Potential Role of Fluoride in the Etiopathogenesis of Alzheimer’s Disease

 , , , , and
, , , , and 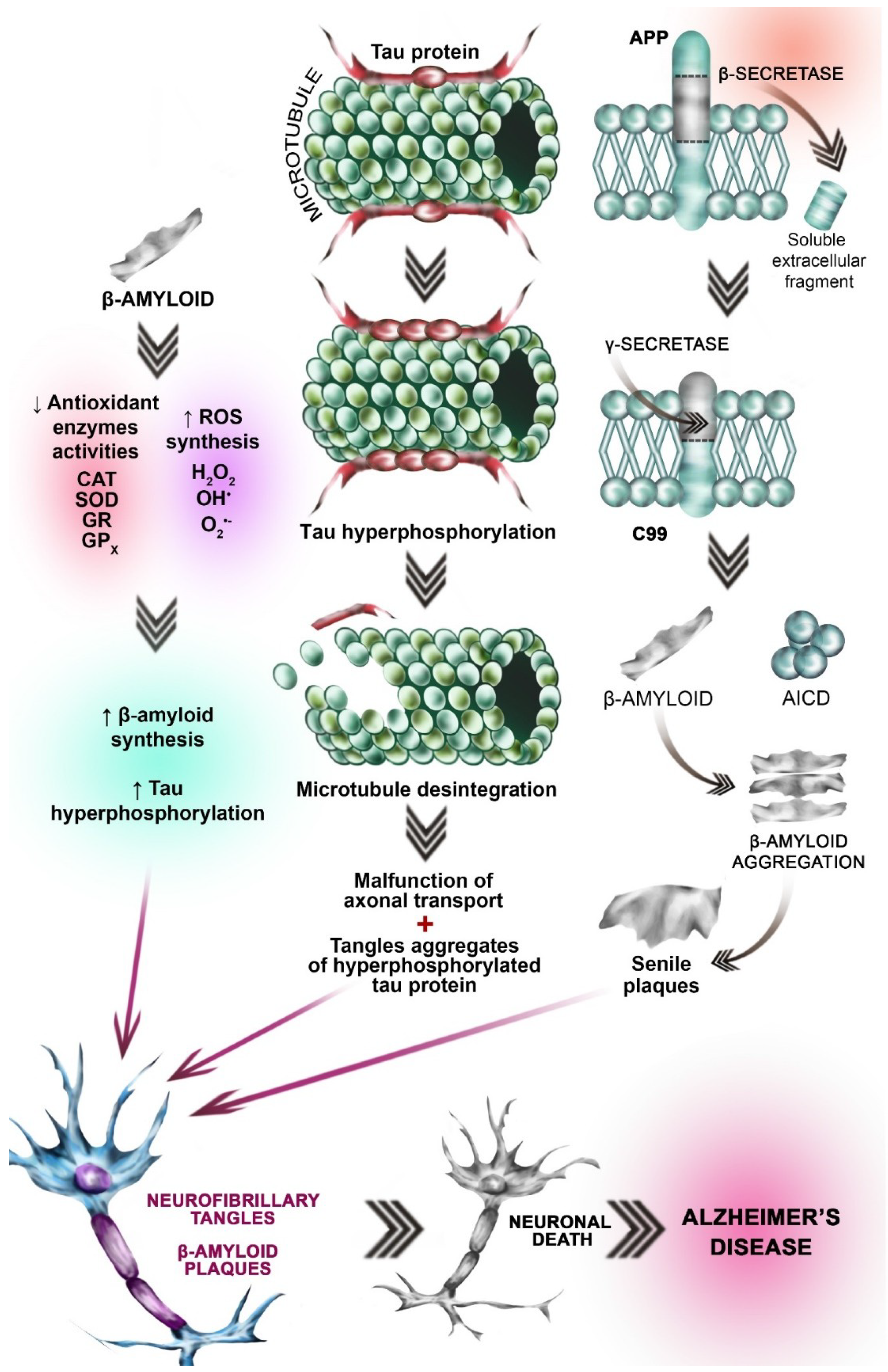{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Role of Environmental Factors in AD Etiopathogenesis
3. Neurobiological Processes Leading to AD
3.1. Cholinergic Hypothesis
3.2. Glutamatergic Hypothesis
3.3. Oxidative Stress Hypothesis
3.4. Amyloid β Aggregation Hypothesis
3.5. Tau (τ) Protein Hyperphosphorylation Hypothesis
4. Fluoride as a Neurotoxic Agent
5. The Role of Fluoride in the Pathogenesis of Alzheimer’s Disease
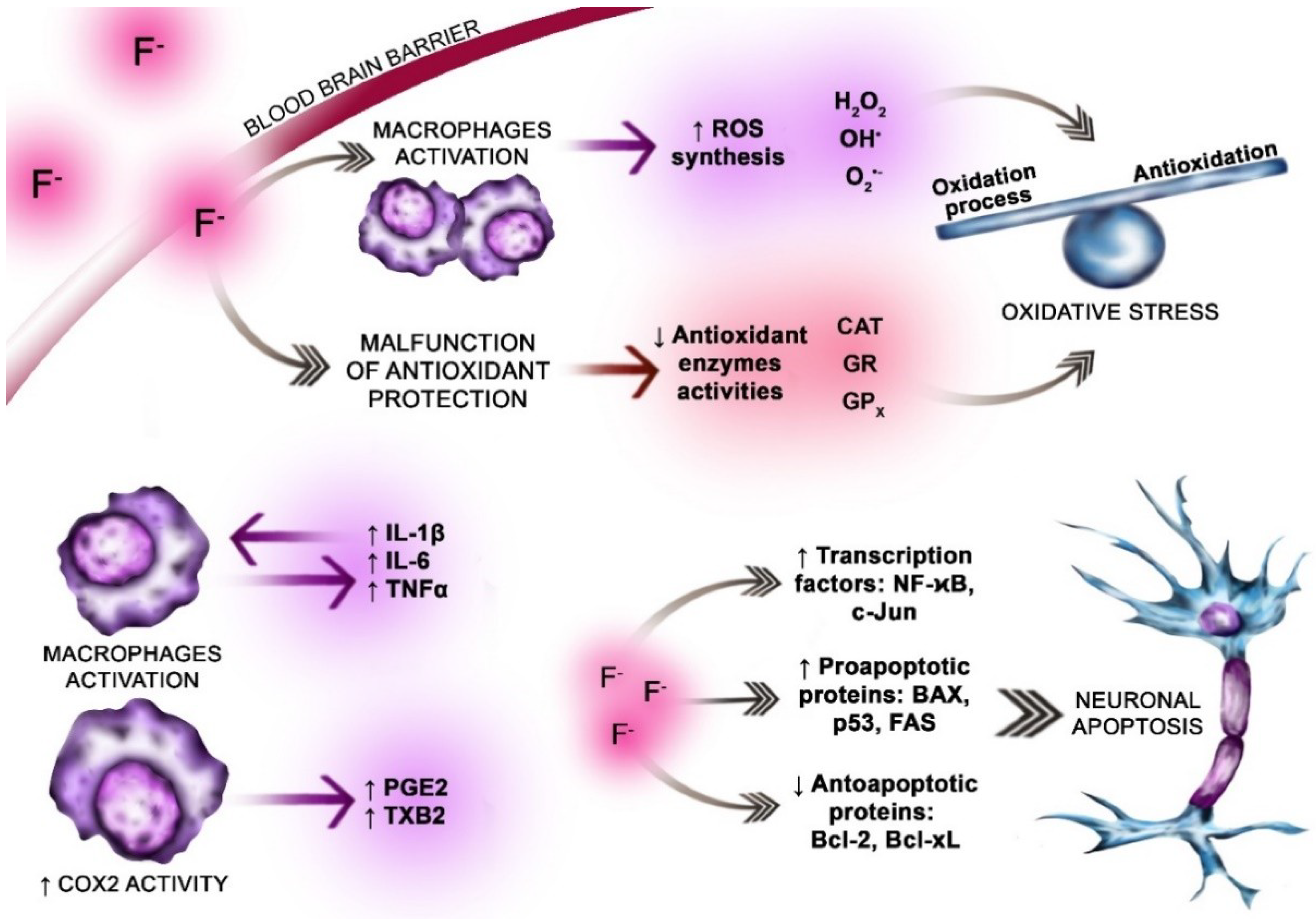
5.1. Oxidative Stress
5.2. The Role of Fluoride
6. Inflammation in AD
6.1. Fluoride vs. Nuclear Factor κB (NF-κB)
6.2. Fluoride vs. Proinflammatory Cytokines
6.3. Fluoride vs. Neuroinflammation Enzymes
6.4. Fluoride vs. Neuroapoptosis
6.5. Fluoride Vs. Mitophagy
6.6. Potential Roles of Alterations in Zinc and Magnesium Concentrations in Relation to Fluoride-Induced Neurodegeneration
6.7. Potential Roles of Xenometals and Xenobiotics in Relation to Fluoride-Induced Neurodegeneration
7. Conclusions
8. Perspectives for Further Research
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hampela, H.; Toschie, N.; Babilonih, C.; Baldaccia, F.; Blackk, K.L.; Bokdel, A.L.W.; Buna, R.S.; Cacciolam, F.; Cavedoa, E.; Chiesaa, P.A.; et al. Revolution of Alzheimer Precision Neurology: Passageway of Systems Biology and Neurophysiology. J. Alzheimers Dis. 2018, 64, S47–S105. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic changes with cognitive status: A review of literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Mantzavinos, V.; Alexiou, A. Biomarkers for Alzheimer’s Disease Diagnosis. Curr. Alzheimer Res. 2017, 14, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Sanabria-Castro, A.; Alvarado-Echeverria, I.; Monge-Bonilla, C. Molecular pathogenesis of Alzheimer’s disease—An update. Ann. Neurosci. 2017, 24, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Campdelacreu, J. Parkinson’s disease and Alzheimer disease: Environmental risk factors. Neurologia 2014, 29, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.B.; Campbell, A.; Itzhaki, R.F.; Savory, J. The significance of environmental factors in etiology of Alzheimer’s disease. J. Alzheimers Dis. 2002, 4, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J. Aluminium alters the permeability of the blood-brain barier to some non-peptides. Neuropharmacology 1985, 24, 407–412. [Google Scholar] [CrossRef]
- Szyperska, A.; Gutowska, I.; Machoy-Mokrzyńska, A.; Rak, J.; Baranowska-Bosiacka, I.; Machoy, Z. A study on hypothesis linking aluminium fluoride to Alzheimers disease: The affinity of amino acids occuring in beta-amyloid to [Al(H2O)6]3+. Fluoride 2017, 50, 468–474. [Google Scholar]
- Martin, R.B. Ternary hydroxide complexes in neutral solutions of Al3+ and F−. Biochem. Biophys. Res. Commun. 1988, 155, 1194–1200. [Google Scholar] [CrossRef]
- Lubkowska, A.; Żyluk, B.; Chlubek, D. Interaction between fluorine and aluminum. Fluoride 2002, 35, 73–77. [Google Scholar]
- Bartus, R.T.; Dean, R.L., 3rd; Beer, B.; Lippa, A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Kaufer, D. Neuropsychiatric aspects of Alzheiemr’s disease. The cholinergic hypothesis revisited. Neurology 1996, 47, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Terry, A.V., Jr.; Buccafusco, J.J. The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: Recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, E.L.; Gattaz, W.F. Cholinergic and glutamatergic alterations beginning at the early stages of Alzheimer disease: Participation of the phospholipase A2 enzyme. Psychopharmacology 2008, 198, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Kato, I.; Koizuka, I. Retrograde-labeling of pretecto-vestibular pathways in cats. Auris Nasus Larynx 2003, 30, S35–S40. [Google Scholar] [CrossRef]
- Ni, R.; Marutle, A.; Nordberg, A. Modulation of α7 nicotinic acetylcholine receptor and fibrillar amyloid-β interactions in Alzheimer’s disease brain. J. Alzheimers Dis. 2013, 33, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Zahid, S.; Mahboob, A.; Farhat, S.M. Cholinergic System and Post-translational Modifications: An Insight on the role in Alzheimer Disease. Curr. Neuropharmacol. 2017, 15, 480–494. [Google Scholar] [CrossRef]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Revett, T.J.; Baker, G.B.; Jhamandas, J.; Kar, S. Glutamate system, amyloid β peptides and tau protein: Functionalinterrelationships and relevance to Alzheimer disease pathology. J. Psychiatry Neurosci. 2013, 38, 6–23. [Google Scholar] [CrossRef]
- Szado, T.; Vanderheyden, V.; Parys, J.B.; De Smedt, H.; Rietdorf, K.; Kotelevets, L.; Chastre, E.; Khan, F.; Landegren, U.; Söderberg, O.; et al. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2427–2432. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, G.G.; Pacheco Moisés, F.P.; Mireles-Ramírez, M.; Flores-Alvarado, L.J.; González-Usigli, H.; Sánchez-González, V.J.; Sánchez-López, A.L.; Sánchez-Romero, L.; Díaz-Barba, E.I.; Santoscoy-Gutiérrez, J.F.; et al. Oxidative Stress: Love and Hate History in Central Nervous System. Adv. Protein Chem. Struct. Biol. 2017, 108, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human diseases. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M. Oxidative stress in Alzheimer disease as a target for therapy. Bratisl. Lek. Listy 2018, 119, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Hua, J.; Wang, K.; Zhang, H.; Zhang, C.; He, Y.; Guo, Z.; Wang, X. Modulating conformation of Aβ-peptide: An effective way to prevent protein-misfolding disease. Inorg. Chem. 2018, 57, 13533–13543. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid peptide cascade. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Doody, R.; Clark, C. Diseasemodifying therapies for Alzheimer disease: Challenges to early intervention. Neurology 2007, 69, 1622–1634. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef]
- Lindwall, G.; Cole, R.D. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar]
- Gong, C.X.; Iqbal, K. Hyperphosphorylation of microtubule-associated protein tau: A promising therapeutic target for Alzheimer disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Köpke, E.; Tung, Y.C.; Shaikh, S.; Alonso, A.C.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar]
- Kuret, J.; Congdon, E.E.; Li, G.; Yin, H.; Yu, X.; Zhong, Q. Evaluating triggers and enhancers of tau fibrillization. Microsc. Res. Tech. 2005, 67, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhao, L.D.; Liu, H.; Li, H.H.; Ren, C.; Zhang, P.; Guo, K.T.; Zhang, H.X.; Geng, D.Q.; Zhang, C.Y. Fluoride induces neuroinflammation and alters Wnt signaling pathway in BV2 microglial cells. Inflammation 2017, 40, 1123–1130. [Google Scholar] [CrossRef]
- Geeraert, F. Kinetics of fluoride penetration in liver and brain. Fluoride 1986, 19, 108–112. [Google Scholar]
- Du, L.; Wan, C.; Cao, X.; Liu, J. The effect of fluorine on the developing human brain. Chin. J. Pathol. 1992, 21, 218–220. [Google Scholar]
- Trivedi, M.H.; Verma, R.J.; Chinoy, N.J. Mitigation of sodium-fluoride induced toxicity in mice brain by black tea infusion. Fluoride 2009, 42, 29–33. [Google Scholar]
- Tang, Q.Q.; Du, J.; Ma, H.H.; Jiang, S.J.; Zhou, X.J. Fluoride and children’s intelligence, a meta-analysis. Biol. Trace Elem. Res. 2008, 126, 115–120. [Google Scholar] [CrossRef]
- Spittle, B. Psychopharmacology of fluoride: A review. Int. Clin. Psychopharmacol. 1994, 9, 79–82. [Google Scholar] [CrossRef]
- Choi, A.L.; Sun, G.; Zhang, Y.; Grandjean, P. Developmental fluoride neurotoxicity: A systematic review and meta-analysis. Environ. Health Perspect. 2012, 120, 1362–1368. [Google Scholar] [CrossRef]
- Ma, J.; Liu, F.; Liu, P.; Dong, Y.Y.; Chu, Z.; Hou, T.Z.; Dang, Y.H. Impact of early developmental fluoride exposure on the peripheral pain sensitivity in mice. Int. J. Dev. Neurosci. 2015, 47, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Goschorska, M.; Gutowska, I.; Olszewska, M.; Baranowska-Bosiacka, I.; Olszowski, T.; Rać, M.; Chlubek, D. Effect of sodium fluoride on catalase activity in THP-1 macrophages. Fluoride 2015, 48, 274–282. [Google Scholar]
- Ghosh, J.; Das, J.; Manna, P.; Sil, P.C. Cytoprotective effect of arjunolic acid in response to sodium fluoride mediated oxidative stress and cell death via necrotic pathway. Toxicol. In Vitro 2008, 22, 1918–1926. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, A.; He, W.; He, P.; Xu, B.; Xia, T.; Chen, X.; Yang, K. Effects of fluoride on the expression of NCAM, oxidative stress and apoptosis in primary cultured hippocampal neurons. Toxicology 2007, 236, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Dec, K.; Łukomska, A.; Maciejewska, D.; Jakubczyk, K.; Baranowska-Bosiacka, I.; Chlubek, D.; Wąsik, A.; Gutowska, I. The influence of fluorine on the disturbances of homeostasis in the central nervous system. Biol. Trace Elem. Res. 2017, 177, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004, 10, S18–S25. [Google Scholar] [CrossRef]
- Shi, Q.; Gibson, G.E. Oxidative stress and transcriptional regulation in Alzheiemer’s disease. Alzheimer Dis. Assoc. Disord. 2007, 21, 276–291. [Google Scholar] [CrossRef]
- Driver, A.S.; Kodavanti, P.R.; Mundy, W.R. Age-realted changes in reactive oxygen species production in rat brains homogenates. Neurotoxicol. Teratol. 2000, 22, 175–181. [Google Scholar] [CrossRef]
- Barja, G. Free radicals and aging. Trends Neurosci. 2004, 27, 595–600. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Castegna, A.; Lauderback, A.; LAuderback, C.M.; Drake, J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol. Aging 2002, 23, 655–664. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Lovell, M.A. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol. Aging 1998, 19, 33–36. [Google Scholar] [CrossRef]
- Ansari, M.A.; Scheff, S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [CrossRef]
- Gałecka, E.; Jacewicz, R.; Mrowicka, M.; Florkowski, A.; Gałecki, P. Antioxidative enzymes—Structure, properties, functions. Polski Merkuriusz Lekarski Organ Polskiego Towarzystwa Lekarskiego 2008, 28, 266–268. [Google Scholar]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Kitao, T.; Kisino, T.; Yamamuro, A.; Maeda, S. Nitric oxide protects macrophages from hydrogen peroxide—Induced apoptosis by inducing the formation of catalase. J. Immunol. 2006, 176, 4675–4681. [Google Scholar] [CrossRef] [PubMed]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative stress in neurodegeneratice diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Xie, C.; Markesbery, W.R. Decreased glutathionetransferase activity in brain and ventricular fluid in Alzheimer’s disease. Neurology 1998, 51, 1562–1566. [Google Scholar] [CrossRef]
- Casado, Á.; Encarnación López-Fernández, M.; Concepción Casado, M.; De La Torre, R. Lipid peroxidation and antioxidant enzyme activities in vascular and Alzheimer dementias. Neurochem. Res. 2008, 33, 450–458. [Google Scholar] [CrossRef]
- Marcus, D.L.; Thomas, C.; Rodriguez, C.; Simberkoff, K.; Tsai, J.S.; Strafaci, J.A.; Freedman, M.L. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer’s disease. Exp. Neurol. 1998, 150, 40–44. [Google Scholar] [CrossRef]
- Gatta, L.; Cardinale, A.; Wannenes, F.; Consoli, C.; Armani, A.; Molinari, F.; Mammi, C.; Stocchi, F.; Torti, M.; Rosano, G.M.; et al. Peripheral blood mononuclear cells from mild cognitive impairment patients show deregulation of Bax and Sod1 mRNAs. Neurosci. Lett. 2009, 453, 36–40. [Google Scholar] [CrossRef]
- Goschorska, M.; Giewoń, K.; Hasior, N.; Gutowska, I.; Baranowska-Bosiacka, I.; Rać, M.; Chlubek, D. The protective properties of selected naturally occurring antioxidants of plant origin against fluoride-induced neurotoxicity. Fluoride 2017, 50, 203–212. [Google Scholar]
- Chlubek, D. Fluoride and oxidative stress. Fluoride 2003, 36, 217–228. [Google Scholar]
- Gutowska, I.; Baranowska-Bosiacka, I.; Goschorska, M.; Kolasa, A.; Łukomska, A.; Jakubczyk, K.; Dec, K.; Chlubek, D. Fluoride as a factor initiating and potentiating inflammation in monocytes/macrophages. Toxicol. In Vitro 2015, 29, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Gutowska, I.; Baranowska-Bosiacka, I.; Baśkiewicz, M.; Millo, B.; Siennicka, A.; Marchlewicz, M.; Wiszniewska, B.; Machaliński, B.; Stachowska, E. Fluoride as a pro-inflammatory factor and inhibitor of ATP bioavailability in differentiated human THP1 monocytic cells. Toxicol. Lett. 2010, 196, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Shuhua, X.; Ziyou, L.; Ling, Y.; Fei, W.; Sun, G. A role of fluoride on free radical generation and oxidative stress in BV-2 microglia cells. Mediat. Inflamm. 2012, 2012, 8. [Google Scholar] [CrossRef]
- Saralakumari, D.; Rao, P.R. Red blood cell glucose metabolism in human chronic fluoride toxicity. Bull. Environ. Contam. Toxicol. 1991, 47, 834–839. [Google Scholar] [CrossRef]
- Güner, S.; Uvar-Bozkurt, S.; Haznedaroğlu, E.; Menteş, A. Dental fluorosis and catalase immunoreactivity of the brain tissues in rats exposed to high fluoride pre- and postnatally. Biol. Trace Elem. Res. 2016, 174, 150–157. [Google Scholar] [CrossRef]
- Pal, S.; Sarkar, C. Protective effect of resveratrol on fluoride induced alteration in protein and nucleic acid metabolism, DNA damage and biogenic amines in rat brain. Environ. Toxicol. Pharmacol. 2014, 38, 684–699. [Google Scholar] [CrossRef]
- Goschorska, M.; Baranowska-Bosiacka, I.; Gutowska, I.; Piotrowska, K.; Metryka, E.; Safranow, K.; Chlubek, D. Effect of acetylcholinesterase inhibitors donepezil and rivastigmine on the activity and expression of cyclooxygenases in a model of the inflammatory action of fluoride on macrophages obtained from the THP-1 monocytes. Toxicology 2018, 406–407, 9–20. [Google Scholar] [CrossRef]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [Green Version]
- Eikelenboom, P.; Rozemuller, A.J.M.; Hoozemans, J.J.M.; Veerhuis, R.; van Gool, W.A. Neuroinflammation and Alzheimer disease: Clinical and therapeutic implications. Alzheimer Dis. Assoc. Disord. 2000, 14, S54–S61. [Google Scholar] [CrossRef] [PubMed]
- Szekely, C.A.; Thorne, J.E.; Zandi, P.P.; Ek, M.; Messias, E.; Breitner, J.C.; Goodman, S.N. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease: A systematic review. Neuroepidemiology 2004, 23, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. An-Inflammation—Centric View of Neurological Disease: Beyond the neuron. Neuron. Front. Cell. Neurosci. 2018, 12, 72. [Google Scholar] [CrossRef] [PubMed]
- Orellana, A.M.; Vasconcelos, A.R.; Leite, J.A.; de Sá Lima, L.; Andreotti, D.Z.; Munhoz, C.D.; Kawamoto, E.M.; Scavone, C. Age-related neuroinflammation and changes in AKT-GSK-3beta and WNT/beta-CATENIN signaling in rat hippocampus. Aging 2015, 7, 1094–1111. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Zhang, Y.; Bouchard, R.; Mahaffey, G. Induction of an inflammatory loop by interleukin-1 beta and tumor necrosis factoralpha involves NF-kappa B and STAT-1 in differentiated human neuroprogenitor cells. PLoS ONE 2013, 8, e69585. [Google Scholar] [CrossRef] [PubMed]
- Simmons, L.J.; Surles-Zeigler, M.C.; Li, Y.; Ford, G.D.; Newman, G.D.; Ford, B.D. Regulation of inflammatory responses by neuregulin-1 in brain ischemia and microglial cells in vitro involves the NF-kappa B pathway. J. Neuroinflamm. 2016, 13, 237. [Google Scholar] [CrossRef]
- Zhang, F.; Jiang, L. Neuroinflammation in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2015, 11, 243–255. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, W.J.; Xu, X.H.; Zhang, Z.G. Effect of fluoride on calcium ion concentration and expression of nuclear transcription factor kappa-B ρ65 in rat hippocampus. Exp. Toxicol. Pathol. 2011, 63, 407–411. [Google Scholar] [CrossRef]
- Goldman, R.; Zor, U. On the mechanism of activation of PLA2 and Ptdlns-PLC by fluorine in murine macrophages. Adv. Prostag. Thromb. Leuktr. Res. 1995, 23, 93–97. [Google Scholar]
- Kamboh, M.I.; Sanghera, D.K.; Ferell, R.E.; DeKosky, S.T. ApoE4-associated Alzheimer’s disease risk is modified by α1-antichymotrypsin polymorphism. Nat. Genet. 1995, 10, 486–488. [Google Scholar] [CrossRef]
- Papassotiropoulos, A.; Bagli, M.; Jessen, F.; Bayer, T.A.; Mayer, W.; Rao, M.L.; Heun, R. A genetic variation of the inflammatory cytokine interleukin-6 delays the initial onset and reduces the risk of sporadic Alzheimer’s disease. Ann. Neurol. 1999, 45, 666–668. [Google Scholar] [CrossRef]
- McCusker, S.M.; Curran, M.D.; Dynan, K.B.; McCullagh, C.D.; Urquhart, D.D.; Middleton, D.; Patterson, C.C.; McIlroy, S.P.; Passmore, A.P. Association between polymorphism in regulatory region of gene encoding tumour necrosis factor alpha and risk of Alzheimer’s disease and vascular dementia. Lancet 2001, 357, 436–439. [Google Scholar] [CrossRef]
- Nicoll, J.A.; Mrak, R.E.; Graham, D.I.; Stewart, J.; Wilcock, G.; MacGowan, S.; Esiri, M.M.; Murray, L.S.; Dewar, D.; Love, S.; et al. Association of interleukin-1 gene polymorphisms with Alzheimer’s disease. Ann. Neurol. 2000, 47, 365–368. [Google Scholar] [CrossRef]
- Takeda, S.; Sato, N.; Ikimura, K.; Nishino, H.; Rakugi, H.; Morishita, R. Increased blood–brain barrier vulnerability to systemic inflammation in an Alzheimer disease mouse model. Neurobiol. Aging 2013, 34, 2064–2070. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Shaftel, S.S.; Griffin, W.S.; O’Banion, M.K. He role of interleukin-1 in neuroinflammation and Alzheimer Disease:an evolving perspective. J. Neuroinflamm. 2008, 5, 7. [Google Scholar] [CrossRef]
- Wang, H.U.; Zhou, B.H.; Cao, J.W.; Zhao, J.; Zhao, W.P.; Tan, P.P. Pro-inflammatory cytokines are involved in fluoride-induced cytotoxity potential in HeLa cells. Biol. Trace Elem. Res. 2017, 175, 98–102. [Google Scholar] [CrossRef]
- Yan, N.; Liu, Y.; Liu, S.; Cao, S.; Wang, F.; Wang, Z.; Xi, S. Fluoride-induced neuron apoptosis and expressions of inflammatory factors by activating microglia in rats brain. Mol. Neurobiol. 2016, 53, 4449–4460. [Google Scholar] [CrossRef]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Glial cell reactions in neurodegenerative diseases: Pathophysiology and therapeutic interventions. Alzheimer Dis. Assoc. Disord. 1998, 12, S1–S6. [Google Scholar] [CrossRef]
- Sun, G.Y.; Horrocks, L.A.; Farooqui, A.A. The roles of NADPH oxidase and phospholipases A2 in oxidative and inflammatory responses in neurodegenerative diseases. J. Neurochem. 2007, 103, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chalimoniuk, M. Sekrecyjna fosfolipaza A2-udział w stresie oksydacyjnym i stanach zapalnych. Postępy Biochemii 2012, 58, 204–208. [Google Scholar] [PubMed]
- Gutowska, I.; Baranowska-Bosiacka, I.; Siennicka, A.; Baśkiewicz, M.; Machaliński, B.; Stachowska, E.; Chlubek, D. Fluoride and generation of proinflammatory factors in human macrophages. Fluoride 2011, 44, 125–134. [Google Scholar]
- Wessel, K.; Resch, K.; Kaever, V. Aluminium fluoride enhances phospholipase A2 activity and eicosanoid synthesis in macrophages. Eicosanoids 1989, 2, 223–227. [Google Scholar] [PubMed]
- Chalbot, S.; Zetterberg, H.; Blennow, K.; Fladby, T.; Grundke-Iqbal, I.; Iqbal, K. Cerebrospinal fluid secretory Ca2+-dependent phospholipase A2 activity is increased in Alzheimer disease. Clin. Chem. 2009, 55, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.T.; Lemere, C.A.; Selkoe, D.J.; Clemens, J.A. Cytosolic phospholipase A2 (PLA2) immunoreactivity is elevated in Alzheiemer’s disease brain. Neurobiol Dis. 1996, 3, 51–63. [Google Scholar] [CrossRef]
- Stephenson, D.; Rash, K.; Smalstig, B.; Roberts, E.; Johnstone, E.; Sharp, J.; Panetta, J.; Little, S.; Kramer, R.; Clemens, J. Cytosolic phospholipase A2 is induced in reactive glia following different forms of neurodegeneration. Glia 1999, 27, 110–128. [Google Scholar] [CrossRef]
- Simonyi, A.; He, Y.; Sheng, W.; Sun, A.Y.; Wood, W.G.; Weisman, G.A.; Sun, G.Y. Targeting NADPH oxidase and phospholipases A2 in Alzheimer’s disease. Mol. Neurobiol. 2010, 41, 73–86. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, A.; Xia, T.; He, P. Effects of fluoride onDNA damage, S-phase cell cycle arrest and the expression of NFkappaB in primary cultured rat hippocampal neurons. Toxicol. Lett. 2008, 179, 1–5. [Google Scholar] [CrossRef]
- Rao, J.S.; Kelshian, V.L.; Klein, S.; Rapoport, S.I. Epigenetic modificationsin frontal cortex from Alzheimer’s disease and bipolar disease patients. Transl. Psychiatry 2012, 2, e132. [Google Scholar] [CrossRef]
- Li, Q.; Ma, C.; Zhang, Z.; Chen, S.; Zhi, W.; Zhang, L.; Zhang, G.; Shi, L.; Cao, F.; Ma, T. Association between cyclooxygenase-2 (COX-2) 8473 T > C polymorphism and cancer risk: A meta-analysis and trial sequential analysis. BMC Cancer 2018, 18, 847. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.K.; Hong, K.; Jang, B.C. Transcriptional and translational regulation of COX-2 expression by cadmium in C6 glioma cells. Int. J. Mol. Med. 2012, 30, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.A.; Iadarola, M.; Yang, H.Y.; Dionne, R.A. Expression of COX-1 and COX-2 in a clinical model of acute inflammation. J. Pain 2007, 8, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Aid, S.; Bosetti, F. The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: Implications for translational research. Trends Pharmacol. Sci. 2009, 30, 174–191. [Google Scholar] [CrossRef] [PubMed]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 903–910. [Google Scholar] [CrossRef]
- Rogers, J.; Kirby, L.C.; Hempelman, S.R.; Berry, D.L.; McGeer, P.L.; Kaszniak, A.W.; Zalinski, J.; Cofield, M.; Mansukhani, L.; Willson, P.; et al. Clinical trial of indomethacin in Alzheimer’s disease. Neurology 1993, 43, 1609–1611. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Schulzer, M.; McGeer, E.G. Arthritis and anti-infalmmatory agents as possible protective factors for Alzheimer’s disease a review of 17 epidemiologic studies. Neurology 1996, 47, 425–434. [Google Scholar] [CrossRef]
- Wang, P.; Guan, PP.; Wang, T.; Yu, X.; Guo, J.J.; Wang, Z.Y. Aggravation of Alzheimer’s disease due to the COX-2 mediated reciprocal regulation of IL-1β and Aβ between glial and neuron cells. Aging Cell. 2014, 13, 605–616. [Google Scholar] [CrossRef]
- Pasinetti, G.M.; Aisen, P.S. Cyclooxygenase-2 expression is increased in frontal cortex of Alzheimer’s disease brain. Neuroscience 1998, 87, 319–324. [Google Scholar] [CrossRef]
- Ho, L.; Purohit, D.; Haroutunian, V.; Luterman, J.D.; Willis, F.; Naslund, J.; Buxbaum, J.D.; Mohs, R.C.; Aisen, P.S.; Pasinetti, G.M. Neuronal Cyclooxygenase-2 expression in the hippocampal formation as a function of a clinical progression of Alzheimer’s disease. Arch. Neurol. 2001, 58, 487–492. [Google Scholar] [CrossRef]
- Yermaova, A.V.; O’Banion, M.K. Down-regulation of neuronal cyclooxygenase-2expression in end stage Alzheimer’s disease. Neurobiol. Aging 2001, 22, 823–836. [Google Scholar] [CrossRef]
- Teissman, P.; Tieu, K.; Choi, D.K.; Wu, D.C.; Naini, A.; Hunot, S.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 5473–5478. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Specking, A.; Duyster, J.; Gebicke-Haerter, P.J.; Wurster, S.; Dieter, P. Effect of fluoride, pertussis and cholera toxin on the release of arachidonic acid and the formation of prostaglandin E2, D2, superoxide and inositol phosphates in rat liver macrophages. Cell Signal. 1991, 3, 599–606. [Google Scholar] [CrossRef]
- Dieter, P.; Fitzke, E. Formation of diacylglycerol, inositol phosphates, arachidonic acid and its metabolites in macrophages. Eur. J. Biochem. 1993, 218, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Barinaga, M. Is apoptosis key in Alzheimer’s disease? Science 1998, 281, 1303–1304. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Nomura, M.; Schuck, R.; Yustaein, J. Cancer’s Achilles’s heel: Apotosis and necroptosis to the rescue. Int. J. Mol. Sci. 2017, 18, 23. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Satoh, T.; Enkoido, Y.; Nishio, C.; Ikeuchi, T.; Hatanaka, H. Generation of reactive oxygen species, release of Lglutamate and activation of caspases are required for oxygeninduced apoptosis of embryonic hippocampal neurons in culture. Brain Res. 1999, 824, 71–80. [Google Scholar] [CrossRef]
- Dickson, D.W. Apoptotic mechanism in Alzheimer neurofibrillary degeneration: Cause or effect. J. Clin. Investig. 2004, 114, 23–27. [Google Scholar] [CrossRef]
- Wei, T.; Ni, Y.; Hou, J.; Chen, C.; Zhao, B.; Xin, W. Hydrogen peroxide induced oxidative damage and apoptosis in cerebellar granule cells: Protection by Ginkgo biloba extract. Pharmacol. Res. 2000, 41, 427–433. [Google Scholar] [CrossRef]
- Temple, M.D.; Perrone, G.G.; Dawes, I.W. Complex cellular responses to reactive oxygen species. Trends Cell Biol. 2005, 15, 319–326. [Google Scholar] [CrossRef]
- Obulesu, M.; Lakshmi, M.J. Apoptosis in Alzheimer disease: An understanding of the physiology, pathology and therapeutic avenues. Neurochem. Res. 2014, 39, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Kopitar-Jerala, N. Innate immune response in brain, NfkappaB signaling and cystatins. Front. Mol. Neurosci. 2015, 8, 73. [Google Scholar] [CrossRef]
- Xu, B.; Xu, Z.; Xia, T.; He, P.; Gao, P.; He, W.; Zhang, M.; Guo, L.; Niu, Q.; Wang, A. Effects of the Fas/Fas-L pathway on fluoride-induced apoptosis in SH-SY5Y cells. Environ. Toxicol. 2011, 26, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Guan, Z.Z.; Gao, Q.; Pei, J.J. Increased level of apoptosis in rat brains and SH-SY5Y cells exposed to excessive fluoride—A mechanism connected with activating JNK phosphorylation. Toxicol. Lett. 2011, 204, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, J.H.; Anderson, A.J.; Cummings, B.J.; Cotman, C.W. Immunohistochemical evidence for apoptosis in Alzheimer’s disease. Neuroreport 1994, 5, 2529–2533. [Google Scholar] [CrossRef] [PubMed]
- Troncoso, J.C.; Sukhov, R.R.; Kawas, C.H.; Kolistos, V.E. In situ labeling of dying cortical neurons in normal aging and in Alzheimer’s disease: Correlation with senile plaques and disease progression. J. Neuropathol. Exp. Med. 1996, 55, 1134–1142. [Google Scholar] [CrossRef]
- Kitamura, Y.; Shimohama, S.; Kamoshima, W.; Ota, T.; Matsuoka, Y.; Nomura, Y.; Smith, M.A.; Perry, G.; Whitehouse, P.J.; Taniguchi, T. Alteration of proteins regulating apoptosis, Bcl-2, Bcl-x, Bax, Bak, Bad, ICH-1 and CPP-32, in Alzheimer’s disease. Brain Res. 1998, 780, 260–269. [Google Scholar] [CrossRef]
- Shimohama, S. Apoptosis in Alzheimer disease—An update. Apoptosis 2000, 5, 9–16. [Google Scholar] [CrossRef]
- Masliah, E.; Mallory, M.; Alford, M.; Tanaka, S.; Hansen, L.A. Caspase dependent DNA-fragmentation might be associated with excitotoxicity in Alzheimer disease. J. Neuropathol. Exp. Neurol. 1998, 1, 319–325. [Google Scholar] [CrossRef]
- Kitamura, Y.; Shimohama, S.; Ota, T.; Matsuoka, Y.; Nomura, Y.; Taniguchi, T. Alteration of transcription factors NFkB and STAT-1 in Alzheimer’s disease brains. Neurosci. Lett. 1997, 237, 17–20. [Google Scholar] [CrossRef]
- Herdegen, T.; Skene, P.; Bähr, M. The c-JUN transcription factor—Bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci. 1997, 20, 227–231. [Google Scholar] [CrossRef]
- Rodolfo, C.; Campello, S.; Cecconi, F. Mithophagy in neurodegenerative diseases. Neurochem. Int. 2017, 117, 156–166. [Google Scholar] [CrossRef]
- Cai, Q.; Tammineni, T. Alteration in mitochondrial quality control in Alzheimer’s disease. Front. Cell. Neurosci. 2016, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Mahaboob, B.P.; Saumya, S.M. Suppression ofmitochondrial oxidative phosphorylation and TCA enzymes in discrete brain regions of mice exposed to high fluoride: Amelioration by Panax ginseng (Ginseng) and Lagerstroemia speciosa (Banaba) extracts. Cell. Mol. Neurobiol. 2013, 33, 453–464. [Google Scholar] [CrossRef]
- Reddy, P.H. Mitochondrial medicine for aging and neurodegenerative diseases. Neruomol. Med. 2008, 10, 291–315. [Google Scholar] [CrossRef]
- Andreini, C.; Bertini, I. A bioinformatics view of zinc enzymes. J. Inorgan. Biochem. 2012, 111, 150–156. [Google Scholar] [CrossRef]
- Ziemińska, E.; Strużyńska, L. Zinc Modulates Naono-Silver Induced Toxicity in Primary Neuronal Cultures. Neurotox. Res. 2016, 29, 325–343. [Google Scholar] [CrossRef]
- Nuttall, J.R.; Oteiza, P.L. Zinc and the aging brain. Genes. Nutr. 2014, 9, 379. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, C.; Martin, T.; Cacho, J.; Breñas, M.T.; Arroyo, T.; Garcia-Berrocal, B.; Navajo, J.A.; González-Buitrago, J.M. Serum zinc, copper, insulin, and lipids in Alzheimer’s disease epsilon 4 apolipoprotein E allele carriers. Eur. J. Clin. Investig. 1999, 29, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 2010, 142, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, A.J.R. Zinc, aging and immunosenescence: An overview. Pathobiol. Aging Age Relat. Dis. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Grabrucker, A.M.; Schmeisser, M.J.; Udvardi, P.T.; Arons, M.; Schoen, M.; Woodling, N.S.; Andreasson, K.I.; Hof, P.R.; Buxbaum, J.D.; Garner, C.C.; et al. Amyloid beta proteininduced zinc sequestration leads to synaptic loss via dysregulation of the ProSAP2/Shank3 scaffold. Mol. Neurodegener. 2011, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Aimo, L.; Cherr, G.N.; Oteiza, P.l. Low extracellular zinc increases neuronal oxidant production through nadph oxidase and nitric oxide synthase activation. Free Radic. Biol. Med. 2010, 48, 1577–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vink, R. Magnesium in the CNS. Recent advances and developments. Magnes. Res. 2016, 29, 95–101. [Google Scholar] [CrossRef]
- Van Den Heuvel, C.; Finnie, J.W.; Blumbergs, P.C.; Manavis, J.; Jones, N.R.; Reilly, P.L.; Pereira, R.A. Upregulation of neuronal amyloid precursor protein (APP) and APP mRNA following magnesium sulphate (MgSO4) therapy in traumatic brain injury. J. Neurotrauma 2000, 17, 1041–1053. [Google Scholar] [CrossRef]
- Wang, P.; Yu, X.; Guan, P.P.; Guo, J.W.; Wang, Y.; Zhang, Y.; Zhao, H.; Wang, Z.Y. Magnesium ion influx reduces neuroinflammation in Aβ precursor protein/Presenilin 1 transgenic mice by suppressing the expression of interleukin-1β. Cell. Mol. Immunol. 2017, 14, 451–464. [Google Scholar] [CrossRef]
- Kirkland, A.E.; Sarlo, G.L.; Holton, K.F. The role of magnesium in neurological disorders. Nutrients 2018, 10, 730. [Google Scholar] [CrossRef]
- Chin-Chan, M.; Navarro-Jepes, J.; Quintanilla-vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.C.; Lokwood, A.H.; Sonawane, B.R. Neurodegenerative disease: An Overwiev of Environmental Risk Factors. Environ. Health Perspect. 2005, 113, 1250–1255. [Google Scholar] [CrossRef] [PubMed]
- Graves, A.B.; White, E.; Koepsell, T.D.; Reifler, B.V.; van Belle, G.; Larson, E.B. The association between aluminum-containing products and Alzheimer’s disease. J. Clin. Epidemiol. 1990, 43, 35–44. [Google Scholar] [CrossRef]
- Wang, Z.; Wei, X.; Yang, J.; Suo, J.; Chen, J.; Liu, X.; Zhao, X. Chronic exposure to aluminum and risk of Alzheimer’s disease: A meta-analysis. Neurosci. Lett. 2016, 610, 200–206. [Google Scholar] [CrossRef]
- Virk, S.A.; Eslick, G.D. Occupational Exposure to Aluminum and Alzheimer’s disease: A Meta-analysis. J. Occup. Environ. Med. 2015, 57, 893–896. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Jiang, L.; Huang, H.; Zeng, S.; Qiu, F.; Yu, M.; Li, X.; Wei, S. Dietary Exposure to Aluminium and Health Risk Assessment in the Residents of Shenzhen, China. PLoS ONE 2014, 9, e89715. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Puri, B.K.; Frye, R.E. The putative role of environmental aluminum in the development of chronic neuropathology in adults and children. How strong is the evidence and what could be the mechanisms involved? Metab. Brain Dis. 2017, 32, 1335–1355. [Google Scholar] [CrossRef]
- Sumathi, T.; Shobana, C.; Thangarajesari, M.; Usha, R. Protective effect of L-Theanine against aluminum induced neurotoxicity in cerebral cortex, hippocampus and cerebellum of rat brain—Histopathological, and biochemical approach. Drug Chem. Toxicol. 2015, 38, 22–31. [Google Scholar] [CrossRef]
- Metryka, E.; Chibowska, K.; Gutowska, I.; Falkowska, A.; Kupnicka, P.; Barczak, K.; Chlubek, D.; Baranowska-Bosiacka, I. Lead (Pb) Exposure Enhances Expression of Factors Associated with Inflammation. Int. J. Mol. Sci. 2018, 19, 1813. [Google Scholar] [CrossRef]
- Chibowska, K.; Baranowska-Bosiacka, I.; Falkowska, A.; Gutowska, I.; Goschorska, M.; Chlubek, D. Effect of Lead (Pb) on Inflammatory Processes in the Brain. Int. J. Mol. Sci. 2016, 17, 2140. [Google Scholar] [CrossRef]
- Baranowska-Bosiacka, I.; Listos, J.; Gutowska, I.; Machoy-Mokrzyńska, A.; Kolasa-Wołosiuk, A.; Tarnowski, M.; Puchałowicz, K.; Prokopowicz, A.; Talarek, S.; Listos, P.; et al. Effects of perinatal exposure to lead (Pb) on purine receptor expression in the brain and gliosis in rats tolerant to morphine analgesia. Toxicology 2016, 339, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Gąssowska, M.; Baranowska-Bosiacka, I.; Moczydłowska, J.; Frontczak-Baniewicz, M.; Gewartowska, M.; Strużyńska, L.; Gutowska, I.; Chlubek, D.; Adamczyk, A. Perinatal exposure to lead (Pb) induces ultrastructural and molecular alterations in synapses of rat offspring. Toxicology 2016, 373, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Łukomska, A.; Baranowska-Bosiacka, I.; Budkowska, M.; Pilutin, A.; Tarnowski, M.; Dec, K.; Dołęgowska, B.; Metryka, E.; Chlubek, D.; Gutowska, I. The effect of low levels of lead (Pb) in the blood on levels of sphingosine-1-phosphate (S1P) and expression of S1P receptor 1 in the brain of the rat in the perinatal period. Chemosphere 2017, 166, 221–229. [Google Scholar] [CrossRef]
- Baranowska-Bosiacka, I.; Strużyńska, L.; Gutowska, I.; Machalińska, A.; Kolasa, A.; Kłos, P.; Czapski, G.A.; Kurzawski, M.; Prokopowicz, A.; Marchlewicz, M.; et al. Perinatal exposure to lead induces morphological, ultrastructural and molecular alterations in the hippocampus. Toxicology 2013, 303, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Mason, L.H.; Harp, J.P.; Han, D.Y. Pb neurotoxicity: Neuropsychological effects of lead toxicity. Biomed. Res. Int. 2014, 840547. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, D.W.; Park, K.S.; Joung, H. Serum trace metal levels in Alzheimer’s disease and normal control groups. Am. J. Alzheimers Dis. Other Demen. 2014, 29, 76–83. [Google Scholar] [CrossRef]
- Gąssowska, M.; Baranowska-Bosiacka, I.; Moczydłowska, J.; Tarnowski, M.; Pilutin, A.; Gutowska, I.; Strużyńska, L.; Chlubek, D.; Adamczyk, A. Perinatal exposure to lead (Pb) promotes Tau phosphorylation in the rat brain in a GSK-3β and CDK5 dependent manner: Relevance to neurological disorders. Toxicology 2016, 347, 17–28. [Google Scholar] [CrossRef]
- Dash, M.; Eid, A.; Subaiea, G.; Chang, J.; Deeb, R.; Masoud, A.; Renehan, W.E.; Adem, A.; Zawia, N.H. Developmental exposure to lead (Pb) alters the expression of the human tau gene and its products in a transgenic animal model. Neurotoxicology 2016, 55, 154–159. [Google Scholar] [CrossRef] [Green Version]
- Bihaqi, S.W.; Zawia, N.H. Enhanced taupathy and AD-like pathology in aged primate brains decades after infantile exposure to Lead (Pb). Neurotoxicology 2013, 39, 95–101. [Google Scholar] [CrossRef]
- Gu, H.; Robison, G.; Hong, L.; Barrea, R.; Wei, X.; Farlow, M.R.; Pushkar, Y.N.; Du, Y.; Zheng, W. Increased β-Amyloid Deposition in Tg-SWDI Transgenic Mouse Brain Following In Vivo Lead Exposure. Toxicol. Lett. 2012, 213, 211–219. [Google Scholar] [CrossRef]
- Wu, J.; Basha, M.R.; Brock, B.; Cox, D.P.; Cardozo-Pelaez, F.; McPherson, C.A.; Harry, J.; Rice, D.C.; Maloney, B.; Chen, D.; et al. Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): Evidence for a developmental origin and environmental link for AD. J. Neurosci. 2008, 28, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Gutowska, I.; Baranowska-Bosiacka, I.; Siwiec, E.; Szczuko, M.; Kolasa, A.; Kondarewicz, A.; Rybicka, M.; Dunaj-Stańczyk, M.; Wiernicki, I.; Chlubek, D.; et al. Lead enhance fluoride influence on apoptosis processes in liver cell line HepG2. Toxicol. Ind. Health 2016, 32, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Kamel, F.; Hoppin, J.A. Association of Pesticide Exposure with Neurologic Dysfunction and Disease. Environ. Health Perspect. 2004, 112, 950–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Santed, F.; Colomina, M.T.; Hernandez, E.H. Organophosphate pesticide exposure and neurodegeneration. Cortex 2016, 74, 417–426. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goschorska, M.; Baranowska-Bosiacka, I.; Gutowska, I.; Metryka, E.; Skórka-Majewicz, M.; Chlubek, D. Potential Role of Fluoride in the Etiopathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 3965. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123965
Goschorska M, Baranowska-Bosiacka I, Gutowska I, Metryka E, Skórka-Majewicz M, Chlubek D. Potential Role of Fluoride in the Etiopathogenesis of Alzheimer’s Disease. International Journal of Molecular Sciences. 2018; 19(12):3965. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123965
Chicago/Turabian StyleGoschorska, Marta, Irena Baranowska-Bosiacka, Izabela Gutowska, Emilia Metryka, Marta Skórka-Majewicz, and Dariusz Chlubek. 2018. "Potential Role of Fluoride in the Etiopathogenesis of Alzheimer’s Disease" International Journal of Molecular Sciences 19, no. 12: 3965. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123965