Biochemical Mechanism of Rhododendrol-Induced Leukoderma


Department of Chemistry, Fujita Health University School of Health Sciences, 1-98 Dengakugakubo, Kutsukake-cho, Toyoake, Aichi 470-1192, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(2), 552; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19020552
Submission received: 30 January 2018
/
Revised: 9 February 2018
/
Accepted: 10 February 2018
/
Published: 12 February 2018
(This article belongs to the Special Issue Melanins and Melanogenesis: From Nature to Applications)
Abstract
:RS-4-(4-hydroxyphenyl)-2-butanol (rhododendrol (RD))—a skin-whitening ingredient—was reported to induce leukoderma in some consumers. We have examined the biochemical basis of the RD-induced leukoderma by elucidating the metabolic fate of RD in the course of tyrosinase-catalyzed oxidation. We found that the oxidation of racemic RD by mushroom tyrosinase rapidly produces RD-quinone, which gives rise to secondary quinone products. Subsequently, we confirmed that human tyrosinase is able to oxidize both enantiomers of RD. We then showed that B16 cells exposed to RD produce high levels of RD-pheomelanin and protein-SH adducts of RD-quinone. Our recent studies showed that RD-eumelanin—an oxidation product of RD—exhibits a potent pro-oxidant activity that is enhanced by ultraviolet-A radiation. In this review, we summarize our biochemical findings on the tyrosinase-dependent metabolism of RD and related studies by other research groups. The results suggest two major mechanisms of cytotoxicity to melanocytes. One is the cytotoxicity of RD-quinone through binding with sulfhydryl proteins that leads to the inactivation of sulfhydryl enzymes and protein denaturation that leads to endoplasmic reticulum stress. The other mechanism is the pro-oxidant activity of RD-derived melanins that leads to oxidative stress resulting from the depletion of antioxidants and the generation of reactive oxygen radicals.
1. Introduction
Rhododendrol (RD, Rhododenol®) is a naturally occurring phenolic compound found in plants such as Acer nikoense and Betula platyphylla [1,2,3,4]. RD was first isolated by Kawaguchi et al. [5] and its structure was determined to be 4-(4-hydroxyphenyl)-2-butanol (Figure 1). Since 2008, a racemic form of RD (RS-RD) had been added to cosmetics as a skin-whitening ingredient by a cosmetic company in Tokyo, Japan. In July 2013, cosmetics containing RD were recalled by that company because a considerable number of consumers developed leukoderma on their face, neck and hands. Among an estimated 800,000 users of RD, 19,605 subjects (as of October, 2016) reported that they developed leukoderma, accounting for 2–2.5% of users. The mechanism by which the cutaneous depigmentation is caused by RD-containing cosmetics was originally considered to be a competitive inhibition of tyrosinase by RD. However, skin biopsy samples taken from depigmented lesions of affected subjects showed fewer or no melanocytes compared with normal skin [6] and 96% of leukoderma lesions were found to occur at or close to the site of RD application [7]. On the other hand, RD did not have cytotoxic effects on keratinocytes and fibroblasts [8]. These data strongly suggested that RD had cytotoxic effects that are direct and specific to melanocytes.
In melanocytes, melanogenesis starts with the tyrosinase-catalyzed oxidation of l-tyrosine to produce dopaquinone [13,14,15,16,17,18]. Subsequent spontaneous (non-enzymatic) and enzyme-catalyzed reactions give rise to the production of eumelanin, while the intervention of l-cysteine (CySH) with dopaquinone leads ultimately to the production of pheomelanin. Tyrosinase—the key enzyme in melanogenesis—is able to oxidize, in addition to l-tyrosine, a number of phenols and catechols to form the corresponding ortho-quinones [15,19]. o-Quinones are highly reactive compounds that exert cytotoxicity through binding with sulfhydryl (thiol) enzymes and/or DNA and by producing reactive oxygen species (ROS) [20,21].
RD has a para-substituted alkylphenol structure. Certain exogenous p-substituted phenols and related compounds are known to cause contact or occupational vitiligo, which is characterized by the irreversible loss of epidermal melanocytes at both exposed and non-exposed areas [22]. Those phenolic substances, e.g., 4-tert-butylphenol [23], 4-methoxyphenol [24] and 4-benzyloxyphenol (monobenzone or hydroquinone monobenzyl ether) [25,26], are good substrates for tyrosinase and produce reactive o-quinones [27] that can bind to biologically relevant thiol compounds such as CySH and glutathione (GSH) through sulfhydryl groups. Covalent binding to bovine serum albumin (BSA) through a cysteinyl residue was confirmed for monobenzone-derived o-quinone [25]. In related studies to develop antimelanoma agents using tyrosinase substrates, we synthesized a number of derivatives of 4-S-cysteaminylphenol (4-S-CAP), among which N-propionyl-4-S-cysteaminylphenol (NPrCAP) was the most active in inhibiting the growth of melanoma cells in vitro and in vivo [28,29]. Our study showed that the phenol NPrCAP is activated by mushroom tyrosinase to an o-quinone that reacts rapidly with CySH, GSH and BSA [30]. The adduct formation with melanocytic proteins was confirmed in vitro and in vivo. In those studies, the covalent binding of BSA (or cellular thiol proteins) was confirmed by HPLC analysis of cysteinylcatechol derivatives after acid hydrolysis.
Since late 2013, we have been examining the biochemical basis of RD-induced leukoderma by elucidating the metabolic fates of RD in the course of tyrosinase-catalyzed oxidation. We reported that the oxidation of racemic RD (Figure 1) by mushroom tyrosinase rapidly produces RD-quinone, which gives rise to secondary quinone products [9]. Subsequently, we confirmed that human tyrosinase is able to oxidize both enantiomers of RD [31]. We then showed that B16 cells exposed to RD produce high levels of RD-pheomelanin and protein-SH adducts of RD-quinone [32]. Furthermore, our recent studies showed that RD-eumelanin—an oxidation product of RD—exhibits a potent pro-oxidant activity [33] that is enhanced by ultraviolet (UV)-A radiation [34]. In this review, we summarize our biochemical findings on the tyrosinase-dependent metabolism of RD and related studies by other research groups. In addition, the involvement of an immunological mechanism in the spread of depigmented lesions beyond the applied areas will be briefly addressed. A review has already been published that covers the literature on this topic until 2015 [35].
2. Early Events of RD Metabolism Catalyzed by Tyrosinase
RD was put on the market as a depigmenting agent because of its potent inhibition of tyrosinase activity. In fact, Sasaki et al. [36] reported that RD suppressed the tyrosinase activity of human melanocytes in culture and inhibited mushroom tyrosinase activity competitively with a Ki value of 24 μM. It was also reported that when using 3′,5′-[3H]-rhododendrol, RD was oxidized by mushroom tyrosinase with a Km value of 0.27 mM, a value comparable with the Km for l-tyrosine (Km = 0.36 mM).
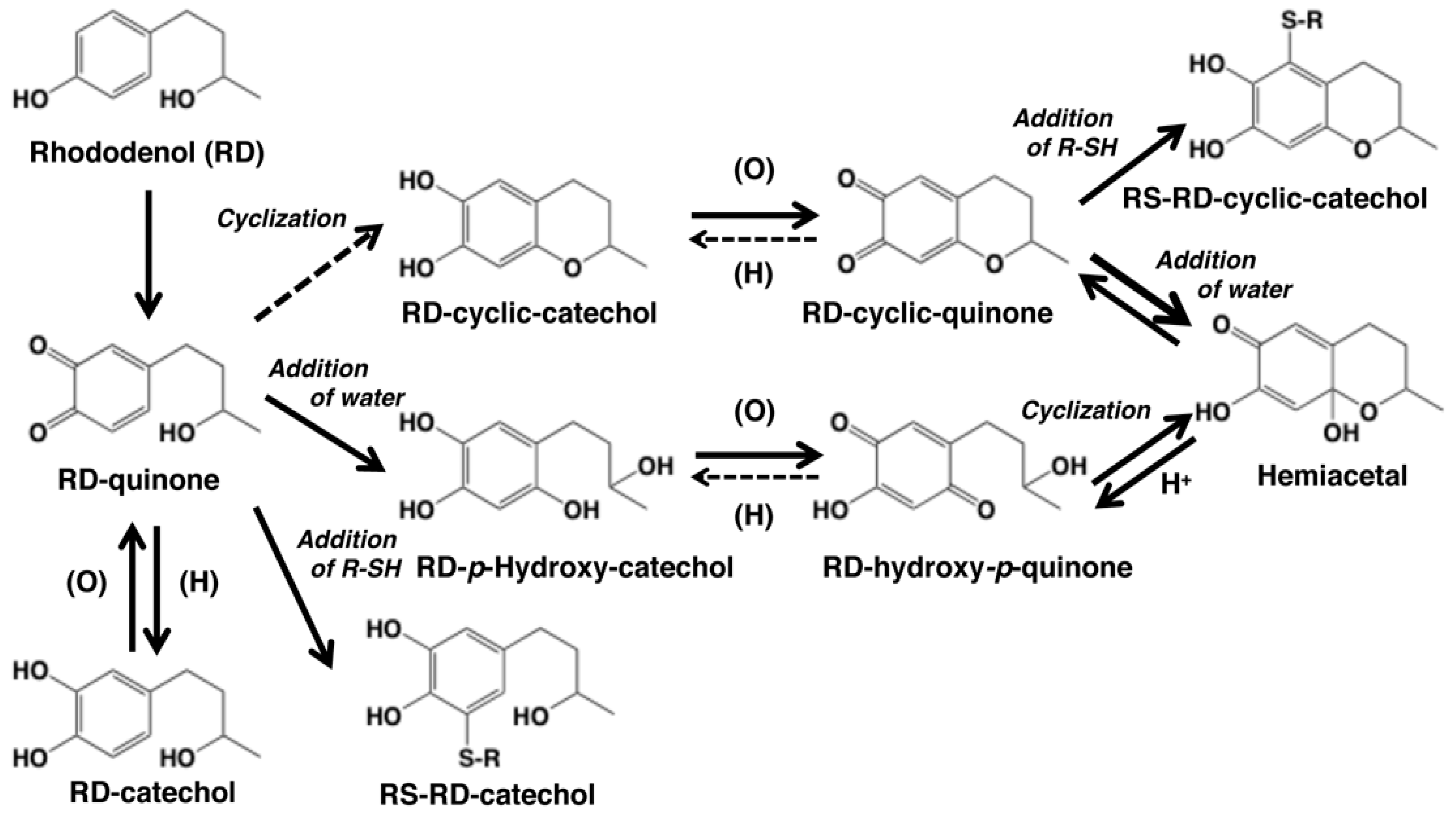
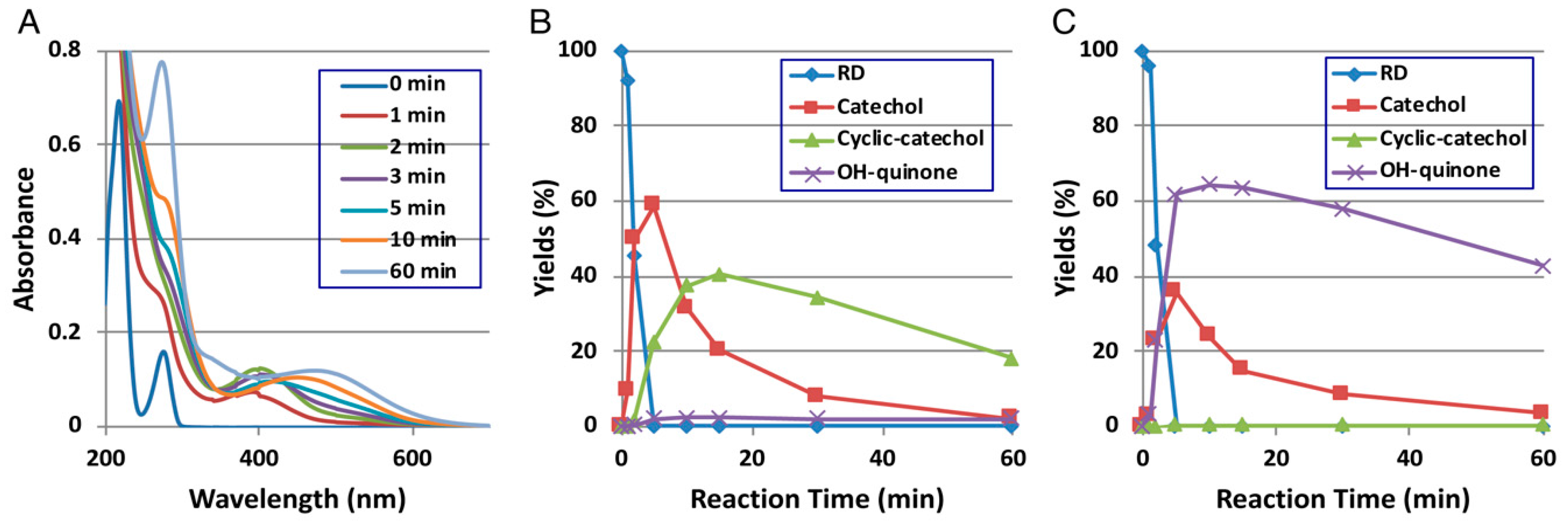
Direct evidence for the production of RD-quinone upon the tyrosinase-catalyzed oxidation of RD was reported by Ito et al. [9], who followed spectral changes in the oxidation of RD by mushroom tyrosinase. The reaction proceeded too rapidly at pH 6.8, and therefore, we slowed down the reaction by performing the oxidation at pH 5.3 (Figure 2A). Within a few minutes, an o-quinone having an absorption maximum near 400 nm appeared, which was replaced in 10 min by a new, distinct absorption peak near 450 nm, suggesting a conversion to secondary o-quinones. This absorption changed further to a spectrum having absorption maxima at 274 and 474 nm.
To identify the oxidation products, we then followed the reaction by HPLC using UV detection at 280 nm to detect both phenols and catechols. The oxidation was stopped by reduction with NaBH4 to convert o-quinones to catechols (Figure 2B). Preparative HPLC afforded two products, one of which was the expected RD-catechol and the other was found to be the RD-cyclic catechol (Figure 1). This identification led to confirmation of the structures of the oxidation products having absorption maxima near 400 nm and 460 nm as RD-quinone and RD-cyclic quinone, respectively. When the oxidation was stopped by acidification with HClO4, another product appeared as a major product (Figure 2C). This compound could not be isolated because of its high instability, but available evidence suggested it to be RD-hydroxy-p-quinone (Figure 2C). It was originally interpreted that RD-cyclic catechol and RD-p-hydroxycatechol arise from an intramolecular addition of the hydroxy group of RD-quinone and an addition reaction of a water molecule to RD-quinone, respectively. Interestingly, upon acidification to pH < 1, RD-cyclic quinone was rapidly converted, through a hemiacetal intermediate, to RD-hydroxy-p-quinone, which had absorption maxima at 270 and 380 nm at pH < 1, similar to those of 4-hydroxydopamine-2,5-quinone (262 and 382 nm).
Kishida et al. [10] investigated the cyclization reaction of RD-quinone using density functional theory-based first principles calculations and found that RD-quinone in the electroneutral structure cannot undergo cyclization, indicating a slow cyclization of RD-quinone at neutral pH. That study also showed that RD-quinone has a preference toward thiol binding rather than cyclization compared to dopaquinone. If the intramolecular addition of the hydroxy group in RD-quinone to form RD-cyclic catechol would be too slow to proceed, another mechanism for the generation of RD-cyclic quinone should be considered. If RD-p-hydroxycatechol would be produced much faster from RD-quinone than RD-cyclic catechol, its oxidized form, RD-hydroxy-p-quinone, may undergo the reverse course of reactions to form RD-cyclic quinone. RD-cyclic quinone and RD-hydroxy-p-quinone appear to exist in equilibrium and gradually undergo dimerization (see [10,35] for similar coupling reactions). The co-existence of RD-cyclic quinone and RD-hydroxy-p-quinone can explain their chemical behaviors as described above.
Thiols such as CySH and GSH have a high reactivity toward o-quinones [15,37]. We examined whether RD-quinone and RD-cyclic quinone are able to react with thiols (R-SH) to form R-SH adducts. This type of addition reaction, thiol binding, was confirmed for CySH, GSH and N-acetylcysteine [9]. In a subsequent study [32], we examined the reactivity of BSA with RD-quinone and RD-cyclic quinone. We found that both quinones bound to BSA effectively through a cysteine residue with yields of about 60%, which is much higher than the binding efficacy (<10%) of dopaquinone [38,39].
Racemic (RS)-RD was used as a topical skin-whitening agent. We then examined whether human tyrosinase is able to oxidize either or both of the enantiomers of RD [31]. Using a chiral HPLC column, racemic RD was resolved optically to R(−)-RD and S(+)-RD enantiomers. In the presence of a catalytic amount of l-dopa, human tyrosinase, which can oxidize l-tyrosine but not d-tyrosine, was found to oxidize both R(−)-RD and S(+)-RD to give RD-catechol and its oxidation products. S(+)-RD was more effectively oxidized than l-tyrosine, while R(-)-RD was as effective as l-tyrosine.
3. RD Metabolism In Vitro and In Vivo
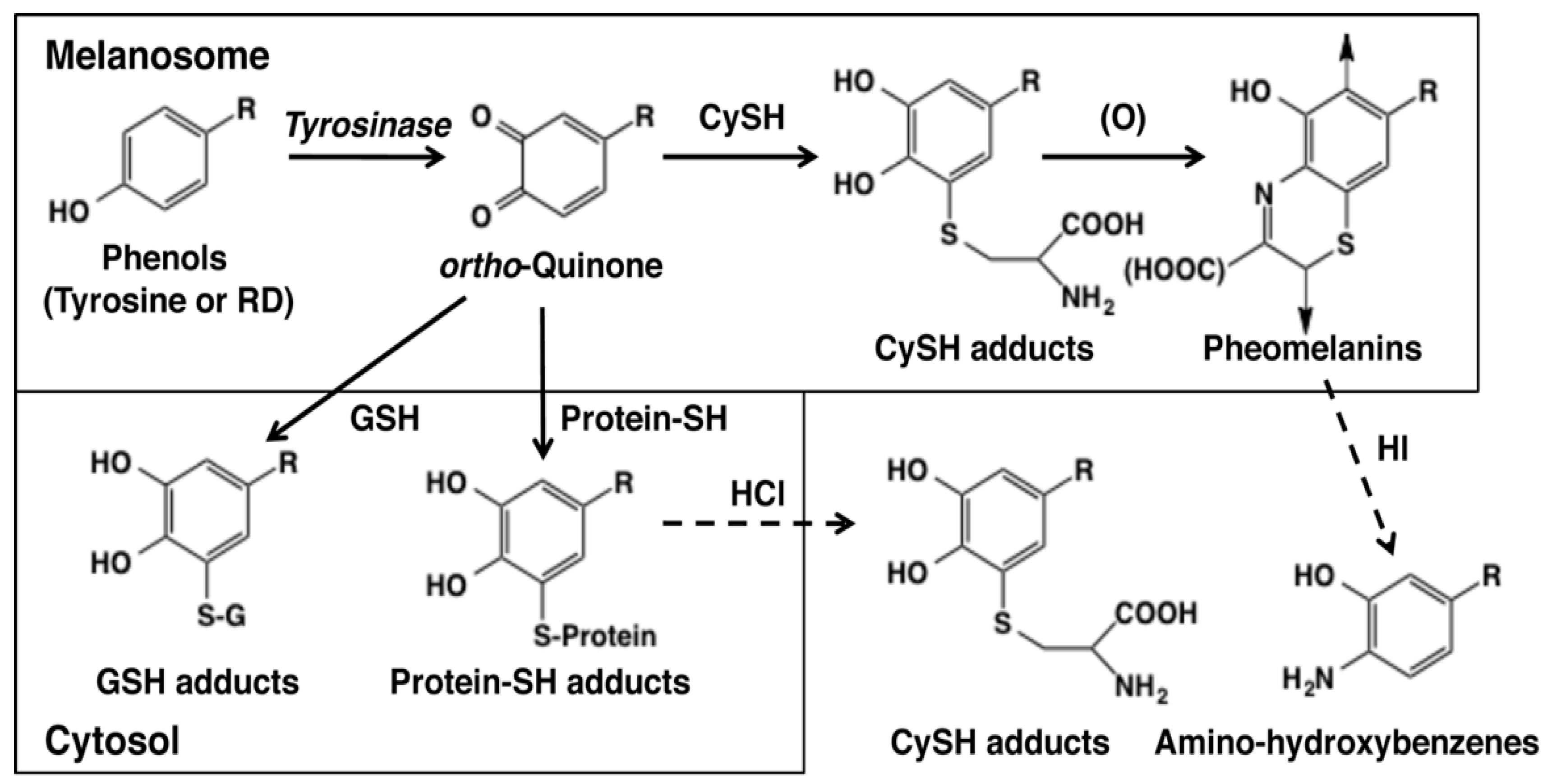
As the early events of RD metabolism by tyrosinase-catalyzed oxidation were clarified, we next examined whether those reactions actually occur in vitro [32]. Figure 3 depicts possible metabolic products in the tyrosinase-catalyzed reactions of l-tyrosine and RD. o-Quinones are produced at the beginning which then bind to CySH that is present abundantly in melanosomes [15,40]. The CySH adducts are oxidized to form pheomelanic pigments (natural pheomelanin and RD-pheomelanin) via benzothiazine intermediates. Pheomelanin is known to exert cytotoxicity to melanocytes through the production of ROS and the depletion of antioxidants [41,42,43]. The amounts of pheomelanic pigments can be analyzed as amino-hydroxybenzene derivatives produced upon hydroiodic acid hydrolysis of pheomelanic pigments [44]. o-Quinones that are produced excessively in melanosomes leak to the cytosol and react with GSH that is present there at high levels to produce GSH adducts. This process should result in the consumption of GSH, possibly leading to oxidative stress. On the other hand, this GSH binding would be a detoxifying process against toxic o-quinones. o-Quinones that have escaped from binding to the small thiols CySH and GSH would then bind to cellular proteins through the thiol group of cysteine residues. This protein binding is considered one of the most important mechanisms of cytotoxicity. The extents of protein binding can be analyzed as CyS-adducts, such as CyS-Dopa or CyS-RD-catechol, after HCl hydrolysis [32,38,39].
Based on the above background, we examined the metabolism of RD in B16F1 mouse melanoma cells in vitro in a collaboration with Toshiharu Yamashita of the Sapporo Medical University [32]. Melanoma cells were exposed to 0.3 mM or 0.5 mM RD (toxic concentration) for three days and metabolites were analyzed by HPLC. The results are summarized in Table 1. Eumelanin levels were reduced to one-eighth that of the control, which accounts for the hypopigmentation of melanocytes exposed to RD. RD-pheomelanin was detected in the RD-exposed cells. Unfortunately, we could not develop a method to detect RD-eumelanin. However, the detection of RD-pheomelanin suggests the production of RD-eumelanin, because the normal course of melanogenesis is known to always produce both eumelanin and pheomelanin (the concept of mixed melanogenesis) [15]. CyS-RD-catechol, an adduct of CySH to RD-quinone, was detected in addition to CyS-Dopa. GS-RD-catechol, an adduct of GSH to RD-quinone, was produced at two-fold greater levels than GS-Dopa, suggesting a high level of excretion of RD-quinone to the cytosol. Adducts to RD-cyclic quinone were not detected. RD-quinone also bound effectively to proteins through the SH group. We observed 20- to 30-fold greater levels of binding of RD-quinone compared to dopaquinone. This high level of protein binding suggests the possibility that the inactivation of SH enzymes and the denaturation of SH proteins are the major mechanism of cytotoxicity of RD-quinone. Lastly, we expected that the cellular levels of GSH and CySH would be much reduced due to binding to RD-quinone. Surprisingly, the levels of GSH and CySH were elevated by exposure to RD roughly two-fold and 10- to 15-fold compared to the control, respectively. This suggests that melanoma cells that have survived the cytotoxicity of RD have a much higher activity of the detoxifying mechanism. This issue will be discussed later.
In a collaborative study with Chikako Nishigori of Kobe University [45], we examined the metabolism of RD in human epidermal melanocytes exposed to UVB irradiation. Higher levels of CyS-RDC and RD-pheomelanin were found in melanocytes treated with RD and UVB compared to the RD only-treated cells.
RD-catechol was detected in human melanocytes with high levels of tyrosinase activity [46]. In our in vitro study using B16 melanoma cells [32], RD-catechol was not detected in cells but was detected in the culture medium.
In addition to the in vitro study, we examined the metabolism of RD in vivo in collaboration with Tamio Suzuki’s group at Yamagata University [47]. In this study, hairless hk14-SCF transgenic mice, which have melanocytes distributed in the epidermis, were treated daily with 30% RD for 28 days. Histological examination indicated a decrease of epidermal melanocytes as early as day 7 and depigmentation in the RD-treated sites appeared on day 14. Eumelanin content was decreased to a 60% level on day 15 while pheomelanin content was increased to some extent. Adducts of RD-quinone to GSH, CySH and protein-SH were also detected in addition to RD-catechol. An earlier in vivo study using brown or black guinea pigs also showed marked decreases of epidermal melanocytes [48].
4. Late Events of RD Metabolism in Relation to the Pro-Oxidant Activity
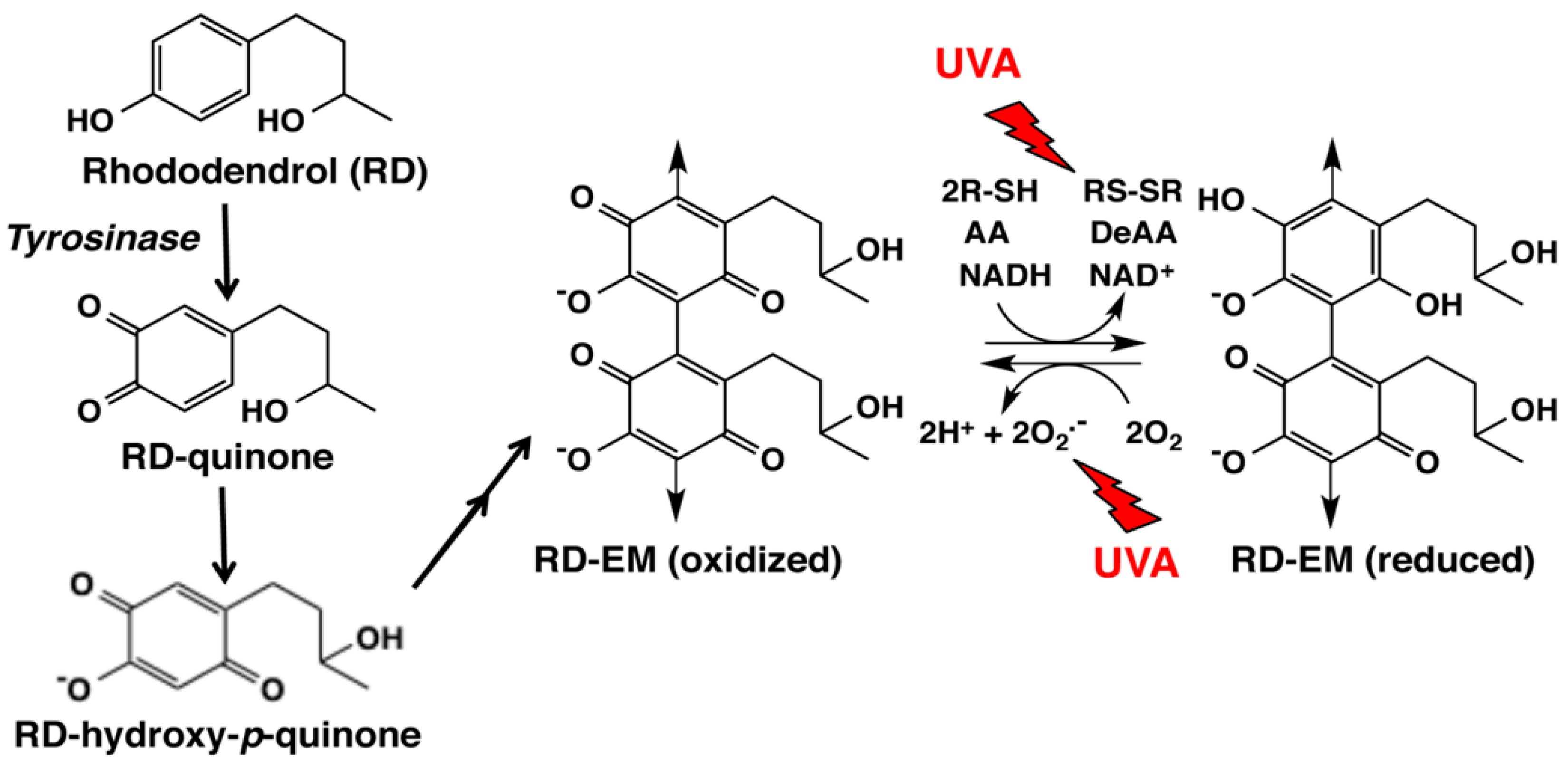
In a recent study, we examined changes of GSH and CySH in B16 melanoma cells exposed to RD for up to 24 h [33]. CySH, but not GSH, was found to decrease during 0.5 to 3 h exposure, due to oxidation to cystine. This selective depletion of CySH may be explained by assuming that this reaction takes place in melanosomes [40]. This pro-oxidant activity was then compared for synthetic melanins prepared from RD or Dopa in the absence or presence of CySH by oxidation with mushroom tyrosinase. RD-eumelanin—a mixture of dimeric and tetrameric RD-hydroxy-p-quinone; Figure 4—was found to exert a pro-oxidant activity as potent as Dopa-pheomelanin (Table 2). GSH, CySH, ascorbic acid and NADH were oxidized by RD-eumelanin with a concomitant production of H2O2 (Figure 4). RD-eumelanin is thus redox-active, possessing both oxidizing and reducing activities. This redox property has been known for many years for melanins [49,50], and a recent study by Kim et al. [51] demonstrated that both pheomelanin and eumelanin are redox-active and that they can rapidly and repeatedly redox-cycle between the oxidized and reduced states.
In our latest study [34], we examined whether this pro-oxidant activity of RD-eumelanin is enhanced by ultraviolet A (UVA) radiation because most RD-induced leukoderma lesions were found in sun-exposed areas [7]. That study also showed that RD-induced leukoderma occurred more frequently in the summer months compared to the spring and autumn months [7]. We found that exposure to a physiological level of UVA (3.5 mW/cm2) [52] induced two- to four-fold increases in the rates of oxidation of GSH, CySH, ascorbic acid and NADH. This oxidation is oxygen-dependent and is accompanied by the production of H2O2. Based on these findings, we propose that RD-eumelanin induces cytotoxicity through its potent pro-oxidant activity that is enhanced by UVA radiation. This study thus suggests UVA radiation as an additional causative factor of RD-induced leukoderma, although the frequent occurrence of RD-induced leukoderma in summer months can be due to the frequent use of RD-containing cosmetics by most of users in sun-exposed area in summer.
5. Melanocyte Toxicity of RD and the Detoxifying Mechanism
Based on the above findings on the early and late events of RD metabolism, we propose the mechanism for the melanocyte toxicity of RD and the detoxifying mechanism against RD as summarized in Figure 5.
5.1. Roles of Tyrosinase
Tyrosinase is able to oxidize a great number of phenols and catechols to form o-quinones. We have shown that mushroom tyrosinase oxidizes not only RD but also other leukoderma-inducing phenols, such as raspberry ketone [53], 4-methoxyphenol, 4-benzyloxyphenol, 4-tert-butylphenol and 4-tert-butylcatechol, to form the corresponding o-quinones [27]. It is apparent that tyrosinase initiates the first step toward the cytotoxicity of RD. In fact, Sasaki et al. [36] showed that treatment with phenylthiourea, a chelator of the copper ions necessary for tyrosinase activity, attenuated RD-induced cytotoxicity in a dose-dependent manner. Similarly, the depletion of tyrosinase by a siRNA against tyrosinase mRNA almost completely rescued melanocytes from damage by RD at concentrations as high as three millimolars. Kasamatsu et al. [46] used 13 different lines of human normal melanocytes with different levels of tyrosinase activity to show that melanocyte damage was related to tyrosinase activity at a certain threshold.
Other studies also indicated a role for tyrosinase in the cytotoxicity of RD to melanocytes. In the in vivo study by Abe et al. [47] (see above), albino mice that lack tyrosinase activity did not show a change in the number of melanocytes when treated with RD for 14 days.
5.2. Cytotoxicity of o-Quinones (RD-Quinone and RD-Cyclic Quinone)
o-Quinones are potent cytotoxic compounds. The cytotoxicity of o-quinones appears to be correlated to the efficacy of binding to cellular proteins. The protein binding leads to the inactivation of SH-enzymes that are essential for cell proliferation and survival such as DNA polymerase [20,21]. The cellular site(s) of adduct formation between RD-quinone and thiol proteins needs to be discussed as it is often mentioned that it is melanosomal proteins that bind to o-quinones. However, the detection of GS-RD-catechol in addition to CyS-RD-catechol is a good indication that a considerable portion of RD-quinone is able to escape from melanosomes to the cytosol where it binds to GSH (Figure 3) [32]. Thus, considering the much lower reactivity of protein-SH compared to CySH and GSH [38,39], most, if not all, of the addition reaction of protein-SH may take place in the cytosol.
How the binding of RD-quinone (2) to protein-SH induces cytotoxicity to melanocytes is a matter of conjecture. A number of studies have shown that RD induces endoplasmic reticulum (ER) stress and the apoptosis that follows [8,36,45,54,55]. Sasaki et al. [36] found that the gene expression level of CCAAT-enhancer-binding protein homologous protein (CHOP), a transcription factor with a major role in unfolded protein response (UPR)-induced cell death, was up-regulated in melanocytes exposed to RD and this up-regulation was tyrosinase-dependent. Also, the release of IL-8 to the culture medium was found to be up-regulated in a tyrosinase-dependent manner. This tyrosinase-dependent cell death induced by RD was found to be apoptotic, as revealed by the increased level of cleaved caspase-3. Lee et al. [54] also reported caspase-3 activation by RD, which is increased by UVB radiation. Arase et al. [55] reported the augmentation of UVB-induced apoptosis of melanocytes exposed to RD and the suppression of NF-κB activation by TNFα that depends on tyrosinase activity. Goto et al. [45] reported that UVB radiation enhanced RD-induced cytotoxicity in melanocytes via the induction of ER stress. An in vivo study by Abe et al. [47] also suggested the involvement of ER stress using biopsy samples from RD exposed mice. In addition, Yang et al. [8] showed that the autophagy-lysosome pathway is involved in RD cytotoxicity using autophagy-deficient and autophagy-enhanced melanocytes.
However, what metabolic event actually elicits the ER stress was not addressed in those studies. One possible cause of ER stress is the accumulation of unfolded proteins [56]. Based on the high level of production of protein-SH adducts to RD-quinone (Table 1), we propose that the accumulation of proteins that are denatured through the binding to RD-quinone should lead to ER-stress and eventually to apoptosis. Other mechanisms of cytotoxicity may also be possible, such as cell damage through antioxidant depletion and/or ROS generation. These mechanisms are not mutually exclusive. This possibility will be discussed later.
If the generation of RD-quinone triggers cytotoxicity to melanocytes, its reduction to RD-catechol within the cells would suppress the cytotoxicity by RD. This was found to be the case. Okubo et al. [57] examined the effect of the forced expression of NAD(P)H quinone dehydrogenase, quinone 1 (NQO1), a major quinone-reducing enzyme in the cytosol (Figure 5) [58]. They found that treatment of B16BL6 mouse melanoma cells or normal human melanocytes with carnoic acid, a transcriptional inducer of the NQO1 gene through activation of the transcription factor, nuclear factor E2-related factor 2 (NRF2), significantly mitigated the cytotoxicity of RD. NRF2 is a master regulator of anti-oxidative responses [59]. Okubo et al. [57] also showed that RD-induced apoptosis, as examined by the cleavage of caspase 3 and poly-ADP ribose polymerase, was strongly suppressed by carnoic acid. RD itself was found to activate the expression of NRF2 and NQO1 in B16 melanoma cells, although the effects of RD were weaker than carnoic acid.
Another possible way for melanocytes to protect themselves from the cytotoxicity of RD-quinone would be to scavenge it through binding with non-protein thiols GSH and CySH (Figure 5). GSH is a major thiol present in the cytosol while CySH is in melanosomes [40]. In fact, we detected GS-RD-catechol and CyS-RD-catechol in B16 melanoma cells exposed to RD (Table 1). In this respect, Kondo et al. [60] showed that GSH maintenance is crucial for the survival of melanocytes after exposure to RD. Pretreatment of normal human epidermal melanocytes with N-acetylcysteine, which leads to an increase in GSH levels, attenuated the cytotoxicity induced by RD, while pretreatment of cells with L-buthionine sulfoximine, a potent and selective inhibitor of GSH synthesis, led to an increased cytotoxicity of RD (and a decrease in GSH). A similar protective role of N-acetylcysteine was also reported by Kim et al. [61]. RD exposure up-regulated the mRNA expression levels of glutamate cysteine ligase (GCL), NQO1 and heme oxygenase 1 (HO-1) in a dose-dependent manner [60]. GCL is a rate-limiting enzyme of GSH synthesis [62] while HO-1 is an enzyme involved in phase II detoxification [63]. Kondo et al. [60] used an NRF2-specific siRNA to show that the RD-induced activation of NRF2 attenuated GSH depletion and promoted melanocyte survival. Cystine-glutamate exchanger (xCT) may act together with GCL to supply GSH (and CySH) that have been depleted through thiol binding to RD-quinone (Figure 5). xCT is a solute carrier family member 11 (SLC7A11) that transports cystine into cells [64].
The binding of GSH to RD-quinone should lead, at least transiently, to the depletion of GSH (and CySH). In fact, in human melanocytes, exposure to 0.1 mM RD for six hours significantly reduced GSH levels [60]. Contrary to our expectation, when we analyzed GSH and CySH levels after exposure of RD to melanoma cells for three days, those levels were significantly increased (Table 1) [32]. The reason for this increase in GSH has now become clear. As mentioned above, the up-regulation of GCL would contribute to the increase in GSH. In fact, GSH levels increased in a dose-dependent manner from the control levels 24 h after RD exposure through the transient decrease at six hours [60].
5.3. Cytotoxicity of Catechols (RD-Catechol and RS-RD-Catechol)
Catechols are known to be cytotoxic to various types of cells, especially to melanocytes and melanoma cells. Two general mechanisms exert the cytotoxicity, one through the production of o-quinones and the other through the production of ROS during autoxidation of catechols or redox-cycling of catechols/o-quinones [20,65]. It may be possible that various catechols produced during the tyrosinase-catalyzed oxidation of RD augment cytotoxic effects on melanocytes. In fact, Okura et al. [66] showed that RD-catechol was approximately 10 times more toxic to B16 melanoma cells and two lines of normal human melanocytes compared to RD and that RD-cyclic catechol was even more toxic than RD-catechol. Another study also found that RD-catechol was much more toxic to human melanocytes [36].
The adduct formation of o-quinones with GSH may not necessarily be a detoxifying event, as some GSH-quinone adducts (such as GS-RD-catechol) retain the ability to redox cycle with the concomitant formation of ROS [67]. However, the protective effects of NQO1 and GSH in detoxifying RD-derived metabolites (as discussed in the previous section) led us to believe that it is RD-quinone rather than RD-catechol that plays a central role in the cytotoxicity to melanocytes exposed to RD.
5.4. Cytotoxicity by the Pro-Oxidant Activity of RD-Eumelanin and RD-Pheomelanin
Several studies have detected ROS generation in melanocytes exposed to RD. For example, Okura et al. [66] showed that RD and RD-catechol produced ROS detected both by flow cytometry and by immunostaining in B16F1 melanoma cells and human melanocytes. Nagata et al. [68] reported a greater level of ROS in B16F10 cells exposed to RD compared to HaCaT keratinocytes. Kim et al. [61] showed approximately 10-fold increased levels of ROS generation in RD-exposed B16F10 cells using a fluorescence assay. Furthermore, Goto et al. [45] reported that ROS generation was synergistically enhanced in normal human melanocytes treated with RD and UVB compared to cells treated with RD alone.
Then how can ROS be generated in RD-treated melanocytes? Natural Dopa-pheomelanin is known to produce superoxide radicals in the dark [42,43], which is accelerated by UV radiation [69]. Our recent study showed that RD-eumelanin is a pro-oxidant as active as Dopa-pheomelanin in oxidizing cellular antioxidants such as GSH, CySH, ascorbic acid and NADH with a concomitant production of H2O2 (Figure 4) [33]. This pro-oxidant activity is significantly enhanced by UVA radiation [34]. RD-pheomelanin possesses a pro-oxidant activity about one-half that of RD-eumelanin. Thus, the production of RD-eumelanin and RD-pheomelanin would lead to both the depletion of antioxidants and the generation of ROS (Figure 5). These pro-oxidant effects of RD-derived melanins can be counter-acted by various antioxidant mechanisms (Figure 5). HO-1 is involved in the survival of H2O2-exposed melanocytes and the susceptibility to vitiligo [63]. It is interesting that up-regulation of the HO-1 gene was approximately 500-fold compared to only three-fold up-regulation of the GCL and NQO1 genes [60]. In addition to NQO1, GCL and HO-1, some other antioxidant enzymes can be up-regulated by the NRF2 signaling pathway (Figure 5) [70]. Thus, glutathione peroxidase (GPx) reduces peroxides in the presence of GSH. In this process, GSH is oxidized to GSSG, which in turn is reduced back to GSH by glutathione reductase (GR) [71]. In this respect, Kim et al. [61] reported that GPx was depleted by the exposure of B16 cells to RD, whose extent was significantly attenuated by N-acetylcysteine. In melanocytes that are surviving the RD-induced toxicity, GSH (and CySH) acts to cope with the cytotoxicity not only through the direct scavenging of RD-quinone but also through the concerted action with GPx (and GR and HO-1) to decompose ROS, in particular H2O2 (Figure 5).
ROS generation during the tyrosinase-catalyzed oxidation of RD was studied by Miyaji et al. [72]. That study detected hydroxyl radical and singlet oxygen during the tyrosinase-catalyzed oxidation of RD using ESR trapping techniques. However, hydroxyl radicals are not directly produced but are derived from H2O2 that is produced from superoxide radicals. Certainly, more studies are necessary to clarify the tyrosinase-depended production of ROS from RD.
6. Immunological Mechanisms
This review is not intended to discuss in depth the immunological aspects of RD-induced leukoderma and its similarity (or dissimilarity) to vitiligo vulgaris. However, immunological reactions are certainly involved in the pathogenesis of the RD-induced leukoderma. For example, Tanemura et al. [6] reported an increase of CD4+ and CD8+ cells infiltrating the dermis of RD-induced leukoderma lesions. Fujiyama et al. [73] observed a high frequency of Melan-A-specific cytotoxic T lymphocytes in RD leukoderma patients. Nishioka et al. [74] found larger numbers of CCR4+ CD8+ cells and higher concentrations of CCL17 and CCL22. These findings may help explain the depigmentation in skin areas not directly exposed to RD and its expansion in some patients [75,76].
How do RD or RD metabolites trigger immunological responses? One possibility is the haptenation theory that was put forth to explain the molecular mechanism of monobenzone-induced skin depigmentation (Figure 5) [26,77]. Phenolic substrates as prohaptens are oxidized by tyrosinase to produce o-quinones, which act as haptens that covalently bind to tyrosinase or other melanosomal (or melanocytic) proteins to generate possible neo-antigens [26,30]. These neo-antigens, in turn, can trigger an immunological response cascade that leads to melanocyte loss to produce depigmentation. However, RD usually causes melanocyte loss only at the applied sites [7], suggesting that immunological responses may not play a major role in the initiation of the RD-induced cytotoxicity.
7. Concluding Remarks
How RD is metabolized by tyrosinase in melanocytes is summarized in Figure 1. RD produces two major toxic metabolites, RD-quinone and RD-melanins. RD-quinone is a highly reactive compound that binds to SH-proteins, leading eventually to cell death (Figure 5). RD-melanins, especially RD-eumelanin, are pro-oxidants that deplete cellular antioxidants with ROS production, leading eventually to cell death. The toxicity of RD-quinone could be prevented through binding to small thiols, GSH and CySH, or through reduction to RD-catechol by NQO1. The toxicity of RD-melanins could be prevented by a concerted action of antioxidant enzymes that are up-regulated by NRF2 (Figure 5).
Then why did only roughly two percent of RD consumers actually develop leukoderma? Figure 5 suggests several factors that lead to RD-induced cytotoxicity, including tyrosinase activity, levels of antioxidant defense mechanism controlled by NRF2 activity, ER-stress response and UV radiation. The activity of tyrosinase is determined by genetic factors [78] and hormonal factors [14,17] as well as by environmental factors, especially its delayed activation by UVB exposure [79]. In this regard, how much RD was applied to the skin by each consumer is important because rather high doses of RD were required to cause cytotoxicity in cultured melanocytes [8,32,36,46] and in an animal model [47,48]. The activity of the ER-stress response may determine whether melanocytes would survive or die [56]. In addition, autophagy may be involved in resistance to the cytotoxicity of RD [8]. Although UVB appears to augment the cytotoxicity of RD [45,54,55], how UVB interacts with RD metabolites directly or indirectly has yet to be examined. Also, the role of UVA in RD-induced cytotoxicity remains to be confirmed by in vitro studies.
Acknowledgements
This work was supported, in part, by a Japan Society for the Promotion of Science (JSPS) grant (No. 26461705) and by a Japan Health, Labour and Welfare Policy Research Grant (H29-Iyaku-Shitei-003). The authors wish to acknowledge collaborations with Toshiharu Yamashita (Sapporo Medical University), Tamio Suzuki (Yamagata University), Chikako Nishigori (Kobe University), Makoto Ojika (Nagoya University) and Ludger Kolbe (Beiersdorf AG).
Author Contributions
Shosuke Ito and Kazumasa Wakamatsu wrote the review manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Tallent, W.H. d-Betuligenol from Rhododendron maximum L. J. Org. Chem. 1964, 29, 988–989. [Google Scholar] [CrossRef]
- Inoue, T.; Ishidate, Y.; Fujita, M.; Kubo, M.; Fukushima, M.; Nagai, M. Studies on the constituents of Aceraceae plants. I. Constituents in the leaves and the stem bark of Acer nikoense Maxim. Yakugaku Zasshi 1978, 98, 41–46. (In Japanese) [Google Scholar] [CrossRef] [PubMed]
- Fuchino, H.; Honishi, S.; Satoh, T.; Yagi, A.; Saitsu, K.; Tatsumi, T.; Tanaka, N. Chemical evaluation of Betula species in Japan. II. Constituents of Betula platyphylia var. japonica. Chem. Pharm. Bull. 1996, 44, 1033–1038. [Google Scholar] [CrossRef]
- Akazawa, H.; Ahihisa, T.; Taguchi, Y.; Banno, N.; Yoneima, R.; Yasukawa, K. Melanogenesis inhibitory and free radical scavenging activities of diarylheptanoids and other phenolic compounds from the bark of Acer nikoense. Biol. Pharm. Bull. 2006, 29, 1970–1972. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Kim, G.; Kaneko, M. Studies on constituents of Rhdodendron fauriei leaves. Yakugaku Zasshi 1942, 62, 4–6. (In Japanese) [Google Scholar]
- Tanemura, A.; Yang, L.; Yang, F.; Nagata, Y.; Wataya-Kaneda, M.; Fukai, K.; Tsuruta, D.; Ohe, R.; Yamakawa, M.; Suzuki, T.; et al. An immune pathological and ultrastructural skin analysis for rhododenol-induced leukoderma patients. J. Dermatol. Sci. 2015, 77, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, Y.; Ito, A.; Suzuki, K.; Suzuki, T.; Tanemura, A.; Nishigori, K.; Ito, M.; Katayama, I.; Sugiura, S.; Matsunaga, K. The first epidemiological report on rhododenol-induced leukoderma in Japan based on a nationwide survey. Jpn. J. Dermatol. 2014, 124, 2095–2109. (In Japanese) [Google Scholar]
- Yang, L.; Yang, F.; Wataya-Kaneda, M.; Tanemura, A.; Tsuruta, D.; Katayama, I. 4-(4-Hydroroxyphenyl)-2-butanol (rhododendrol) activates the autophagy-lysosome pathway in melanocytes: Insights into the mechanisms of rhododendrol-induced leukoderma. J. Dermatol. Sci. 2015, 77, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Yamashita, T.; Ojika, M.; Wakamatsu, K. Tyrosinase-catalyzed oxidation of rhododendrol produces 2-methylchromane-6,7-dione, the putative ultimate toxic metabolite: Implications for melanocyte toxicity. Pigment Cell Melanoma Res. 2014, 27, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Kishida, R.; Kasai, H.; Aspera, S.M.; Arevalo, R.L.; Nakanishi, H. Density functional theory-based First principles calculations of rhododendrol-quinone reactions: Preference to thiol binding over cyclization. J. Phys. Soc. Jpn. 2017, 86, 024804. [Google Scholar] [CrossRef]
- Palumbo, A.; d’Ischia, M.; Misuraca, G.; Prota, G. Skin depigmentation by hydroquinone: A chemical and biochemical insight. Pigment Cell Melanoma Res. 1992, 3, 299–303. [Google Scholar] [CrossRef]
- De Lucia, M.; Panzella, A.; Napolitano, A.; d’Ischia, M. Oxidative chemistry of the natural antioxidant hydroxytyrosol: Hydrogen peroxide-dependent hydroxylation and hydroquinone/o-quinone coupling pathways. Tetrahedron 2006, 62, 1273–1278. [Google Scholar] [CrossRef]
- Cooksey, C.J.; Garratt, P.J.; Land, E.J.; Pavel, S.; Ramsden, C.A.; Riley, P.A.; Smit, N.P.M. Evidence of the indirect formation of the catecholic intermediate substrate responsible for the autoactivation kinetics of tyrosinase. J. Biol. Chem. 1997, 272, 26226–26235. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Wakamatsu, K. Chemistry of mixed melanogenesis—Pivotal roles of dopaquinone. Photochem. Photobiol. 2008, 84, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Hearing, V.J. Determination of melanin synthetic pathways. J. Investig. Dermatol. 2011, 131, E8–E11. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Zmijewski, M.A.; Pawelek, J. l-Tyrosine and l-dihydroxyphenylalanine as hormone-like regulators of melanocyte functions. Pigment Cell Melanoma Res. 2012, 25, 14–27. [Google Scholar] [CrossRef] [PubMed]
- d’Ischia, M.; Wakamatsu, K.; Napolitano, A.; Briganti, S.; Garcia-Borron, J.-C.; Kovacs, D.; Meredith, P.; Pezzella, A.; Picardo, M.; Sarna, T.; et al. Melanins and melanogenesis: Methods, standards, protocols. Pigment Cell Melanoma Res. 2013, 26, 616–633. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, C.A.; Riley, R.A. Tyrosinase: The four oxidation states of the active site and their relevance to enzymatic activation, oxidation and inactivation. Bioorg. Med. Chem. 2014, 22, 2388–2395. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.G.; Tiffany, S.M.; Bell, W.R., Jr.; Gutknecht, W.F. Autoxidation versus covalent binding of quinones as the mechanism of toxicity of dopamine, 6-hydroxydopamine, and related compounds toward C1300 neuroblastoma cells in vitro. Mol. Pharmacol. 1978, 14, 644–653. [Google Scholar] [PubMed]
- Bolton, J.L.; Trush, M.A.; Penning, T.M.; Dryhurst, G.; Monks, T.J. Role of quinones in toxicology. Chem. Res. Toxicol. 2000, 13, 135–160. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, V.; Klarquist, J.; Reust, M.J.; Koshoffer, A.; McKee, M.D.; Boissy, R.E.; Le Poole, I.C. Monobenzyl ether of hydroquinone and 4-tertiary butyl phenol activate markedly different physiological responses in melanocytes: Relevance to skin depigmentation. J. Investig. Dermatol. 2010, 130, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Thörneby-Andersson, K.; Sterner, O.; Hansson, C. Tyrosinase-mediated formation of a reactive quinone from the depigmenting agents, 4-tert-butylphenol and 4-tert-butylcatechol. Pigment Cell Res. 2000, 13, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Naish, S.; Cooksey, C.J.; Riley, P.A. Initial mushroom tyrosinase-catalysed oxidation product of 4-hydroxyanisole is 4-methoxy-ortho-benzoquinone. Pigment Cell Res. 1988, 1, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Manini, P.; Napolitano, A.; Westerhof, W.; Riley, P.A.; d’Ischia, M.A. Reactive ortho-quinone generated by tyrosinase-catalyzed oxidation of the skin depigmenting agent monobenzone: Self-coupling and thiol-conjugation reactions and possible implications for melanocyte toxicity. Chem. Res. Toxicol. 2009, 22, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Van den Boorn, J.G.; Picavet, D.I.; van Swieten, P.F.; van Veen, H.A.; Konijnenberg, D.; van Veelen, P.A.; van Capel, T.; Jong, E.C.; Reits, E.A.; Drijhout, J.W.; et al. Skin-depigmenting agent monobenzone induces potent T-cell autoimmunity toward pigmented cells by tyrosinase haptenation and melanosome autophagy. J. Investig. Dermatol. 2011, 131, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Wakamatsu, K. A convenient screening method to differentiate phenolic skin whitening tyrosinase inhibitors from leukoderma-inducing phenols. J. Dermatol. Sci. 2015, 80, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Tandon, M.; Thomas, P.D.; Shokravi, M.; Singh, S.; Chang, D.; Jimbow, K. Synthesis and antitumor effect of the melanogenesis-based antimelanoma agent, N-propionyl-4-S-cysteaminylphenol. Biochem. Pharmacol. 1998, 55, 2023–2029. [Google Scholar] [CrossRef]
- Ishii-Osai, Y.; Yamashita, T.; Tamura, Y.; Sato, N.; Ito, A.; Honda, H.; Wakamatsu, K.; Ito, S.; Nakayaka, E.; Okura, M.; et al. N-Propionyl-4-S-cysteaminylphenol induces apoptosis in B16F1 cells and mediates tumor-specific T-cell immune responses in a mouse melanoma model. J. Dermatol. Sci. 2012, 67, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Nishigaki, A.; Ishii-Osai, Y.; Ojika, M.; Wakamatsu, K.; Yamashita, T.; Tamura, Y.; Ito, A.; Honda, H.; Nakayama, A.; et al. Mechanism of putative neo-antigen formation from N-propionyl-4-S-cysteaminylphenol, a tyrosinase substrate, in melanoma models. Biochem. Pharmacol. 2012, 84, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Gerwat, W.; Kolbe, L.; Yamashita, T.; Ojika, M.; Wakamatsu, K. Human tyrosinase is able to oxidize both enantiomers of rhododendrol. Pigment Cell Melanoma Res. 2014, 27, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Okura, M.; Nakanishi, Y.; Ojika, M.; Wakamatsu, K.; Yamashita, T. Tyrosinase-catalyzed metabolism of rhododendrol (RD) in B16 melanoma cells: Production of RD-pheomelanin and covalent binding with thiol proteins. Pigment Cell Melanoma Res. 2015, 28, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Okura, M.; Wakamatsu, K.; Yamashita, T. The potent pro-oxidant activity of rhododendrol-eumelanin induces cysteine depletion in B16 melanoma cells. Pigment Cell Melanoma Res. 2017, 30, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Agata, M.; Okochi, K.; Wakamatsu, K. The potent pro-oxidant activity of rhododendrol-eumelanin is enhanced by ultraviolet A radiation. Pigment Cell Melanoma Res. 2018. accepted for publication. [Google Scholar]
- Tokura, Y.; Fujiyama, T.; Ikeya, S.; Tatsuno, K.; Aoshima, M.; Kasuya, A.; Ito, T. Biochemical, cytological, and immunological mechanisms of rhododendrol-induced leukoderma. J. Dermatol. Sci. 2015, 77, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Kondo, M.; Sato, K.; Umeda, M.; Kawabata, K.; Takahashi, Y.; Suzuki, T.; Matsunaga, K.; Inoue, S. Rhododendrol, a depigmentation-inducing phenolic compound, exerts melanocyte cytotoxicity via a tyrosinase-dependent mechanism. Pigment Cell Melanoma Res. 2014, 27, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Tse, D.C.; McCreery, R.L.; Adams, R.N. Potential oxidative pathways of brain catecholamines. J. Med. Chem. 1976, 19, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Ito, S.; Fujita, K. Tyrosinase-catalyzed binding of 3,4-dihydroxyphenylalanine with proteins through the sulfhydryl group. Biochim. Biophys. Acta 1986, 881, 415–421. [Google Scholar] [CrossRef]
- Ito, S.; Kato, T.; Fujita, K. Covalent binding of catechols to proteins through the sulphydryl group. Biochem. Pharmacol. 1988, 37, 1707–1710. [Google Scholar] [CrossRef]
- Potterf, S.B.; Virador, V.; Wakamatsu, K.; Furumura, M.; Santis, C.; Ito, S.; Hearing, V.J. Cysteine transport in melanosomes from murine melanocytes. Pigment Cell Res. 1999, 12, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Mitra, D.; Luo, X.; Morgan, A.; Wang, J.; Hoang, M.P.; Lo, J.; Guerrero, C.R.; Lennerz, J.K.; Mihm, M.C.; Wargo, J.A.; et al. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature 2012, 491, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, A.; Panzella, L.; Monfrecola, G.; d’Ischia, M. Pheomelanin-induced oxidative stress: Bright and dark chemistry bridging red hair phenotype and melanoma. Pigment Cell Melanoma Res. 2014, 27, 721–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panzella, L.; Leone, L.; Greco, G.; Vitiello, G.; D’Errico, G.; Napolitano, A.; d’Ischia, M. Red human hair pheomelanin is a potent pro-oxidant mediating UV-independent contributory mechanisms of melanomagenesis. Pigment Cell Melanoma Res. 2014, 27, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Ito, S.; Rees, J.L. The usefulness of 4-amino-3-hydroxyphenylalanine as a specific marker of pheomelanin. Pigment Cell Res. 2002, 15, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Goto, N.; Tsujimoto, M.; Nagai, H.; Masaki, T.; Ito, S.; Wakamatsu, K.; Nishigori, C. 4-(4-Hydroxyphenyl)-2-butanol (rhododendrol)-induced melanocyte cytotoxicity is enhanced by UVB exposure through generation of oxidative stress. Exp. Dermatol. 2018. in revision. [Google Scholar]
- Kasamatsu, S.; Hachiya, A.; Nakamura, S.; Yasuda, Y.; Fujimori, T.; Takano, K.; Moriwaki, S.; Hase, T.; Suzuki, T.; Matsunaga, K. Depigmentation caused by application of the active brightening material, rhododendrol, is related to tyrosinase activity at a certain threshold. J. Dermatol. Sci. 2014, 76, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Okamura, K.; Kawaguchi, M.; Hozumi, Y.; Aoki, H.; Kunisada, T.; Ito, S.; Wakamatsu, K.; Matsunaga, K.; Suzuki, T. Rhododenol-induced leukoderma in a mouse model mimicking Japanese skin. J. Dermatol. Sci. 2016, 81, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, Y.; Takahashi, Y.; Sakaguchi, H.; Matsunaga, K.; Suzuki, T. Depigmentation of the skin induced by 4-(4-hydroxyphenyl)-2-butanol is spontaneously re-pigmented in brown and black guinea pigs. J. Toxicol. Sci. 2014, 9, 615–623. [Google Scholar] [CrossRef]
- Korytowski, W.; Kalynaraman, B.; Menon, I.A.; Sarna, T.; Sealy, R.C. Reaction of superoxide anions with melanins: Electron spin resonance and spin trapping studies. Biochim. Biophys. Acta 1986, 882, 145–153. [Google Scholar] [CrossRef]
- Korytowski, W.; Plas, B.; Sarna, T.; Kalyanaraman, B. Photoinduced generation of hydrogen peroxide and hydroxyl radicals in melanins. Photochem. Photobiol. 1987, 45, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Panzella, L.; Micillo, R.; Bentley, W.E.; Napolitano, A.; Payne, G.F. Reverse engineering applied to red human hair pheomelanin reveals redox-buffering as a pro-oxidant mechanism. Sci. Rep. 2015, 5, 18447. [Google Scholar] [CrossRef] [PubMed]
- Haywood, R.M.; Andrady, C.; Kassouf, N.; Sheppard, N. Intensity-dependent direct solar radiation- and UVA-induced radical damage to human skin and DNA, lipids and proteins. Photochem. Photobiol. 2011, 87, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Hinoshita, M.; Suzuki, E.; Ojika, M.; Wakamatsu, K. Tyrosinase-catalyzed oxidation of the leukoderma-inducing agent raspberry ketone produces (E)-4-(3-oxo-1-butenyl)-1,2-benzoquinone: Implications for melanocyte toxicity. Chem. Res. Toxicol. 2017, 30, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Joo, Y.H.; Park, M.; Kim, J.-H.; Shin, H.-J.; Park, N.-H.; Lee, J.H.; Park, Y.-H.; Shin, S.S.; Lee, H.-K. Different effects of five depigmentary compounds, rhododendrol, raspberry ketone, monobenzone, rucinol and AP736 on melanogenesis and viability of human epidermal melanocytes. Exp. Dermatol. 2016, 25, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Arase, N.; Yang, L.; Tanemura, A.; Yang, F.; Suenaga, L.; Arase, H.; Katayama, I. The effect of rhododendrol inhibition of NF-κB on melanocytes in the presence of tyrosinase. J. Dermatol. Sci. 2016, 83, 157–159. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Okubo, A.; Yasuhira, S.; Shibazaki, M.; Takahashi, K.; Akasaka, T.; Masuda, T.; Maesawa, C. NAD(P)H dehydrogenase, quinone 1 (NQO1), protects melanin-producing cells from cytotoxicity of rhododendrol. Pigment Cell Melanoma Res. 2016, 29, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Yan, C.; Ross, D. NAD(P)H:quinone oxidoreductase 1 (NQO1) in the sensitivity and resistance to antitumor quinones. Biochem. Pharmacol. 2012, 83, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; von Knethen, A. Nrf2, the master regulator of anti-oxidative responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Kawabata, K.; Sato, K.; Yamaguchi, S.; Hachiya, A.; Takahashi, Y.; Inoue, S. Glutathione maintenance is crucial for survival of melanocytes after exposure to rhododendrol. Pigment Cell Melanoma Res. 2016, 29, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Baek, H.S.; Lee, M.; Park, H.; Shin, S.S.; Choi, D.W.; Lim, K.-M. Rhododenol and raspberry ketone impair the normal proliferation of melanocytes through reactive oxygen species-dependent activation of GADD45. Toxicol. In Vitro 2016, 32, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Li, K.; Liu, L.; Zhang, Y.; Zhou, Z.; Li, C.; Gao, T. Heme oxygenase-1 protects human melanocytes from H2O2-induced oxidative stress via the Nrf2-ARE pathway. J. Investig. Dermatol. 2011, 131, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Mann, G.E. Redox status in mammalian cells and stem cells during culture in vitro: Critical roles of Nrf2 and cystine transporter activity in the maintenance of redox balance. Redox Biol. 2014, 2, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Felim, A.; Urios, A.; Neudorffer, A.; Herrera, G.; Blanco, M.; Largeron, M. Bacterial plate assays and electrochemical methods: An efficient tandem for evaluating the ability of catechol-thioether metabolites of MDMA (“ecstasy”) to induce toxic effects through redox-cycling. Chem. Res. Toxicol. 2007, 20, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Okura, M.; Yamashita, T.; Ishii-Osai, Y.; Yoshikawa, M.; Sumikawa, Y.; Wakamatsu, K.; Ito, S. Effects of rhododendrol and its metabolic products on melanocytic cell growth. J. Dermatol. Sci. 2015, 80, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Monks, T.J.; Hanzlik, R.P.; Cohen, G.M.; Ross, D.; Graham, D.G. Contemporary issues in toxicology. Quinone chemistry and toxicity. Toxicol. Appl. Pharmacol. 1992, 112, 2–16. [Google Scholar] [CrossRef]
- Nagata, T.; Ito, S.; Itoga, K.; Hanazawa, H.; Masaki, H. The mechanism of melanocytes-specific cytotoxicity induced by phenol compounds having a prooxidant effect, relating to the appearance of leukoderma. Biomed. Res. Int. 2015, 2015, 479798. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.; Hong, L.; Garguilo, J.; Pawlak, A.; Edwards, G.S.; Nemanich, R.J.; Sarna, T.; Simon, J.D. Photoionization thresholds of melanins obtained from free electron laser-photoelectron emission microscopy, femtosecond transient absorption spectroscopy and electron paramagnetic resonance measurements of oxygen photoconsumption. Photochem. Photobiol. 2006, 82, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol. Med. 2013, 65, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Miyaji, A.; Gabe, Y.; Kohno, M.; Baba, T. Generation of hydroxyl radicals and singlet oxygen during oxidation of rhododendrol and rhododendrol-catechol. J. Clin. Biochem. Nutr. 2017, 60, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Fujiyama, T.; Ikeya, S.; Ito, T.; Tatsuno, K.; Aoshima, M.; Kasuya, A.; Sakabe, J.; Suzuki, T.; Tokura, Y. Melanocyte-specific cytotoxic T lymphocytes in patients with rhododendrol-induced leukoderma. J. Dermatol. Sci. 2015, 77, 190–192. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, M.; Tanemura, A.; Yang, L.; Tanaka, A.; Arase, N.; Katayama, I. Possible involvement of CCR4+CD8+ T cells and elevated plasma CCL22 and CCL17 in patients with Rhododenol-induced leukoderma. J. Dermatol. Sci. 2015, 77, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Nishigori, C.; Aoyama, Y.; Ito, A.; Suzuki, K.; Suzuki, T.; Tanemura, A.; Ito, M.; Katayama, I.; Oiso, N.; Kagohashi, Y.; et al. Guide for medical professionals (i.e., dermatologists) for the management of Rhododenol-induced leukoderma. J. Dermatol. 2015, 42, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.E. Chemical-induced vitiligo. Dermatol. Clin. 2017, 35, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Westerhof, W.; Manini, P.; Napolitano, A.; d’Ischia, M. The haptenation theory of vitiligo and melanoma rejection: A close-up. Exp. Dermatol. 2011, 20, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Sturm, R.A. Molecular genetics of human pigmentation diversity. Hum. Mol. Genet. 2009, 18, R9–R17. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Miyamura, Y.; Wolber, R.; Smuda, C.; Reihold, W.; Liu, H.; Kolbe, L.; Hearing, V.J. Regulation of human skin pigmentation in situ by repetitive UV exposure: Molecular characterization of responses to UVA and/or UVB. J. Investig. Dermatol. 2010, 130, 1685–1696. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Scheme showing the tyrosinase-catalyzed oxidation of rhododendrol (RD) in the absence or presence of thiols R-SH. The oxidation of RD gives RD-quinone as an immediate product, which gives rise to RD-cyclic quinone and RD-hydroxy-p-quinone [9]. These quinones are produced by redox exchange of the corresponding catechols, RD-cyclic catechol and RD-p-hydroxycatechol, which are produced through an intramolecular cyclization of RD-quinone and the addition of a water molecule to RD-quinone, respectively. However, the feasibility of the cyclization was questioned recently by Kishida et al. [10]. RD-cyclic quinone produces RD-hydroxy-p-quinone instantaneously upon acidification via the hemiacetal intermediate. The reverse reaction from RD-hydroxy-p-quinone to RD-cyclic quinone appears to proceed rapidly at neutral pH to explain the presence of the latter quinone in the oxidation mixture. These quinones exist in an equilibrium. RD-hydroxy-p-quinone decays gradually through a coupling to form RD-eumelanin (see [11,12] for similar coupling reactions of hydroxy-p-quinones). RD-catechol can be produced through a redox exchange between RD-quinone and other catechol products. Tyrosinase-catalyzed oxidation of RD and RD-cyclic catechol in the presence of thiols (R-SH) affords the thiol adducts, RS-RD-catechol and RS-RD-cyclic catechol, respectively. Adapted from Ito et al. [9] with some modifications.
Figure 1.
Scheme showing the tyrosinase-catalyzed oxidation of rhododendrol (RD) in the absence or presence of thiols R-SH. The oxidation of RD gives RD-quinone as an immediate product, which gives rise to RD-cyclic quinone and RD-hydroxy-p-quinone [9]. These quinones are produced by redox exchange of the corresponding catechols, RD-cyclic catechol and RD-p-hydroxycatechol, which are produced through an intramolecular cyclization of RD-quinone and the addition of a water molecule to RD-quinone, respectively. However, the feasibility of the cyclization was questioned recently by Kishida et al. [10]. RD-cyclic quinone produces RD-hydroxy-p-quinone instantaneously upon acidification via the hemiacetal intermediate. The reverse reaction from RD-hydroxy-p-quinone to RD-cyclic quinone appears to proceed rapidly at neutral pH to explain the presence of the latter quinone in the oxidation mixture. These quinones exist in an equilibrium. RD-hydroxy-p-quinone decays gradually through a coupling to form RD-eumelanin (see [11,12] for similar coupling reactions of hydroxy-p-quinones). RD-catechol can be produced through a redox exchange between RD-quinone and other catechol products. Tyrosinase-catalyzed oxidation of RD and RD-cyclic catechol in the presence of thiols (R-SH) affords the thiol adducts, RS-RD-catechol and RS-RD-cyclic catechol, respectively. Adapted from Ito et al. [9] with some modifications.

Figure 2.
Time course of the tyrosinase-catalyzed oxidation of RD. (A) UV/Vis spectral changes over time at pH 5.3; (B) HPLC following oxidation at pH 5.3, the reaction being stopped by the addition of NaBH4 followed by the addition of HClO4; (C) HPLC following oxidation at pH 5.3, the reaction being stopped by the addition of HClO4 alone. Adapted from Ito et al. [9].
Figure 2.
Time course of the tyrosinase-catalyzed oxidation of RD. (A) UV/Vis spectral changes over time at pH 5.3; (B) HPLC following oxidation at pH 5.3, the reaction being stopped by the addition of NaBH4 followed by the addition of HClO4; (C) HPLC following oxidation at pH 5.3, the reaction being stopped by the addition of HClO4 alone. Adapted from Ito et al. [9].

Figure 3.
Metabolic pathways for phenols (such as RD or l-tyrosine) catalyzed by tyrosinase in melanin-producing cells [32]. The oxidation of RD or tyrosine with tyrosinase gives RD-quinone or dopaquinone as an immediate product in melanosomes. These quinones bind CySH to give CyS-RD-catechol or CyS-Dopa, respectively. They are oxidized to RD-pheomelanin and Dopa-pheomelanin. RD-eumelanin and Dopa-eumelanin are also produced in melanosomes [15]. RD-pheomelanin and Dopa-pheomelanin can be analyzed as amino-hydroxybutylbenzene and amino-hydroxyphenylalanine, respectively, after HI hydrolysis [32,44]. When leaked to the cytosol, these quinones bind to GSH to give GS-RD-catechol or GS-Dopa, respectively. CyS-catechols and GS-catechols can be analyzed as such by HPLC. Quinones that have escaped from binding to CySH or GSH react with protein-SH to form protein-S-RD-catechol or protein-S-Dopa. These protein adducts can be analyzed as CyS-RD-catechol or CyS-Dopa, respectively, after HCl hydrolysis [32,38,39].
Figure 3.
Metabolic pathways for phenols (such as RD or l-tyrosine) catalyzed by tyrosinase in melanin-producing cells [32]. The oxidation of RD or tyrosine with tyrosinase gives RD-quinone or dopaquinone as an immediate product in melanosomes. These quinones bind CySH to give CyS-RD-catechol or CyS-Dopa, respectively. They are oxidized to RD-pheomelanin and Dopa-pheomelanin. RD-eumelanin and Dopa-eumelanin are also produced in melanosomes [15]. RD-pheomelanin and Dopa-pheomelanin can be analyzed as amino-hydroxybutylbenzene and amino-hydroxyphenylalanine, respectively, after HI hydrolysis [32,44]. When leaked to the cytosol, these quinones bind to GSH to give GS-RD-catechol or GS-Dopa, respectively. CyS-catechols and GS-catechols can be analyzed as such by HPLC. Quinones that have escaped from binding to CySH or GSH react with protein-SH to form protein-S-RD-catechol or protein-S-Dopa. These protein adducts can be analyzed as CyS-RD-catechol or CyS-Dopa, respectively, after HCl hydrolysis [32,38,39].

Figure 4.
Tyrosinase-catalyzed oxidation of RD giving rise to RD-eumelanin (RD-EM). Our previous study [9] showed that the immediate product is RD-quinone, which undergoes the addition of a water molecule and is oxidized to form RD-hydroxy-p-quinone. RD-hydroxy-p-quinone gradually dimerizes to form RD-EM [9,11,12]. The oxidized form of RD-EM is able to oxidize thiols (R-SH), ascorbic acid (AA) and NADH to disulfides (RS-SR), dehydroascorbic acid (DeAA) and NAD+, respectively, while the reduced form of RD-EM is able to reduce molecular oxygen producing superoxide radicals. It is likely that RD-EM is a mixture of dimers and tetramers [33]. Our latest study showed that the pro-oxidant activity of RD-EM is enhanced by UVA radiation [34].
Figure 4.
Tyrosinase-catalyzed oxidation of RD giving rise to RD-eumelanin (RD-EM). Our previous study [9] showed that the immediate product is RD-quinone, which undergoes the addition of a water molecule and is oxidized to form RD-hydroxy-p-quinone. RD-hydroxy-p-quinone gradually dimerizes to form RD-EM [9,11,12]. The oxidized form of RD-EM is able to oxidize thiols (R-SH), ascorbic acid (AA) and NADH to disulfides (RS-SR), dehydroascorbic acid (DeAA) and NAD+, respectively, while the reduced form of RD-EM is able to reduce molecular oxygen producing superoxide radicals. It is likely that RD-EM is a mixture of dimers and tetramers [33]. Our latest study showed that the pro-oxidant activity of RD-EM is enhanced by UVA radiation [34].

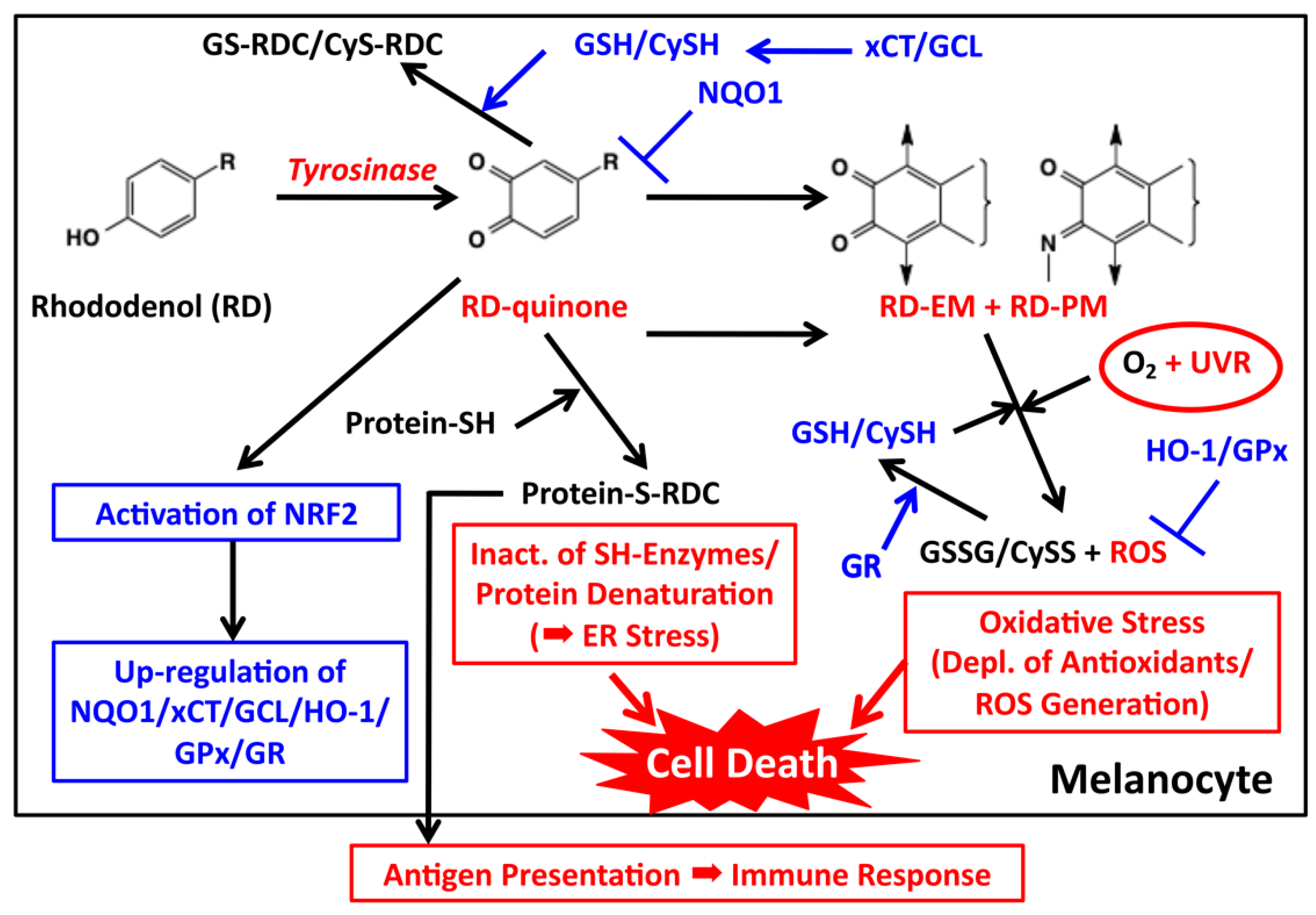
Figure 5.
Proposed biochemical mechanism of RD-induced leukoderma. Toxic events are shown in red while detoxifying events are shown in blue. Tyrosinase catalyzes the oxidation of RD to form two toxic metabolites, RD-quinone and RD-melanin. RD-quinone reacts with CySH and GSH to give the adducts, CyS-RD-catechol (CyS-RDC) and GS-RD-catechol (GS-RDC). RD-quinone also reacts with protein-SH to give the protein adduct, protein-S-RD-catechol (protein-S-RDC). This reaction leads to the inactivation of SH enzymes and the unfolding of SH proteins, which lead to cytotoxicity and ER stress, respectively. This protein binding may produce antigens, which lead eventually to an immune response. RD-quinone is then further metabolized to RD-eumelanin and RD-pheomelanin. These melanins, especially RD-eumelanin, exert a potent pro-oxidant activity oxidizing antioxidants and producing ROS. This pro-oxidant activity can be enhanced by UVA radiation. UVB might also be involved in this process. These reactions lead to oxidative stress.
Figure 5.
Proposed biochemical mechanism of RD-induced leukoderma. Toxic events are shown in red while detoxifying events are shown in blue. Tyrosinase catalyzes the oxidation of RD to form two toxic metabolites, RD-quinone and RD-melanin. RD-quinone reacts with CySH and GSH to give the adducts, CyS-RD-catechol (CyS-RDC) and GS-RD-catechol (GS-RDC). RD-quinone also reacts with protein-SH to give the protein adduct, protein-S-RD-catechol (protein-S-RDC). This reaction leads to the inactivation of SH enzymes and the unfolding of SH proteins, which lead to cytotoxicity and ER stress, respectively. This protein binding may produce antigens, which lead eventually to an immune response. RD-quinone is then further metabolized to RD-eumelanin and RD-pheomelanin. These melanins, especially RD-eumelanin, exert a potent pro-oxidant activity oxidizing antioxidants and producing ROS. This pro-oxidant activity can be enhanced by UVA radiation. UVB might also be involved in this process. These reactions lead to oxidative stress.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Concentrations of Tyrosine- and RD-derived Metabolites in B16 Melanoma Cells Cultured for Three Days a.
Table 1.
Concentrations of Tyrosine- and RD-derived Metabolites in B16 Melanoma Cells Cultured for Three Days a.
| Metabolite b | RD Not Added | RD 0.3 mM | RD 0.5 mM |
|---|---|---|---|
| Eumelanin | 17 μg | 1.8 μg | 2.1 μg |
| Pheomelanin | 0.71 μg | 0.57 μg | 0.47 μg |
| RD-pheomelanin | 0.00 μg | 0.51 μg | 0.61 μg |
| CyS-Dopa | 0.17 nmol | 0.33 nmol | 0.36 nmol |
| CyS-RD-catechol | 0.00 nmol | 0.09 nmol | 0.15 nmol |
| GS-Dopa | 0.24 nmol | 0.08 nmol | 0.12 nmol |
| GS-RD-catechol | 0.00 nmol | 0.17 nmol | 0.26 nmol |
| Protein-S-Dopa | 0.16 nmol | 0.11 nmol | 0.10 nmol |
| Protein-S-RD-catechol | 0.00 nmol | 2.2 nmol | 3.1 nmol |
| GSH | 25 nmol | 38 nmol | 59 nmol |
| CySH | 1.3 nmol | 13 nmol | 20 nmol |
a Summarized from the data in Ito et al. [32]; b Concentrations are per one million cells. Averages are from three experiments.
Table 2.
Oxidation of various antioxidants by synthetic melanin a.
| Antioxidant b | Control | RD-EM | RD-PM | Dopa-EM | Dopa-PM |
|---|---|---|---|---|---|
| GSH, 60 min | 0.7 | 67.3 | 29.7 | 33.0 | 62.3 |
| CySH, 30 min | 10.7 | 90.3 | 41.3 | 79.7 | 80.0 |
| AA, 30 min | 3.9 | 83.1 | 71.0 | 75.9 | 46.4 |
| NADH, 120 min | 5.7 | 58.7 | 37.7 | 85.5 | 84.8 |
a Summarized from the data in Ito et al. [33]. Melanins were prepared by tyrosinase oxidation of precursors (RD or Dopa in the absence/presence of CySH). Oxidation was carried out with one molar equivalent of melanin (as a precursor) for the indicated time. Control contained tyrosinase alone; b Data are percent consumption of the antioxidant. Averages are from three experiments. Variations were less than five percent in most cases. Most of GSH or CySH were oxidized to GSSG or CySSCy.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ito, S.; Wakamatsu, K. Biochemical Mechanism of Rhododendrol-Induced Leukoderma. Int. J. Mol. Sci. 2018, 19, 552. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19020552
AMA Style
Ito S, Wakamatsu K. Biochemical Mechanism of Rhododendrol-Induced Leukoderma. International Journal of Molecular Sciences. 2018; 19(2):552. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19020552
Chicago/Turabian StyleIto, Shosuke, and Kazumasa Wakamatsu. 2018. "Biochemical Mechanism of Rhododendrol-Induced Leukoderma" International Journal of Molecular Sciences 19, no. 2: 552. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19020552
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.