Cells. PLoS ONE 2017, 12, e0169646. [Google Scholar] [CrossRef]
- Shah, S.; Qiao, L. Resting B cells expand a CD4+CD25+Foxp3+ Treg population via TGF-β3. Eur. J. Immunol. 2008, 38, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Molnarfi, N.; Bjarnadóttir, K.; Benkhoucha, M.; Juillard, C.; Lalive, P.H. Activation of human B cells negatively regulates TGF-β1 production. J. Neuroinflamm. 2017, 14, 13. [Google Scholar] [CrossRef]
- Dedobbeleer, O.; Stockis, J.; van der Woning, B.; Coulie, P.G.; Lucas, S. Cutting Edge: Active TGF-β1 Released from GARP/TGF-β1 Complexes on the Surface of Stimulated Human B Lymphocytes Increases Class-Switch Recombination and Production of IgA. J. Immunol. 2017, 199, 391–396. [Google Scholar] [CrossRef] [Green Version]
- Reboldi, A.; Arnon, T.I.; Rodda, L.B.; Atakilit, A.; Sheppard, D.; Cyster, J.G. IgA production requires B cell interaction with subepithelial dendritic cells in Peyers patches. Science 2016, 352, aaf4822. [Google Scholar] [CrossRef]
- Kelly, A.; Gunaltay, S.; McEntee, C.P.; Shuttleworth, E.E.; Smedley, C.; Houston, S.A.; Fenton, T.M.; Levison, S.; Mann, E.R.; Travis, M.A. Human monocytes and macrophages regulate immune tolerance via integrin αvβ8-mediated TGFβ activation. J. Exp. Med. 2018, 215, 2725–2736. [Google Scholar] [CrossRef]
- Fenton, T.M.; Kelly, A.; Shuttleworth, E.E.; Smedley, C.; Atakilit, A.; Powrie, F.; Campbell, S.; Nishimura, S.L.; Sheppard, D.; Levison, S.; et al. Inflammatory cues enhance TGFβ activation by distinct subsets of human intestinal dendritic cells via integrin αvβ8. Mucosal Immunol. 2017, 10, 624–634. [Google Scholar] [CrossRef]
- Basten, A.; Silveira, P.A. B-cell tolerance: mechanisms and implications. Curr. Opin. Immunol. 2010, 22, 566–574. [Google Scholar] [CrossRef]
- Tsubata, T. B-cell tolerance and autoimmunity [version 1; referees: 2 approved]. F1000Research 2017, 6, 391. [Google Scholar] [CrossRef]
- Wolf, S.D.; Dittel, B.N.; Hardardottir, F.; Janeway, C.A. Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J. Exp. Med. 1996, 184, 2271–2278. [Google Scholar] [CrossRef]
- Candando, K.M.; Lykken, J.M.; Tedder, T.F. B10 cell regulation of health and disease. Immunol. Rev. 2014, 259, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Berthelot, J.-M.; Jamin, C.; Amrouche, K.; Le Goff, B.; Maugars, Y.; Youinou, P. Regulatory B cells play a key role in immune system balance. Jt. Bone Spine 2013, 80, 18–22. [Google Scholar] [CrossRef]
- Tian, J.; Zekzer, D.; Hanssen, L.; Lu, Y.; Olcott, A.; Kaufman, D.L. Lipopolysaccharide-activated B cells down-regulate Th1 immunity and prevent autoimmune diabetes in nonobese diabetic mice. J. Immunol. 2001, 167, 1081–1089. [Google Scholar] [CrossRef]
- Parekh, V.V.; Prasad, D.V.R.; Banerjee, P.P.; Joshi, B.N.; Kumar, A.; Mishra, G.C. B Cells Activated by Lipopolysaccharide, But Not by Anti-Ig and Anti-CD40 Antibody, Induce Anergy in CD8+ T Cells: Role of TGF-β1. J. Immunol. 2003, 170, 5897–5911. [Google Scholar] [CrossRef]
- Lee, K.M.; Stott, R.T.; Zhao, G.; SooHoo, J.; Xiong, W.; Lian, M.M.; Fitzgerald, L.; Shi, S.; Akrawi, E.; Lei, J.; et al. TGF-β-producing regulatory B cells induce regulatory T cells and promote transplantation tolerance. Eur. J. Immunol. 2014, 44, 1728–1736. [Google Scholar] [CrossRef] [Green Version]
- Bjarnadóttir, K.; Benkhoucha, M.; Merkler, D.; Weber, M.S.; Payne, N.L.; Bernard, C.C.A.; Molnarfi, N.; Lalive, P.H. B cell-derived transforming growth factor-β1 expression limits the induction phase of autoimmune neuroinflammation. Sci. Rep. 2016, 6, 34594. [Google Scholar] [CrossRef] [Green Version]
- Komai, T.; Inoue, M.; Okamura, T.; Morita, K.; Iwasaki, Y.; Sumitomo, S.; Shoda, H.; Yamamoto, K.; Fujio, K. Transforming Growth Factor-β and Interleukin-10 Synergistically Regulate Humoral Immunity via Modulating Metabolic Signals. Front. Immunol. 2018, 9, 1364. [Google Scholar] [CrossRef]
- Wallace, C.H.; Wu, B.X.; Salem, M.; Ansa-Addo, E.A.; Metelli, A.; Sun, S.; Gilkeson, G.; Shlomchik, M.J.; Liu, B.; Li, Z. B lymphocytes confer immune tolerance via cell surface GARP-TGF-β complex. JCI Insight 2018, 3, e99863. [Google Scholar] [CrossRef]
- Kessel, A.; Haj, T.; Peri, R.; Snir, A.; Melamed, D.; Sabo, E.; Toubi, E. Human CD19+CD25high B regulatory cells suppress proliferation of CD4+ T cells and enhance Foxp3 and CTLA-4 expression in T-regulatory cells. Autoimmun. Rev. 2012, 11, 670–677. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, X.; Qin, M.; Wang, X. Changes in peripheral CD19+Foxp3+ and CD19+TGFβ+ regulatory B cell populations in rheumatoid arthritis patients with interstitial lung disease. J. Thorac. Dis. 2015, 7, 471–477. [Google Scholar] [CrossRef]
- Lee, J.H.; Noh, J.; Noh, G.; Choi, W.S.; Cho, S.; Lee, S.S. Allergen-Specific Transforming Growth Factor-β-Producing CD19(+)CD5(+) Regulatory B-Cell (Br3) Responses in Human Late Eczematous Allergic Reactions to Cow’s Milk. J. Interfaces Cytokine Res. 2011, 31, 441–449. [Google Scholar] [CrossRef]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Núñez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef]
- Kurashima, Y.; Kiyono, H. Mucosal Ecological Network of Epithelium and Immune Cells for Gut Homeostasis and Tissue Healing. Annu. Rev. Immunol. 2017, 35, 119–147. [Google Scholar] [CrossRef]
- Biancheri, P.; Giuffrida, P.; Docena, G.H.; MacDonald, T.T.; Corazza, G.R.; Di Sabatino, A. The role of transforming growth factor (TGF)-β in modulating the immune response and fibrogenesis in the gut. Cytokine Growth Factor Rev. 2014, 25, 45–55. [Google Scholar] [CrossRef]
- Coombes, J.L.; Siddiqui, K.R.R.; Arancibia-Cárcamo, C.V.; Hall, J.; Sun, C.-M.; Belkaid, Y.; Powrie, F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β– and retinoic acid–dependent mechanism. J. Exp. Med. 2007, 204, 1757–1764. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of Colonic Regulatory T Cells by Indigenous Clostridium Species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef]
- Perruche, S.; Zhang, P.; Liu, Y.; Saas, P.; Bluestone, J.A.; Chen, W. CD3-specific antibody–induced immune tolerance involves transforming growth factor-β from phagocytes digesting apoptotic T cells. Nat. Med. 2008, 14, 528–535. [Google Scholar] [CrossRef]
- Nowarski, R.; Jackson, R.; Flavell, R.A. The Stromal Intervention: Regulation of Immunity and Inflammation at the Epithelial-Mesenchymal Barrier. Cell 2017, 168, 362–375. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Bäckhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 2016, 22, 1079–1089. [Google Scholar] [CrossRef]
- Lee, G.R. The balance of Th17 versus Treg Cells in autoimmunity. Int. J. Mol. Sci. 2018, 19, 730. [Google Scholar] [CrossRef]
- Mantis, N.J.; Rol, N.; Corthésy, B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. 2011, 4, 603–611. [Google Scholar] [CrossRef]
- Park, S.R.; Lee, J.H.; Kim, P.H. Smad3 and Smad4 mediate transforming growth factor-beta1-induced IgA expression in murine B lymphocytes. Eur. J. Immunol. 2001, 31, 1706–1715. [Google Scholar] [CrossRef]
- Lin, Y.C.; Stavnezer, J. Regulation of transcription of the germ-line Ig alpha constant region gene by an ATF element and by novel transforming growth factor-beta 1-responsive elements. J. Immunol. 1992, 149, 2914–2925. [Google Scholar]
- Zhang, Y.; Derynck, R. Transcriptional regulation of the transforming growth factor-β-inducible mouse germ line Ig alpha constant region gene by functional cooperation of Smad, CREB, and AML family members. J. Biol. Chem. 2000, 275, 16979–16985. [Google Scholar] [CrossRef]
- Gutzeit, C.; Magri, G.; Cerutti, A. Intestinal IgA production and its role in host-microbe interaction. Immunol. Rev. 2014, 260, 76–85. [Google Scholar] [CrossRef] [Green Version]
- Iliev, I.D.; Spadoni, I.; Mileti, E.; Matteoli, G.; Sonzogni, A.; Sampietro, G.M.; Foschi, D.; Caprioli, F.; Viale, G.; Rescigno, M. Human intestinal epithelial cells promote the differentiation of tolerogenic dendritic cells. Gut 2009, 58, 1481–1489. [Google Scholar] [CrossRef]
- Scott, C.L.; Aumeunier, A.M.; Mowat, A.M. Intestinal CD103+ dendritic cells: master regulators of tolerance? Trends Immunol. 2011, 32, 412–419. [Google Scholar] [CrossRef]
- Sun, C.-M.; Hall, J.A.; Blank, R.B.; Bouladoux, N.; Oukka, M.; Mora, J.R.; Belkaid, Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 2007, 204, 1775–1785. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, M.; Komatsu, N.; Kawamoto, S.; Suzuki, K.; Kanagawa, O.; Honjo, T.; Hori, S.; Fagarasan, S. Preferential Generation of Follicular B Helper T Cells from Foxp3+ T Cells in Gut Peyer’s Patches. Science 2009, 323, 1488–1492. [Google Scholar] [CrossRef]
- Hirota, K.; Turner, J.-E.; Villa, M.; Duarte, J.H.; Demengeot, J.; Steinmetz, O.M.; Stockinger, B. Plasticity of TH17 cells in Peyer’s patches is responsible for the induction of T cell–dependent IgA responses. Nat. Immunol. 2013, 14, 372–379. [Google Scholar] [CrossRef]
- Uematsu, S.; Fujimoto, K.; Jang, M.H.; Yang, B.-G.; Jung, Y.-J.; Nishiyama, M.; Sato, S.; Tsujimura, T.; Yamamoto, M.; Yokota, Y.; et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat. Immunol. 2008, 9, 769–776. [Google Scholar] [CrossRef]
- Seo, G.-Y.; Jang, Y.-S.; Kim, H.-A.; Lee, M.-R.; Park, M.-H.; Park, S.-R.; Lee, J.-M.; Choe, J.; Kim, P.-H. Retinoic acid, acting as a highly specific IgA isotype switch factor, cooperates with TGF-β1 to enhance the overall IgA response. J. Leukoc. Biol. 2013, 94, 325–335. [Google Scholar] [CrossRef]
- Carlino, J.A.; Higley, H.R.; Creson, J.R.; Avis, P.D.; Ogawa, Y.; Ellingsworth, L.R. Transforming growth factor β1 systemically modulates granuloid, erythroid, lymphoid, and thrombocytic cells in mice. Exp. Hematol. 1992, 20, 943–950. [Google Scholar]
- Onichtchouk, D.; Chen, Y.-G.; Dosch, R.; Gawantka, V.; Delius, H.; Massagué, J.; Niehrs, C. Silencing of TGF-β signalling by the pseudoreceptor BAMBI. Nature 1999, 401, 480–485. [Google Scholar] [CrossRef]
- Postigo, J.; Iglesias, M.; Álvarez, P.; Jesús Augustin, J.; Buelta, L.; Merino, J.; Merino, R. Bone Morphogenetic Protein and Activin Membrane-Bound Inhibitor, a Transforming Growth Factor β Rheostat That Controls Murine Treg Cell/Th17 Cell Differentiation and the Development of Autoimmune Arthritis by Reducing Interleukin-2 Signaling. Arthritis Rheumatol. 2016, 68, 1551–1562. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Ligand | Type I Receptor | Type II Receptor | R-Smad | Co-Smad | I-Smad |
|---|---|---|---|---|---|
| TGFβ1, TGFβ2, TGFβ3 | ALK-5 | TβRII | Smad-2, -3 | Smad-4 | Smad-7 |
| BMP-2 to 7, BMP-8A, BMP-8B, BMP-9, BMP-10 | ALK-1, -2, -3, -6 | BMPRII/IIB ActRII/IIB | Smad-1, -5, -8 | Smad-4 | Smad-6, -7 |
| Activins | ALK-4 (ActRIB), ALK-7 | ActRII/IIB | Smad-2, -3 | Smad-4 | Smad-7 |
| GDFs | ALK-2, -3, -6 | BMPRII, ActRIIA/B | Smad-1, -5, -8 | Smad-4 | Smad-6, -7 |
| ALK-4, -5, -7 | Smad-2, -3 | ||||
| Nodal | ALK-4, -7 | ActRII/IIB | Smad-2, -3 | Smad-4 | Smad-7 |
| AMH | ALK-2, -3, -6 | AMHRII | Smad-1, -5, -8 | Smad-4 | Smad-6, -7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamayo, E.; Alvarez, P.; Merino, R. TGFβ Superfamily Members as Regulators of B Cell Development and Function—Implications for Autoimmunity. Int. J. Mol. Sci. 2018, 19, 3928. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123928
Tamayo E, Alvarez P, Merino R. TGFβ Superfamily Members as Regulators of B Cell Development and Function—Implications for Autoimmunity. International Journal of Molecular Sciences. 2018; 19(12):3928. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123928
Chicago/Turabian StyleTamayo, Esther, Pilar Alvarez, and Ramón Merino. 2018. "TGFβ Superfamily Members as Regulators of B Cell Development and Function—Implications for Autoimmunity" International Journal of Molecular Sciences 19, no. 12: 3928. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123928