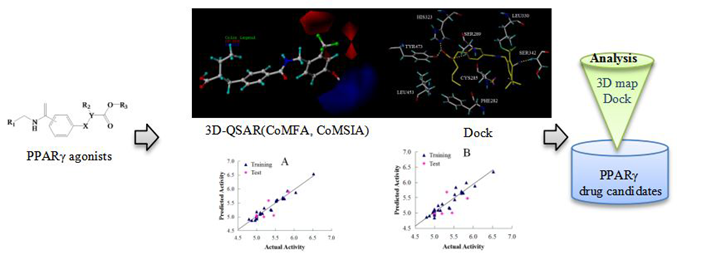
Molecular Modeling Study for the Design of Novel Peroxisome Proliferator-Activated Receptor Gamma Agonists Using 3D-QSAR and Molecular Docking


Abstract
:
1. Introduction
2. Results and Discussion
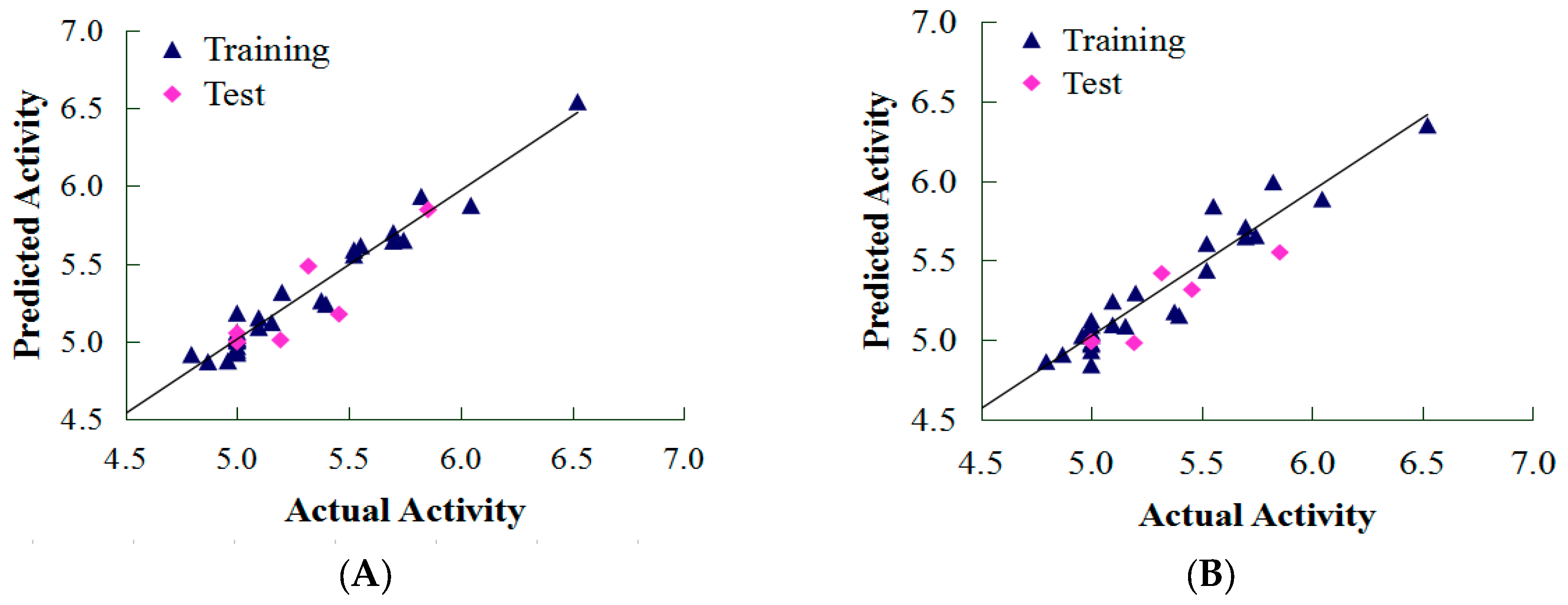
2.1. CoMFA and CoMSIA Results
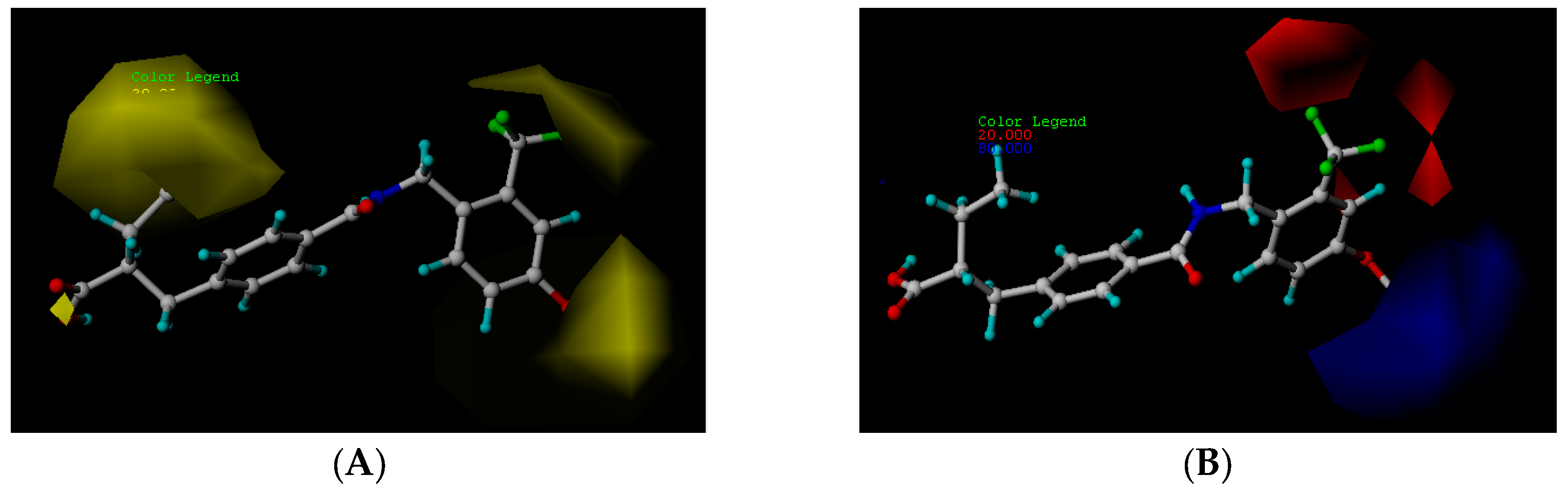
2.2. CoMFA Contour Map Analysis
2.2.1. Steric Contour Map
2.2.2. Electrostatic Contour Map
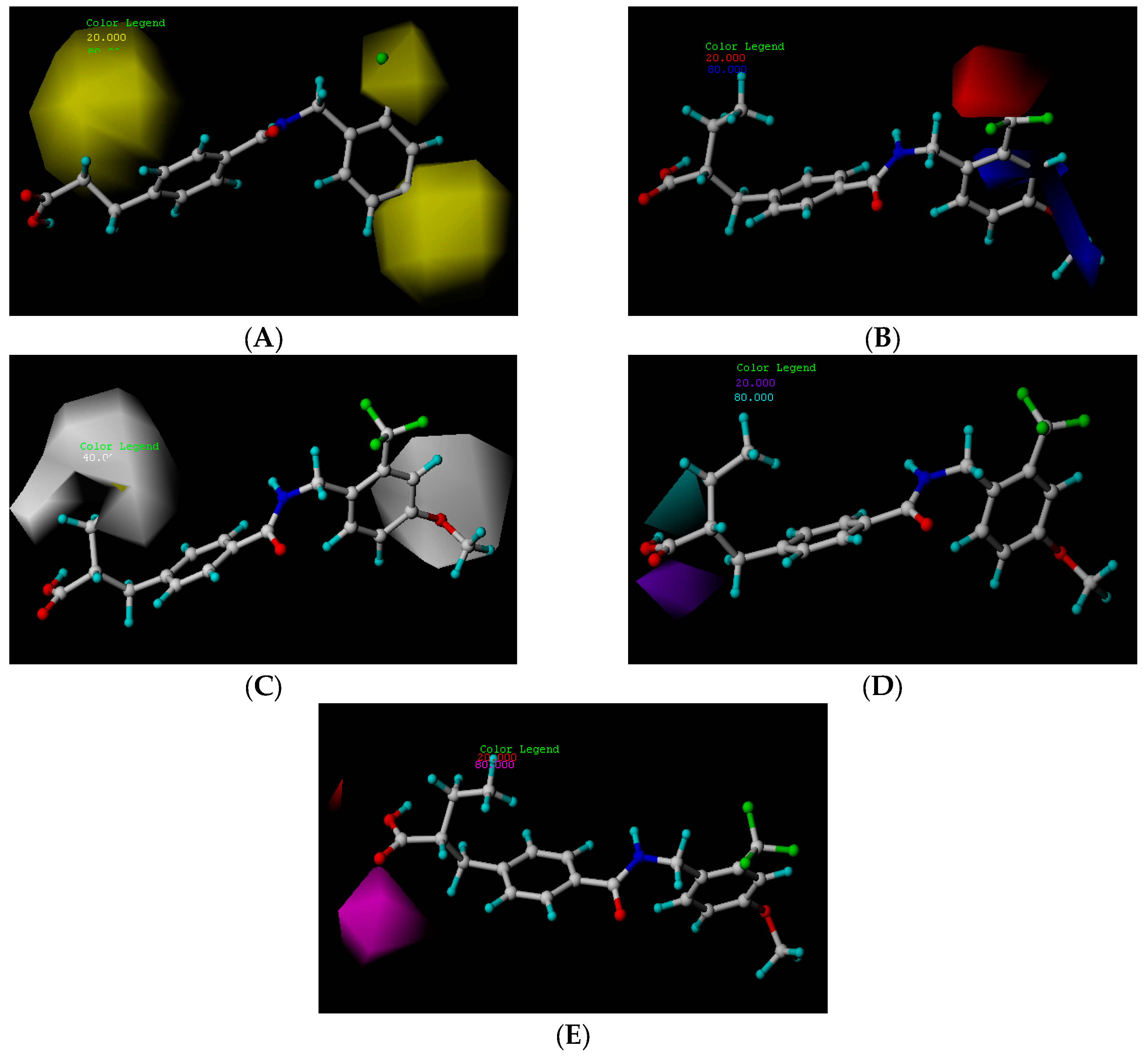
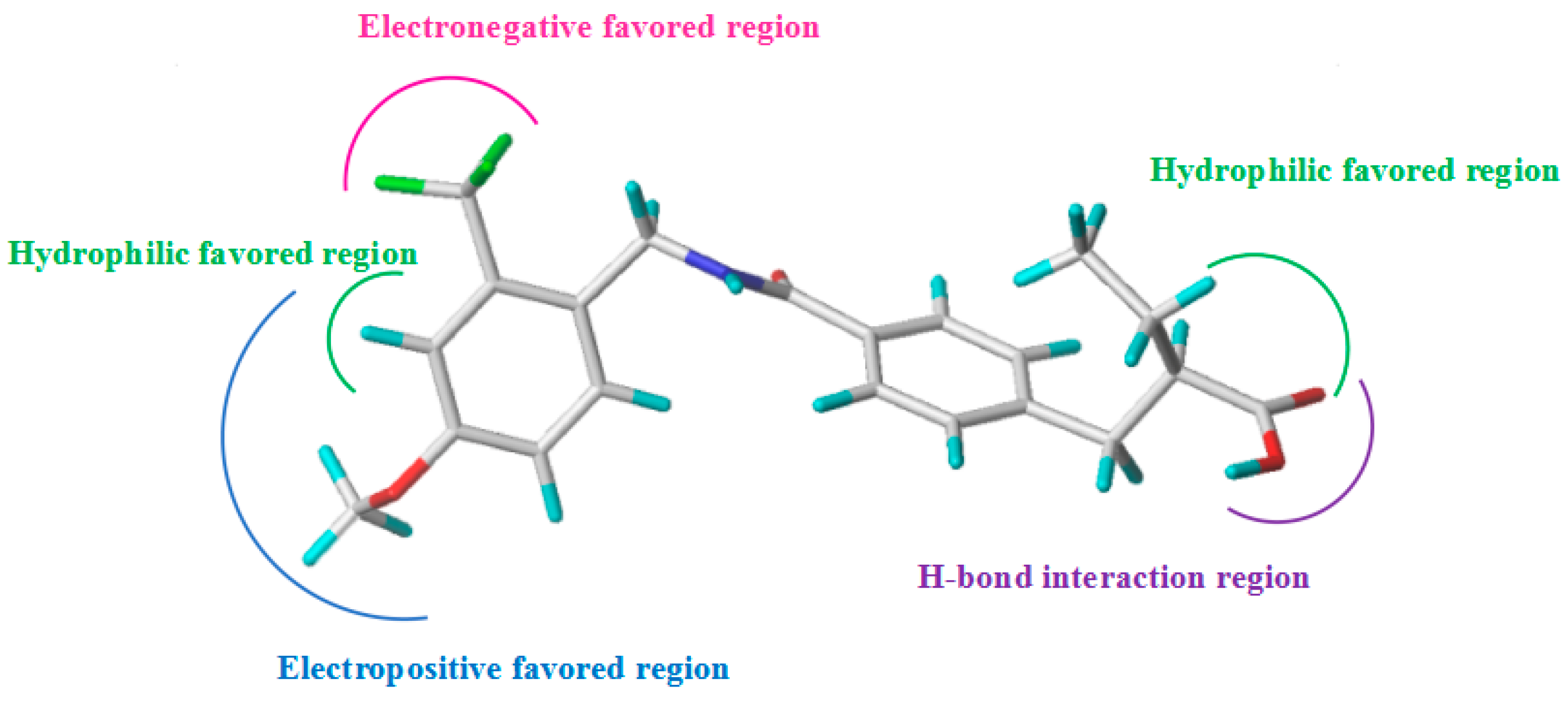
2.3. CoMSIA Contour Map Analysis
2.3.1. Hydrophobic Contour Map
2.3.2. Hydrogen Bond Donor Map
2.3.3. Hydrogen Bond Acceptor Map
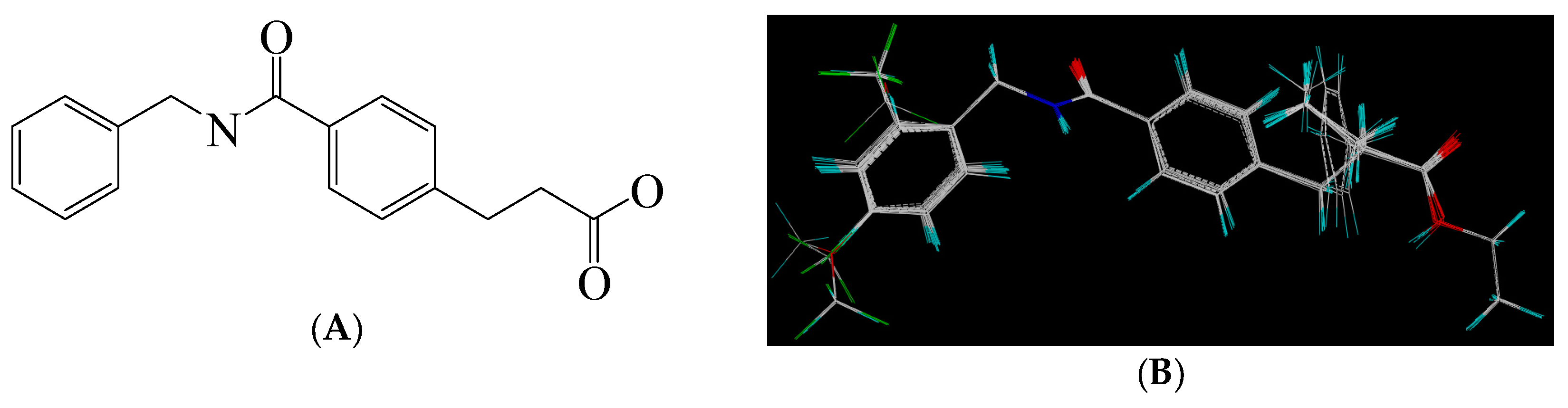
2.4. Design of More Potent Compounds
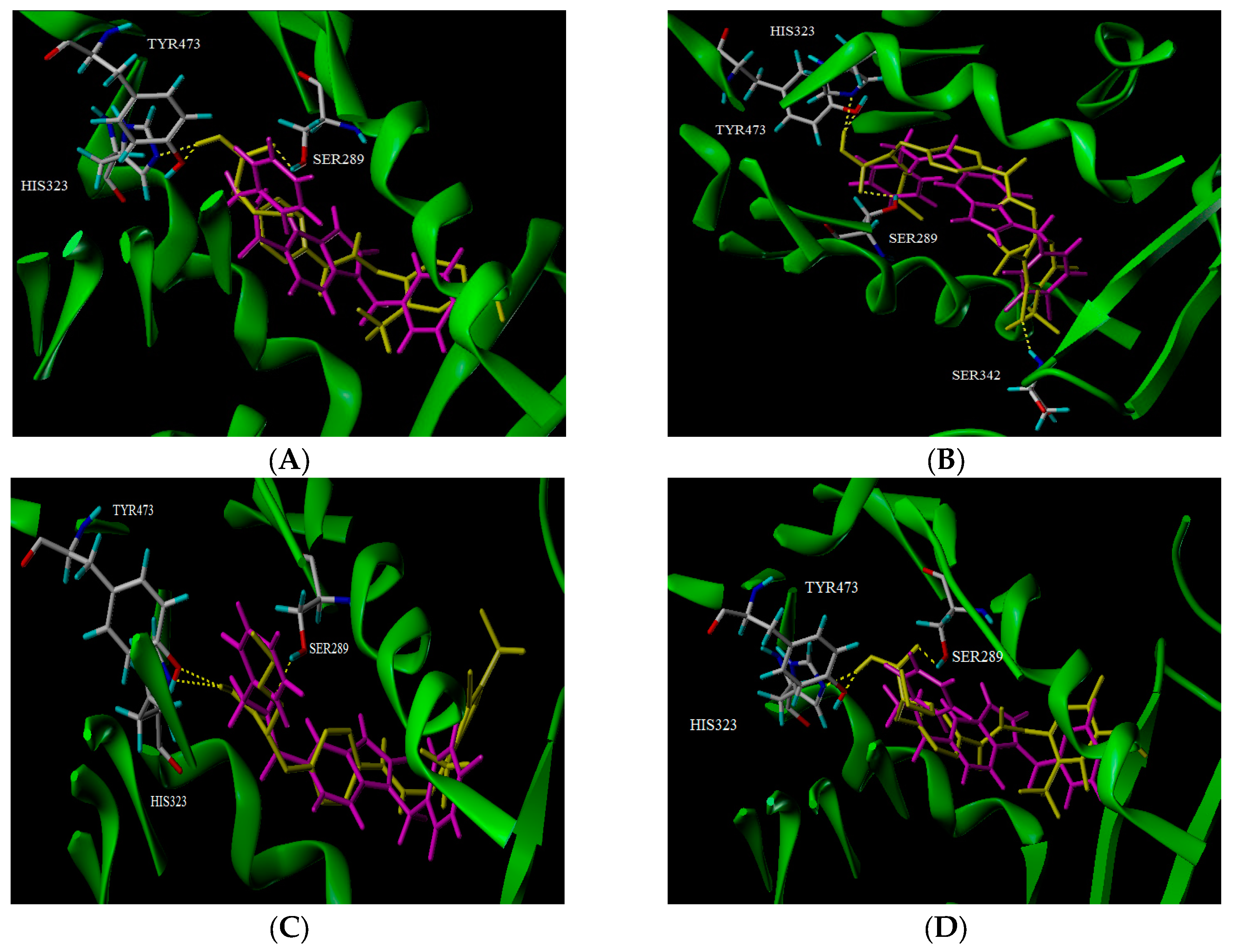
2.5. Docking Analysis
3. Materials and Methods
3.1. Data Set
3.2. Molecular Modeling and Alignment
3.3. CoMFA Method
3.4. CoMSIA Method
3.5. Internal Validation and Partial Least Squares (PLS) Analysis
3.6. External Validation of the QSAR Model
3.7. Molecular Docking
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Spurr, S.; Bally, J.; Bullin, C.; Trinder, K. Type 2 Diabetes in Canadian Aboriginal Adolescents: Risk Factors and Prevalence. J. Pediatr. Nurs. 2017, 36, 111–117. [Google Scholar] [CrossRef] [PubMed]
- American diabetes association. Economic Costs of Diabetes in the U.S. in 2012. Diabetes Care 2013, 36, 1033–1046. [Google Scholar]
- Seuring, T.; Archangelidi, O.; Suhrcke, M. The Economic Costs of Type 2 Diabetes: A Global Systematic Review. PharmacoEconomics 2015, 33, 811–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, J.M.; Moore, L.B.; Smith-Oliver, T.A.; Wilkison, W.O.; Willson, T.M.; Kliewer, S.A. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPARγ). J. Biol. Chem. 1995, 270, 12953–12956. [Google Scholar] [CrossRef] [PubMed]
- Willson, T.M.; Cobb, J.E.; Cowan, D.J.; Wiethe, R.W.; Correa, I.D.; Prakash, S.R.; Beck, K.D.; Moore, L.B.; Kliewer, S.A.; Lehmann, J.M. The Structure-Activity Relationship between Peroxisome Proliferator-Activated Receptor γ Agonism and the Antihyperglycemic Activity of Thiazolidinediones. J. Med. Chem. 1996, 39, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Barroso, I.; Gurnell, M.; Crowley, V.E.; Agostini, M.; Schwabe, J.W.; Soos, M.A.; Maslen, G.L.; Williams, T.D.; Lewis, H.; Schafer, A.J.; et al. Dominant negative mutations in human ppar gamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 1999, 402, 880–883. [Google Scholar] [CrossRef] [PubMed]
- Flavell, D.M.; Ireland, H.; Stephens, J.W.; Hawe, E.; Acharya, J.; Mather, H.; Hurel, S.J.; Humphries, S.E. Peroxisome proliferator-activated receptor α gene variation influences age of onset and progression of type 2 diabetes. Diabetes 2005, 54, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Jay, M.A.; Ren, J. Peroxisome proliferator-activated receptor (PPAR) in metabolic syndrome and type 2 diabetes mellitus. Curr. Diabetes Rev. 2007, 3, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.W. The Clinical Significance of PPAR Gamma Agonism. Curr. Mol. Med. 2005, 5, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Soodvilai, S. Renal and vascular mechanisms of thiazolidinedione-induced fluid retention. PPAR Res. 2008, 2008. [Google Scholar] [CrossRef] [PubMed]
- Blöcher, R.; Lamers, C.; Wittmann, S.K.; Merk, D.P.; Hartmann, M.; Weizel, L.; Diehl, O.; Brüggerhoff, A.; Boß, M.; Kaiser, A.; et al. N-benzylbenzamides: A novel merged scaffold for orally available dual soluble epoxide hydrolase/peroxisome proliferator-activated receptor γ modulators. J. Med. Chem. 2015, 59, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Sahu, N.K.; Sharma, M.C.; Mourya, V.; Kohli, D.V. QSAR studies of some side chain modified 7-chloro-4-aminoquinolines as antimalarial agents. Arab. J. Chem. 2014, 7, 701–707. [Google Scholar] [CrossRef]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular Similarity Indices in a Comparative Analysis (CoMSIA) of Drug Molecules to Correlate and Predict Their Biological Activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.; Roy, K. On Selection of Training and Test Sets for the Development of Predictive QSAR models. Qsar. Comb. Sci. 2010, 25, 235–251. [Google Scholar] [CrossRef]
- Roy, K.; Paul, S. Exploring 2D and 3D QSARs of 2,4-Diphenyl-1,3-oxazolines for Ovicidal Activity Against Tetranychus urticae. Qsar. Comb. Sci. 2009, 28, 406–425. [Google Scholar] [CrossRef]
- Patel, P.; Chintha, C.; Ghate, M.; Bhatt, H.; Vyas, V.K. 3D QSAR study of 4H-chromen-1,2,3,4-tetrahydropyrimidine-5-carboxylate derivatives as potential anti-mycobacterial agents. Med. Chem. Res. 2014, 23, 2955–2963. [Google Scholar] [CrossRef]
- Wang, F.F.; Zhou, B. Toward the identification of a reliable 3D-QSAR model for the protein tyrosine phosphatase 1B inhibitors. J. Mol. Struct. 2018, 1158, 75–87. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, H.; Xiao, Z.; Wang, F.; Wang, X.; Wang, Y. Combined 3D-QSAR, molecular docking and molecular dynamics study on derivatives of peptide epoxyketone and tyropeptin-boronic acid as inhibitors against the β5 subunit of human 20s proteasome. Int. J. Mol. Sci. 2011, 12, 1807–1835. [Google Scholar] [CrossRef] [PubMed]
- Damre, M.V.; Gangwal, R.P.; Dhoke, G.V.; Lalit, M.; Sharma, D.; Khandelwal, K.; Sangamwar, A.T. 3D-QSAR and molecular docking studies of amino-pyrimidine derivatives as PknB inhibitors. J. Taiwan Inst. Chem. E 2014, 45, 354–364. [Google Scholar] [CrossRef]
- Xie, X.Q.; Chen, J.Z. Data Mining a Small Molecule Drug Screening Representative Subset from NIH Pub Chem. J. Chem. Inf. Model. 2008, 48, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Ståhle, L.; Wold, S. Partial least squares analysis with cross-validation for the two-class problem: A Monte Carlo study. J. Chemometr. 1987, 1, 185–196. [Google Scholar] [CrossRef]
- Xiao, A.; Zhang, Z.; An, L.; Xiang, Y. 3D-QSAR and docking studies of 3-arylquinazolinethione derivatives as selective estrogen receptor modulators. J. Mol. Model. 2008, 14, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.; Cramer, R.D.; Opdenbosch, N.V. Validation of the general purpose tripos 5.2 force field. J. Comput. Chem. 1989, 10, 982–1012. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Ambure, P.S.; Gangwal, R.P.; Sangamwar, A.T. 3D-QSAR and molecular docking analysis of biphenyl amide derivatives as p38α mitogen-activated protein kinase inhibitors. Mol. Divers. 2012, 16, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Shi, J.; Zhao, G.; Zhou, X.E.; Suinopowell, K.; Melcher, K.; Xu, H.E. Identification of a novel selective pparγ ligand with a unique binding mode and improved therapeutic profile in vitro. Sci. Rep. 2017, 7, 41487. [Google Scholar] [CrossRef] [PubMed]
- Van Westen, G.J.; Overington, J.P.A. ligand’s-eye view of protein similarity. Nat. Methods 2013, 10, 116–117. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLS Statistics | ONC | q2 | r2 | SEE | F | Cotribution (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| S | E | H | D | A | ||||||
| CoMFA | ||||||||||
| S | 8 | 0.576 | 0.930 | 0.132 | 29.872 | 100 | – | – | – | – |
| E | 7 | 0.497 | 0.937 | 0.122 | 40.470 | – | 100 | – | – | – |
| SE | 6 | 0.750 | 0.958 | 0.097 | 76.113 | 51.6 | 48.4 | – | – | – |
| CoMSIA | ||||||||||
| S | 5 | 0.472 | 0.804 | 0.205 | 17.183 | 100 | – | – | – | – |
| E | 7 | 0.428 | 0.911 | 0.145 | 27.952 | – | 100 | – | – | – |
| H | 10 | 0.506 | 0.958 | 1.108 | 36.771 | – | – | 100 | – | – |
| D | 2 | −0.051 | 0.168 | 0.395 | 2.418 | – | – | – | 100 | – |
| A | 1 | −0.083 | 0.030 | 0.418 | 0.777 | – | – | – | – | 100 |
| SE | 10 | 0.61 | 0.949 | 0.120 | 29.708 | 34.0 | 66.0 | – | – | – |
| SH | 3 | 0.412 | 0.823 | 0.186 | 35.545 | 36.1 | – | 63.9 | – | – |
| SD | 10 | 0.505 | 0.935 | 0.135 | 23.071 | 48.3 | – | – | 51.7 | – |
| SA | 4 | 0.493 | 0.803 | 0.201 | 22.415 | 84.9 | – | – | – | 15.1 |
| EH | 5 | 0.479 | 0.891 | 0.153 | 34.255 | – | 59.1 | 40.9 | – | – |
| ED | 9 | 0.352 | 0.932 | 0.134 | 25.959 | – | 88.1 | – | 11.9 | – |
| EA | 6 | 0.433 | 0.876 | 0.167 | 23.640 | – | 89.1 | – | – | 10.9 |
| HD | 10 | 0.537 | 0.965 | 0.100 | 43.744 | – | – | 75.1 | 24.9 | – |
| HA | 10 | 0.525 | 0.958 | 0.109 | 36.225 | – | – | 90.6 | – | 9.4 |
| DA | 2 | −0.036 | 0.186 | 0.391 | 2.740 | – | – | – | 81.0 | 19.0 |
| SEH | 5 | 0.541 | 0.916 | 0.134 | 45.732 | 17.9 | 51.9 | 30.2 | – | – |
| SED | 10 | 0.6 | 0.954 | 0.113 | 33.508 | 32.0 | 55.9 | – | 12.1 | – |
| SEA | 9 | 0.607 | 0.944 | 0.121 | 32.005 | 34.9 | 60.4 | – | – | 4.7 |
| SHD | 5 | 0.448 | 0.913 | 0.137 | 43.915 | 28.9 | – | 51.0 | 20.1 | – |
| SHA | 5 | 0.426 | 0.909 | 0.140 | 41.769 | 33.8 | – | 59.6 | – | 6.6 |
| SDA | 4 | 0.501 | 0.800 | 0.202 | 22.018 | 57.8 | – | – | 32.2 | 9.9 |
| EHD | 5 | 0.478 | 0.885 | 0.157 | 32.229 | 53.1 | – | 35.9 | 11.0 | – |
| EHA | 5 | 0.492 | 0.889 | 0.154 | 33.757 | – | 56.8 | 38.9 | – | 4.3 |
| EDA | 9 | 0.368 | 0.941 | 0.125 | 30.157 | – | 79.6 | – | 11.8 | 8.6 |
| HDA | 10 | 0.532 | 0.964 | 0.100 | 43.071 | – | – | 70.1 | 23.5 | 6.4 |
| SEHD | 5 | 0.545 | 0.912 | 0.137 | 43.732 | 16.1 | 47.7 | 27.1 | 9.1 | – |
| SEHA | 5 | 0.55 | 0.915 | 0.135 | 45.170 | 17.3 | 50.5 | 28.9 | – | 3.3 |
| SEDA | 10 | 0.593 | 0.954 | 0.113 | 33.520 | 30.3 | 52.9 | – | 11.7 | 5.1 |
| SHDA | 5 | 0.449 | 0.905 | 0.143 | 39.940 | 28.1 | – | 49.5 | 17.5 | 4.9 |
| EHDA | 5 | 0.486 | 0.884 | 0.159 | 32.044 | – | 51.6 | 35.0 | 9.7 | 3.7 |
| SEHDA | 5 | 0.551 | 0.912 | 0.138 | 43.388 | 15.7 | 46.7 | 26.5 | 8.2 | 2.8 |

| NO. | R1 | R2 | R3 | CoMFA Predicted | CoMSIA Predicted |
|---|---|---|---|---|---|
| N1 | CN | C(Me)3 | Me | 6.942 | 7.170 |
| N2 | CN | C(Me)3 | OMe | 6.765 | 7.155 |
| N3 | CN | C(Me)3 | ET | 6.900 | 7.155 |
| N4 | CN | CH(Me)2 | OMe | 6.982 | 7.105 |
| N5 | CN | C(Me)3 | – | 6.822 | 7.083 |
| N6 | CN | CH(Me)2 | Me | 7.064 | 7.080 |
| N7 | CN | CH(Me)2 | ET | 7.103 | 7.070 |
| N8 | COOH | C(Me)3 | Me | 6.820 | 7.063 |
| N9 | CN | CH(Me)2 | – | 7.036 | 6.999 |
| N10 | Cl | C(Me)3 | Me | 6.742 | 6.986 |
| N11 | CHO | C(Me)3 | Me | 6.878 | 6.916 |
| N12 | COOH | CH(Me)2 | Me | 7.073 | 6.902 |
| N13 | CHO | C(Me)3 | c-Pr | 6.815 | 6.886 |
| N14 | Cl | CH(Me)2 | Me | 6.781 | 6.843 |
| N15 | – | C(Me)3 | – | 6.648 | 6.798 |

| NO. | R1 | R2 | Substitution | Actual pEC50 | Pred-pEC50 | |
|---|---|---|---|---|---|---|
| CoMFA | CoMSIA | |||||
| 01a * |  | Et | para | 5.000 | 4.996 | 4.992 |
| 02b |  | Et | para | 5.745 | 5.650 | 5.655 |
| 03c * |  | Et | para | 5.319 | 5.483 | 5.420 |
| 04b |  | Et | para | 4.796 | 4.915 | 4.867 |
| 05c |  | Et | para | 4.870 | 4.869 | 4.911 |
| 06b |  | Et | para | 5.000 | 4.941 | 4.845 |
| 07b |  | Et | para | 5.000 | 5.015 | 5.029 |
| 08c |  | Et | para | 5.000 | 5.039 | 5.093 |
| 09c |  | Et | para | 5.000 | 5.019 | 5.053 |
| 10b * |  | Et | para | 5.456 | 5.174 | 5.317 |
| 11c |  | Et | para | 5.097 | 5.090 | 5.097 |
| 12b |  | Et | para | 4.959 | 4.874 | 5.028 |
| 13c |  | Et | para | 5.000 | 4.924 | 5.125 |
| 14b |  | Et | para | 5.377 | 5.261 | 5.178 |
| 15c |  | Et | para | 5.201 | 5.315 | 5.297 |
| 16b |  | Et | para | 5.523 | 5.588 | 5.607 |
| 17c |  | Et | para | 5.699 | 5.645 | 5.712 |
| 18b |  | Et | para | 5.000 | 5.060 | 4.984 |
| 19c |  | Et | para | 5.155 | 5.121 | 5.089 |
| 20b |  | Et | para | 5.000 | 5.182 | 5.047 |
| 21c |  | Et | para | 5.398 | 5.239 | 5.156 |
| 22b * |  | Et | para | 5.854 | 5.846 | 5.551 |
| 23b |  | Et | para | 5.553 | 5.615 | 5.841 |
| 24c |  | Et | para | 6.523 | 6.542 | 6.349 |
| 25b * |  | Et | meta | 5.000 | 5.052 | 4.991 |
| 26c * |  | Et | meta | 5.194 | 5.008 | 4.983 |
| 27b |  | H | para | 5.000 | 4.966 | 4.934 |
| 28c |  | H | para | 5.000 | 5.002 | 4.974 |
| 29b |  | Me | para | 5.097 | 5.151 | 5.245 |
| 30b |  | Pr | para | 6.046 | 5.875 | 5.886 |
| 31c |  | Pr | para | 5.824 | 5.933 | 5.993 |
| 32b |  | Phenyl | para | 5.699 | 5.699 | 5.648 |
| 33c |  | Phenyl | para | 5.523 | 5.556 | 5.439 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jian, Y.; He, Y.; Yang, J.; Han, W.; Zhai, X.; Zhao, Y.; Li, Y. Molecular Modeling Study for the Design of Novel Peroxisome Proliferator-Activated Receptor Gamma Agonists Using 3D-QSAR and Molecular Docking. Int. J. Mol. Sci. 2018, 19, 630. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19020630
Jian Y, He Y, Yang J, Han W, Zhai X, Zhao Y, Li Y. Molecular Modeling Study for the Design of Novel Peroxisome Proliferator-Activated Receptor Gamma Agonists Using 3D-QSAR and Molecular Docking. International Journal of Molecular Sciences. 2018; 19(2):630. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19020630
Chicago/Turabian StyleJian, Yaning, Yuyu He, Jingjing Yang, Wei Han, Xifeng Zhai, Ye Zhao, and Yang Li. 2018. "Molecular Modeling Study for the Design of Novel Peroxisome Proliferator-Activated Receptor Gamma Agonists Using 3D-QSAR and Molecular Docking" International Journal of Molecular Sciences 19, no. 2: 630. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19020630