Therapeutic Vulnerabilities in FLT3-Mutant AML Unmasked by Palbociclib

 and
and
Abstract
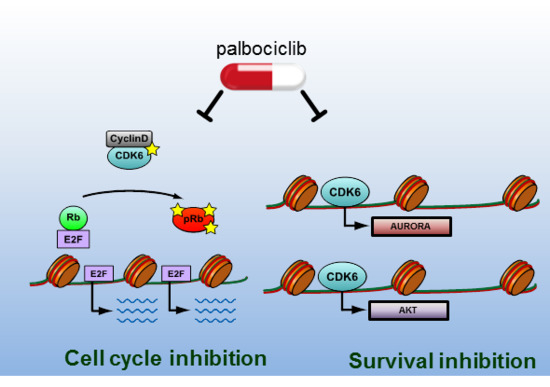
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
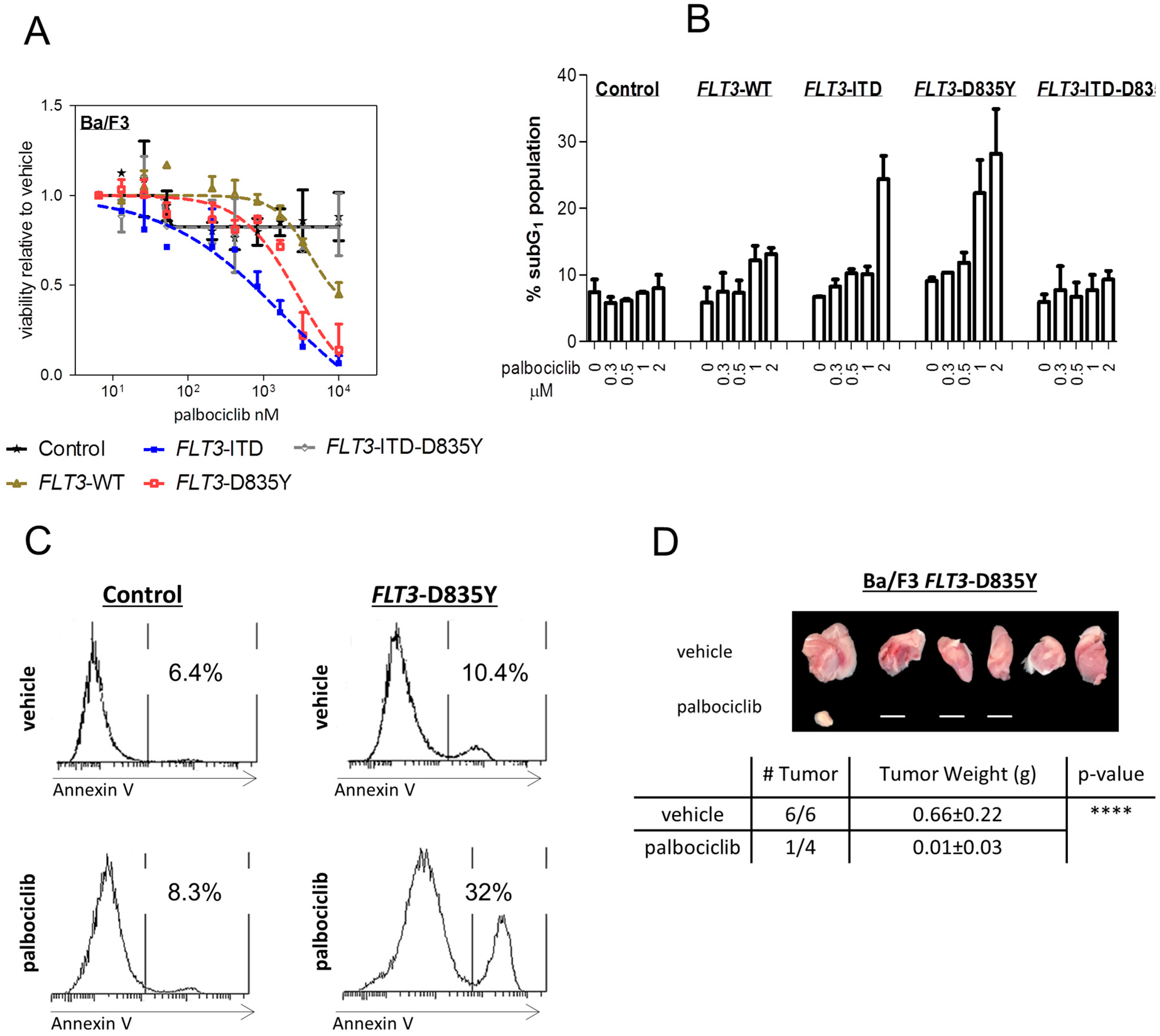
2.1. FLT3 Kinase Domain Mutation Renders Cells Sensitive to the CDK4/6 Inhibitor Palbociclib
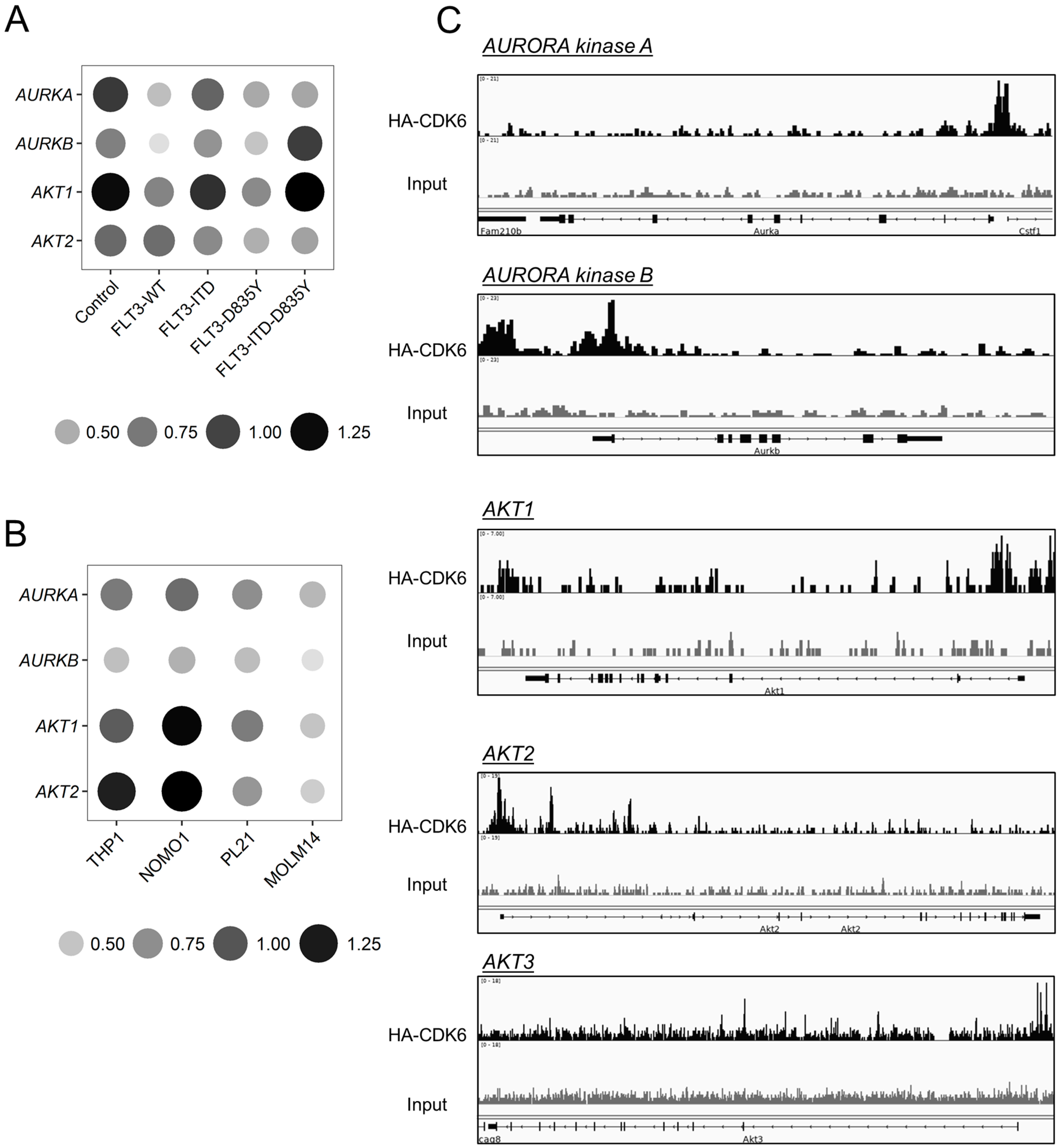
2.2. CDK6 Regulates Expression of AKT and AURORA
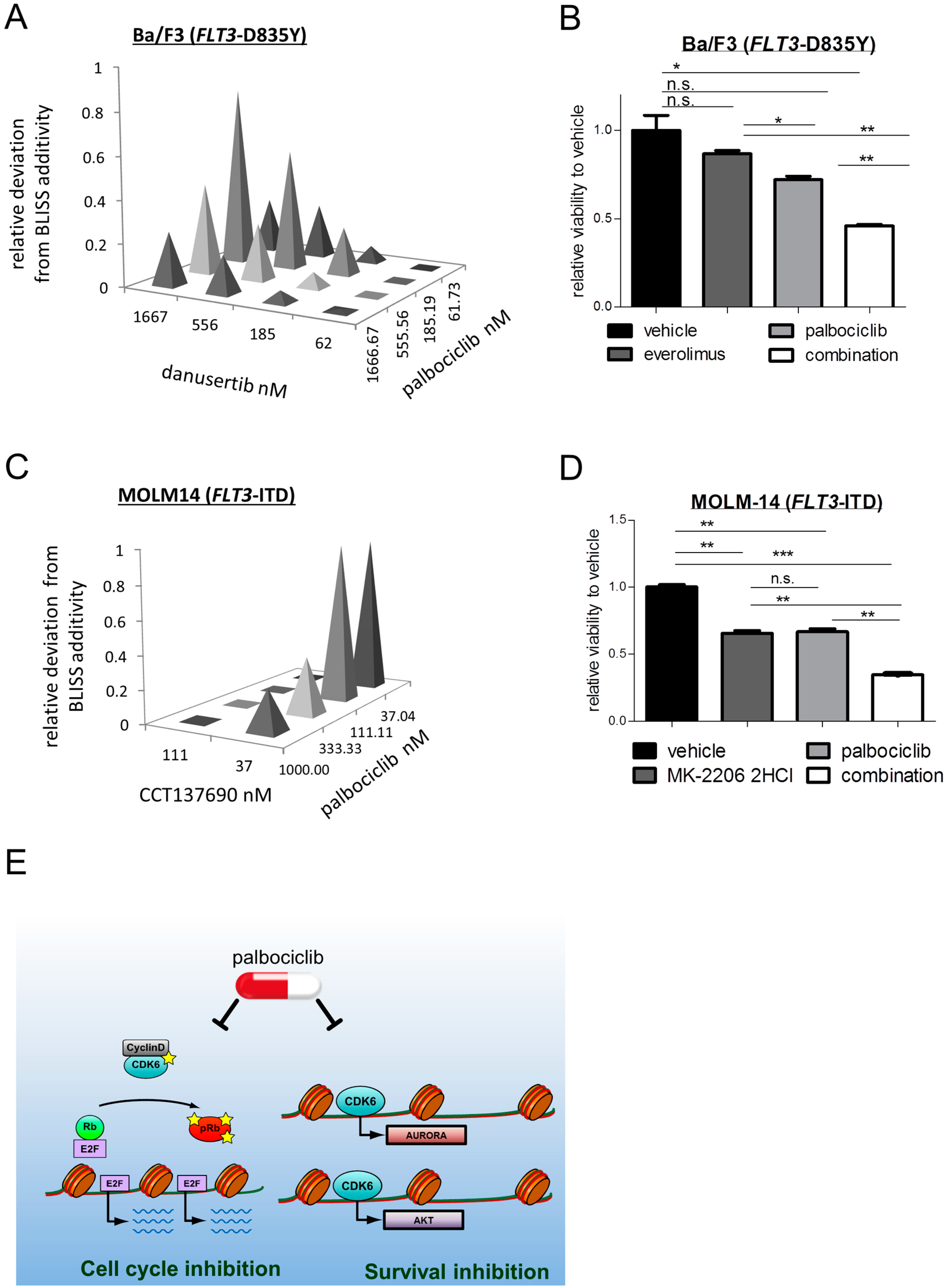
2.3. Palbociclib Synergizes with AKT- and with AURORA Kinase Inhibitors
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Generation of Ba/F3 Cells Expressing FLT3–ITD, FLT3–D835Y, FLT3–WT, and FLT3–ITD–D835Y
4.3. Cell Growth Measurement
4.4. Apoptosis Measurement
4.5. Transplantation Studies
4.6. Quantitative Real-Time PCR
4.7. Immunoblotting
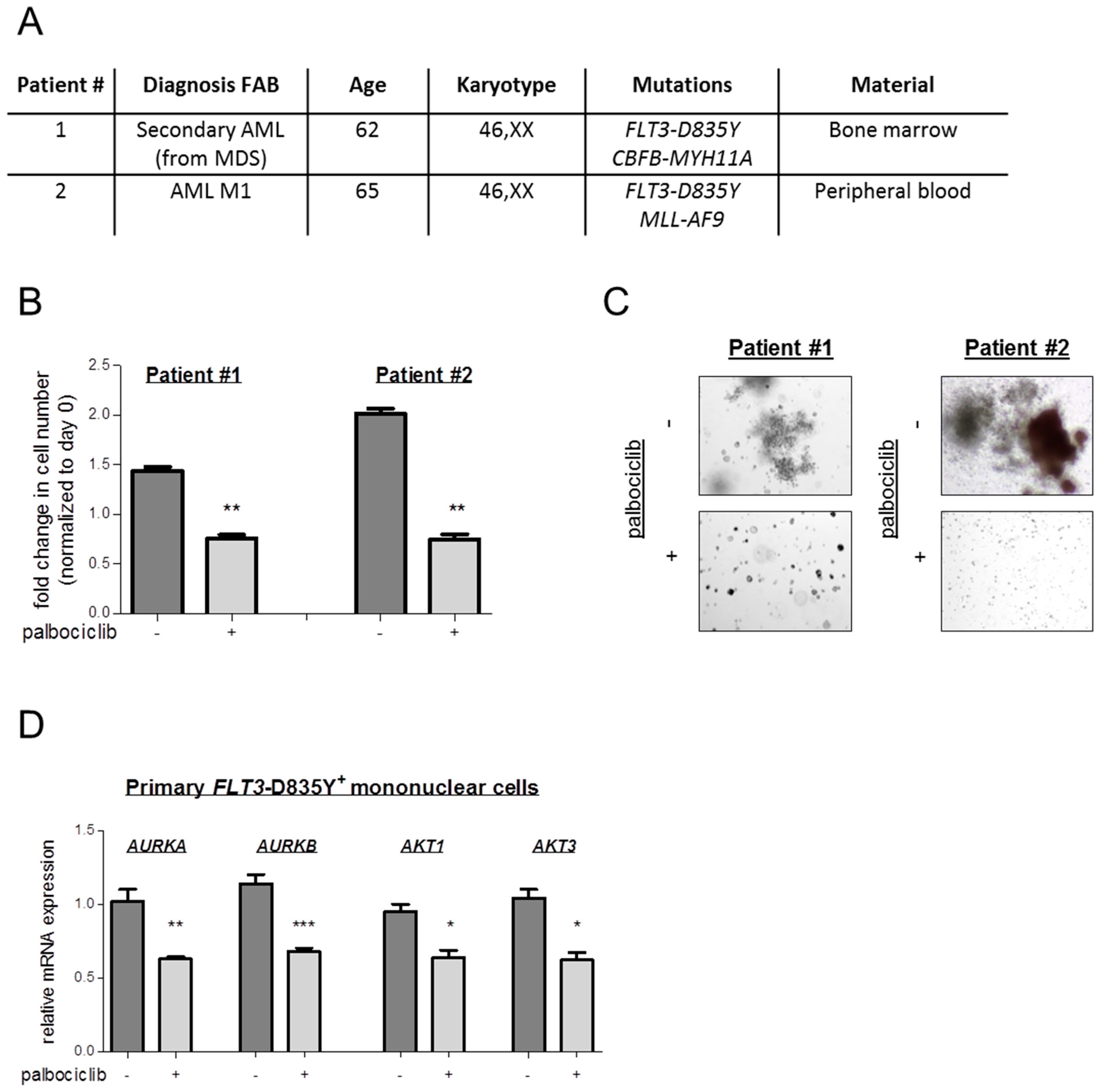
4.8. Studies on Primary Patient-Derived Cells
4.9. Chromatin Immunoprecipitation (ChIP)
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 7-AAD | 7-aminoactinomycin D |
| AML | Acute myeloid leukemia |
| AURK | AURORA kinase |
| ATP | Adenosine triphosphate |
| CDK | Cyclin-dependent kinase |
| ChIP | Chromatin-Immunoprecipitation |
| CTG | Cell-Titer Glo |
| FAB | French-American-British |
| FACS | Fluorescence-activated cell sorting analysis |
| FLT3 | Fms-like tyrosine kinase 3 |
| GFP | Green fluorescence protein |
| IC50 | Half maximal inhibitory concentration |
| ITD | Internal tandem duplications in the juxtamembrane domain |
| mRNA | Messenger RNA |
| OS | Overall survival |
| p | phospho |
| S.E.M. | Standard error of the mean |
| SCF | Stem cell factor |
| SPF | Special pathogen free |
| TKD | Tyrosine kinase domain |
| TKI | Tyrosine kinase inhibitor |
| WHO | World Health Organization |
| WT | Wild-type |
References
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Meshinchi, S.; Appelbaum, F.R. Structural and functional alterations of FLT3 in acute myeloid leukemia. Clin. Cancer Res. 2009, 15, 4263–4269. [Google Scholar] [CrossRef] [PubMed]
- Larrosa-Garcia, M.; Baer, M.R. FLT3 Inhibitors in Acute Myeloid Leukemia: Current Status and Future Directions. Mol. Cancer Ther. 2017, 16, 991–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, R.M.; Manley, P.W.; Larson, R.A.; Capdeville, R. Midostaurin: Its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018, 2, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.J.; Cools, J.; Curley, D.P.; Yu, J.-C.; Lokker, N.A.; Giese, N.A.; Gilliland, D.G. Variable sensitivity of FLT3 activation loop mutations to the small molecule tyrosine kinase inhibitor MLN518. Blood 2004, 104, 2867–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perl, A.E.; Altman, J.K.; Cortes, J.; Smith, C.; Litzow, M.; Baer, M.R.; Claxton, D.; Erba, H.P.; Gill, S.; Goldberg, S.; et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: A multicentre, first-in-human, open-label, phase 1-2 study. Lancet. Oncol. 2017, 18, 1061–1075. [Google Scholar] [CrossRef]
- Leung, A.Y.H.; Man, C.-H.; Kwong, Y.-L. FLT3 inhibition: A moving and evolving target in acute myeloid leukaemia. Leukemia 2013, 27, 260–268. [Google Scholar] [CrossRef]
- Man, C.H.; Fung, T.K.; Ho, C.; Han, H.H.C.; Chow, H.C.H.; Ma, A.C.H.; Choi, W.W.L.; Lok, S.; Cheung, A.M.S.; Eaves, C.; et al. Sorafenib treatment of FLT3-ITD(+) acute myeloid leukemia: Favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood 2012, 119, 5133–5143. [Google Scholar] [CrossRef]
- Alvarado, Y.; Kantarjian, H.M.; Luthra, R.; Ravandi, F.; Borthakur, G.; Garcia-Manero, G.; Konopleva, M.; Estrov, Z.; Andreeff, M.; Cortes, J.E. Treatment with FLT3 inhibitor in patients with FLT3-mutated acute myeloid leukemia is associated with development of secondary FLT3-tyrosine kinase domain mutations. Cancer 2014, 120, 2142–2149. [Google Scholar] [CrossRef]
- Smith, C.C.; Lasater, E.A.; Lin, K.C.; Wang, Q.; McCreery, M.Q.; Stewart, W.K.; Damon, L.E.; Perl, A.E.; Jeschke, G.R.; Sugita, M.; et al. Crenolanib is a selective type I. pan-FLT3 inhibitor. Proc. Natl. Acad. Sci. USA 2014, 111, 5319–5324. [Google Scholar] [CrossRef]
- Piloto, O.; Wright, M.; Brown, P.; Kim, K.-T.; Levis, M.; Small, D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood 2007, 109, 1643–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindblad, O.; Cordero, E.; Puissant, A.; Macaulay, L.; Ramos, A.; Kabir, N.N.; Sun, J.; Vallon-Christersson, J.; Haraldsson, K.; Hemann, M.T.; et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene 2016, 35, 5119–5131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohi, M.G.; Boulton, C.; Gu, T.-L.; Sternberg, D.W.; Neuberg, D.; Griffin, J.D.; Gilliland, D.G.; Neel, B.G. Combination of rapamycin and protein tyrosine kinase (PTK) inhibitors for the treatment of leukemias caused by oncogenic PTKs. Proc. Natl. Acad. Sci. USA 2004, 101, 3130–3135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisberg, E.; Liu, Q.; Zhang, X.; Nelson, E.; Sattler, M.; Liu, F.; Nicolais, M.; Zhang, J.; Mitsiades, C.; Smith, R.W.; et al. Selective Akt Inhibitors Synergize with Tyrosine Kinase Inhibitors and Effectively Override Stroma-Associated Cytoprotection of Mutant FLT3-Positive AML Cells. PLoS ONE 2013, 8, e56473. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Blagg, J.; Linardopoulos, S.; Pearson, A.D.J. Aurora kinase inhibitors: Novel small molecules with promising activity in acute myeloid and Philadelphia-positive leukemias. Leukemia 2010, 24, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Ikezoe, T.; Yang, J.; Nishioka, C.; Tasaka, T.; Taniguchi, A.; Kuwayama, Y.; Komatsu, N.; Bandobashi, K.; Togitani, K.; Koeffler, H.P.; et al. A novel treatment strategy targeting Aurora kinases in acute myelogenous leukemia. Mol. Cancer Ther. 2007, 6, 1851–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farag, S.S. The potential role of Aurora kinase inhibitors in haematological malignancies. Br. J. Haematol. 2011, 155, 561–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uras, I.Z.; Walter, G.J.; Scheicher, R.; Bellutti, F.; Prchal-Murphy, M.; Tigan, A.S.; Valent, P.; Heidel, F.H.; Kubicek, S.; Scholl, C.; et al. Palbociclib treatment of FLT3-ITD+ AML cells uncovers a kinase-dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood 2016. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-T.; Baird, K.; Ahn, J.-Y.; Meltzer, P.; Lilly, M.; Levis, M.; Small, D. Pim-1 is up-regulated by constitutively activated FLT3 and plays a role in FLT3-mediated cell survival. Blood 2005, 105, 1759–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam, M.; Pogacic, V.; Bendit, M.; Chappuis, R.; Nawijn, M.C.; Duyster, J.; Fox, C.J.; Thompson, C.B.; Cools, J.; Schwaller, J. Targeting PIM kinases impairs survival of hematopoietic cells transformed by kinase inhibitor-sensitive and kinase inhibitor-resistant forms of Fms-like tyrosine kinase 3 and BCR/ABL. Cancer Res. 2006, 66, 3828–3835. [Google Scholar] [CrossRef] [PubMed]
- Green, A.S.; Maciel, T.T.; Hospital, M.-A.; Yin, C.; Mazed, F.; Townsend, E.C.; Pilorge, S.; Lambert, M.; Paubelle, E.; Jacquel, A.; et al. Pim kinases modulate resistance to FLT3 tyrosine kinase inhibitors in FLT3-ITD acute myeloid leukemia. Sci. Adv. 2015, 1, e1500221. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Wang, Q.; Chin, C.-S.; Salerno, S.; Damon, L.E.; Levis, M.J.; Perl, A.E.; Travers, K.J.; Wang, S.; Hunt, J.P.; et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 2012, 485, 260–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bavetsias, V.; Large, J.M.; Sun, C.; Bouloc, N.; Kosmopoulou, M.; Matteucci, M.; Wilsher, N.E.; Martins, V.; Reynisson, J.; Atrash, B.; et al. Imidazo[4,5-b]pyridine derivatives as inhibitors of Aurora kinases: Lead optimization studies toward the identification of an orally bioavailable preclinical development candidate. J. Med. Chem. 2010, 53, 5213–5228. [Google Scholar] [CrossRef] [PubMed]
- Bavetsias, V.; Crumpler, S.; Sun, C.; Avery, S.; Atrash, B.; Faisal, A.; Moore, A.S.; Kosmopoulou, M.; Brown, N.; Sheldrake, P.W.; et al. Optimization of imidazo[4,5-b]pyridine-based kinase inhibitors: Identification of a dual FLT3/Aurora kinase inhibitor as an orally bioavailable preclinical development candidate for the treatment of acute myeloid leukemia. J. Med. Chem. 2012, 55, 8721–8734. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Faisal, A.; Gonzalez de Castro, D.; Bavetsias, V.; Sun, C.; Atrash, B.; Valenti, M.; de Haven Brandon, A.; Avery, S.; Mair, D.; et al. Selective FLT3 inhibition of FLT3-ITD+ acute myeloid leukaemia resulting in secondary D835Y mutation: A model for emerging clinical resistance patterns. Leukemia 2012, 26, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Carpinelli, P.; Ceruti, R.; Giorgini, M.L.; Cappella, P.; Gianellini, L.; Croci, V.; Degrassi, A.; Texido, G.; Rocchetti, M.; Vianello, P.; et al. PHA-739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Mol. Cancer Ther. 2007, 6, 3158–3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, E.A.; Bebbington, D.; Moore, J.; Rasmussen, R.K.; Ajose-Adeogun, A.O.; Nakayama, T.; Graham, J.A.; Demur, C.; Hercend, T.; Diu-Hercend, A.; et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat. Med. 2004, 10, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, M.G.; Ecsedy, J.A.; Chakravarty, A.; Silverman, L.; Zhang, M.; Hoar, K.M.; Stroud, S.G.; Chen, W.; Shinde, V.; Huck, J.J.; et al. Characterization of Alisertib (MLN8237), an Investigational Small-Molecule Inhibitor of Aurora A Kinase Using Novel In Vivo Pharmacodynamic Assays. Clin. Cancer Res. 2011, 17, 7614–7624. [Google Scholar] [CrossRef] [Green Version]
- Sedrani, R.; Cottens, S.; Kallen, J.; Schuler, W. Chemical modification of rapamycin: The discovery of SDZ RAD. Transplant. Proc. 1998, 30, 2192–2194. [Google Scholar] [CrossRef]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.-S.; et al. MK-2206, an Allosteric Akt Inhibitor, Enhances Antitumor Efficacy by Standard Chemotherapeutic Agents or Molecular Targeted Drugs In vitro and In vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef] [Green Version]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Grundler, R.; Thiede, C.; Miething, C.; Steudel, C.; Peschel, C.; Duyster, J. Sensitivity toward tyrosine kinase inhibitors varies between different activating mutations of the FLT3 receptor. Blood 2003, 102, 646–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Placke, T.; Faber, K.; Nonami, A.; Putwain, S.L.; Salih, H.R.; Heidel, F.H.; Krämer, A.; Root, D.E.; Barbie, D.A.; Krivtsov, A.V.; et al. Requirement for CDK6 in MLL-rearranged acute myeloid leukemia. Blood 2014, 124, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, K.; Xie, Y.; Burcu, M.; Linn, D.E.; Qiu, Y.; Baer, M.R. Pim-1 kinase phosphorylates and stabilizes 130 kDa FLT3 and promotes aberrant STAT5 signaling in acute myeloid leukemia with FLT3 internal tandem duplication. PLoS ONE. 2013, 8, e74653. [Google Scholar] [CrossRef] [PubMed]
- Siendones, E.; Barbarroja, N.; Torres, L.A.; Buendía, P.; Velasco, F.; Dorado, G.; Torres, A.; López-Pedrera, C. Inhibition of Flt3-activating mutations does not prevent constitutive activation of ERK/Akt/STAT pathways in some AML cells: A possible cause for the limited effectiveness of monotherapy with small-molecule inhibitors. Hematol. Oncol. 2007, 25, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Pikman, Y.; Alexe, G.; Roti, G.; Conway, A.S.; Furman, A.; Lee, E.S.; Place, A.E.; Kim, S.; Saran, C.; Modiste, R.; et al. Synergistic Drug Combinations with a CDK4/6 Inhibitor in T-cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2017, 23, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, M.A.; Digiacomo, G.; Fumarola, C.; Alfieri, R.; Quaini, F.; Falco, A.; Madeddu, D.; La Monica, S.; Cretella, D.; Ravelli, A.; et al. Combined Inhibition of CDK4/6 and PI3K/AKT/mTOR Pathways Induces a Synergistic Anti-Tumor Effect in Malignant Pleural Mesothelioma Cells. Neoplasia 2017, 19, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Cretella, D.; Ravelli, A.; Fumarola, C.; La Monica, S.; Digiacomo, G.; Cavazzoni, A.; Alfieri, R.; Biondi, A.; Generali, D.; Bonelli, M.; et al. The anti-tumor efficacy of CDK4/6 inhibition is enhanced by the combination with PI3K/AKT/mTOR inhibitors through impairment of glucose metabolism in TNBC cells. J. Exp. Clin. Cancer Res. 2018, 37, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanArsdale, T.; Boshoff, C.; Arndt, K.T.; Abraham, R.T. Molecular Pathways: Targeting the Cyclin D-CDK4/6 Axis for Cancer Treatment. Clin. Cancer Res. 2015. [Google Scholar] [CrossRef]
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Huang, B.C.; Zhang, K.; et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef]
- Malumbres, M.; Sotillo, R.; Santamaría, D.; Galán, J.; Cerezo, A.; Ortega, S.; Dubus, P.; Barbacid, M. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 2004, 118, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.G.; Deshpande, A.; Schlichting, N.; Hinds, E.A.; Mao, C.; Dose, M.; Hu, G.-F.; Van Etten, R.A.; Gounari, F.; Hinds, P.W. CDK6 kinase activity is required for thymocyte development. Blood 2011, 117, 6120–6131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uras, I.Z.; Scheicher, R.M.; Kollmann, K.; Glösmann, M.; Prchal-Murphy, M.; Tigan, A.S.; Fux, D.A.; Altamura, S.; Neves, J.; Muckenthaler, M.U.; et al. Cdk6 contributes to cytoskeletal stability in erythroid cells. Haematologica 2017, 102, 995–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bliss, C.I. The toxicity of poisons applied jointly. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.; Gralnick, H.R.; Sultan, C. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br. J. Haematol. 1976, 33, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.; Gralnick, H.R.; Sultan, C. Proposals for the classification of the myelodysplastic syndromes. Br. J. Haematol. 1982, 51, 189–199. [Google Scholar] [CrossRef]
- Vardiman, J.W. The World Health Organization (WHO) classification of tumors of the hematopoietic and lymphoid tissues: An overview with emphasis on the myeloid neoplasms. Chem. Biol. Interact. 2010, 184, 16–20. [Google Scholar] [CrossRef]
- Kollmann, K.; Heller, G.; Schneckenleithner, C.; Warsch, W.; Scheicher, R.; Ott, R.G.; Schäfer, M.; Fajmann, S.; Schlederer, M.; Schiefer, A.-I.; et al. A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell 2013, 24, 167–181. [Google Scholar] [CrossRef]
- Scheicher, R.; Hoelbl-kovacic, A.; Bellutti, F.; Tigan, A.; Prchal-murphy, M.; Heller, G.; Schneckenleithner, C.; Sabine, Z.; Zuber, J.; Malumbres, M.; et al. CDK6 as a key regulator of hematopoietic and leukemic stem cell activation. Blood 2015, 125, 90–102. [Google Scholar] [CrossRef]
- Bellutti, F.; Tigan, A.-S.; Nebenfuehr, S.; Dolezal, M.; Zojer, M.; Grausenburger, R.; Hartenberger, S.; Kollmann, S.; Doma, E.; Prchal-Murphy, M.; et al. CDK6 antagonizes P53-induced responses during tumorigenesis. Cancer Discov. 2018, 8, 884–897. [Google Scholar] [CrossRef]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uras, I.Z.; Maurer, B.; Nebenfuehr, S.; Zojer, M.; Valent, P.; Sexl, V. Therapeutic Vulnerabilities in FLT3-Mutant AML Unmasked by Palbociclib. Int. J. Mol. Sci. 2018, 19, 3987. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123987
Uras IZ, Maurer B, Nebenfuehr S, Zojer M, Valent P, Sexl V. Therapeutic Vulnerabilities in FLT3-Mutant AML Unmasked by Palbociclib. International Journal of Molecular Sciences. 2018; 19(12):3987. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123987
Chicago/Turabian StyleUras, Iris Z., Barbara Maurer, Sofie Nebenfuehr, Markus Zojer, Peter Valent, and Veronika Sexl. 2018. "Therapeutic Vulnerabilities in FLT3-Mutant AML Unmasked by Palbociclib" International Journal of Molecular Sciences 19, no. 12: 3987. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123987