Bias-Exchange Metadynamics Simulation of Membrane Permeation of 20 Amino Acids

 , , , ,
, , , ,
Abstract
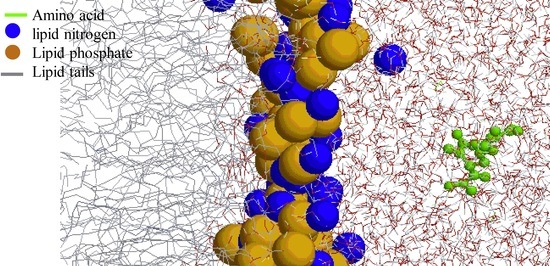
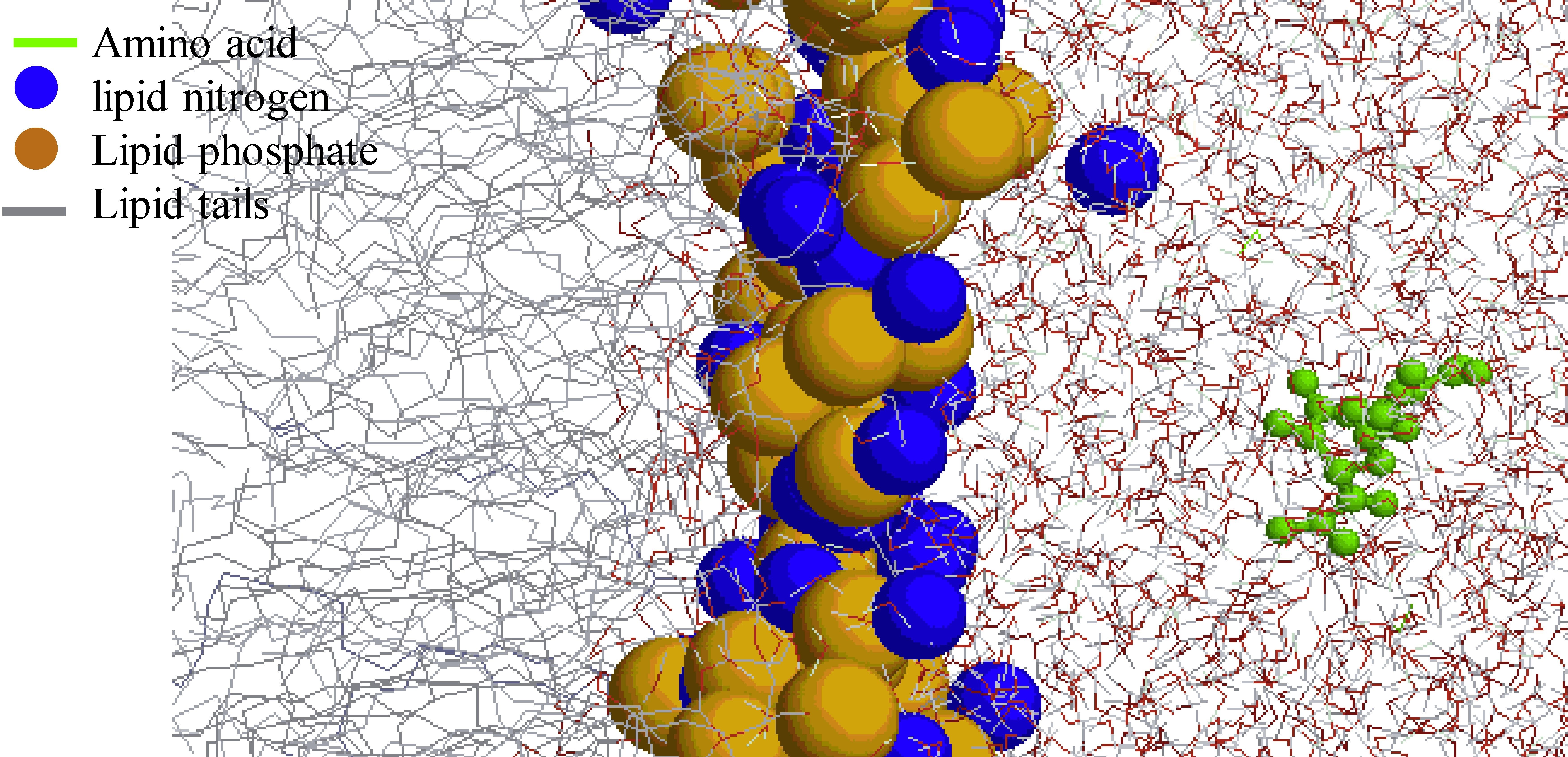
:
1. Introduction
2. Results
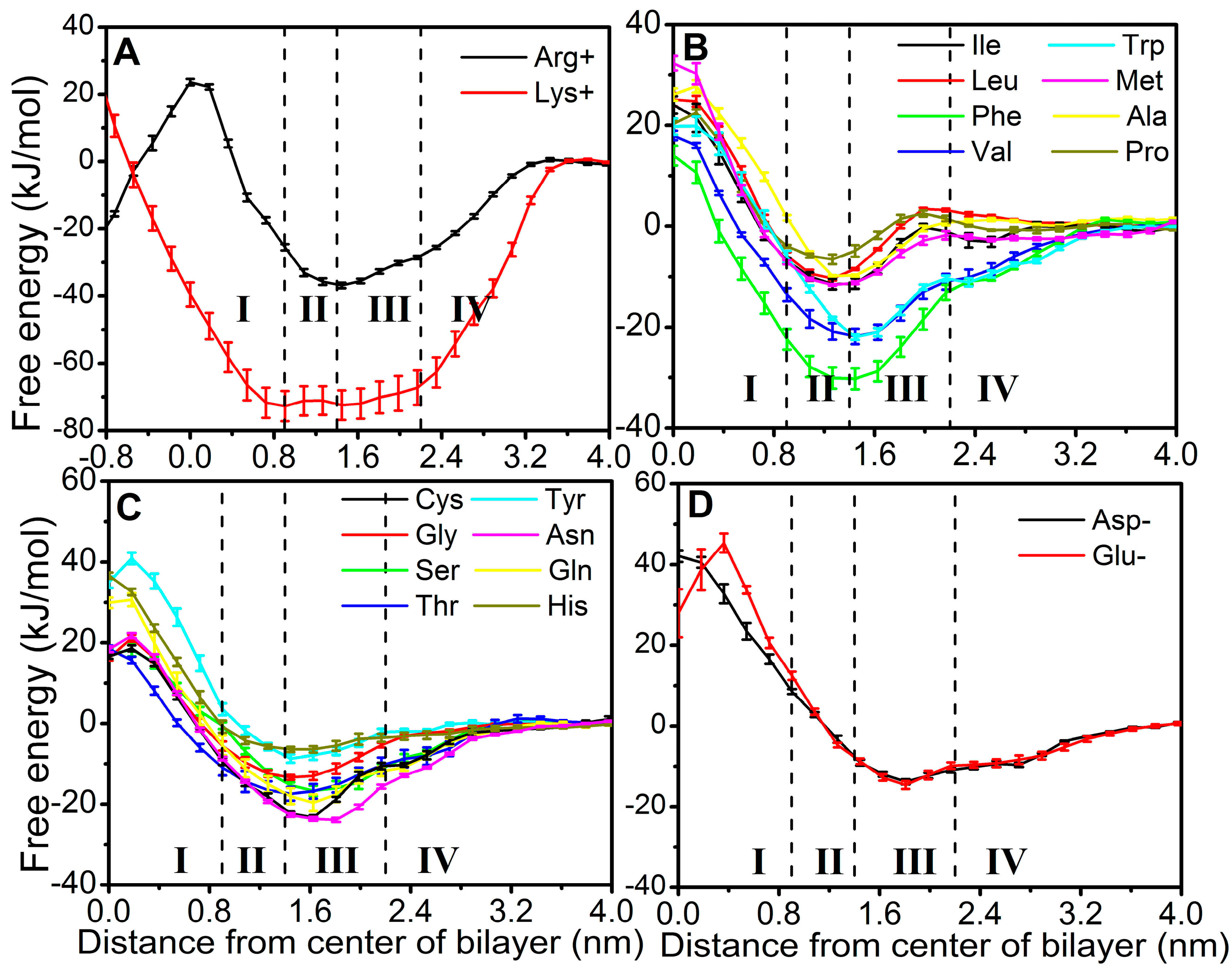
2.1. Examination of Convergence
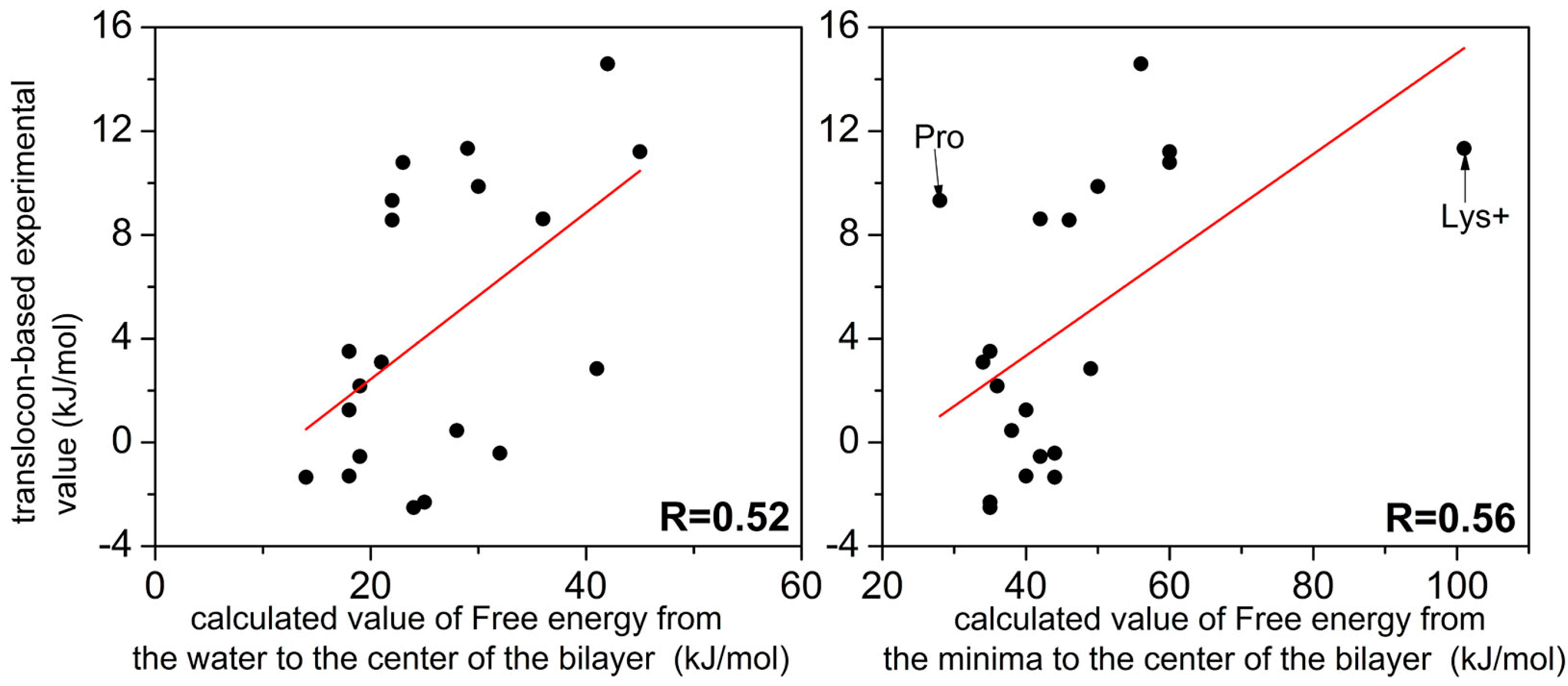
2.2. FEPs as a Function of z for 20 Natural Amino Acids
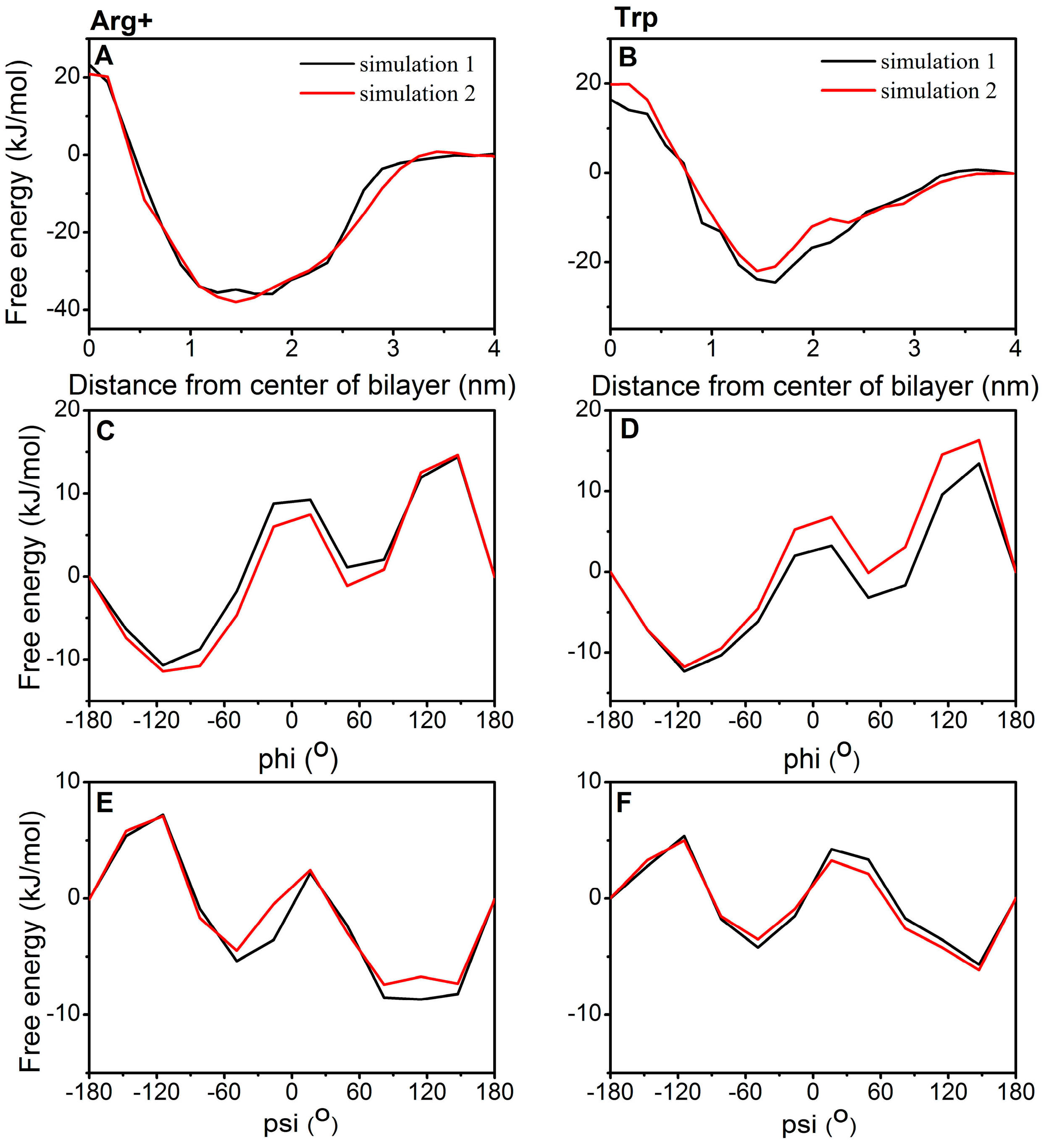
2.3. Torsion Angle Dependence
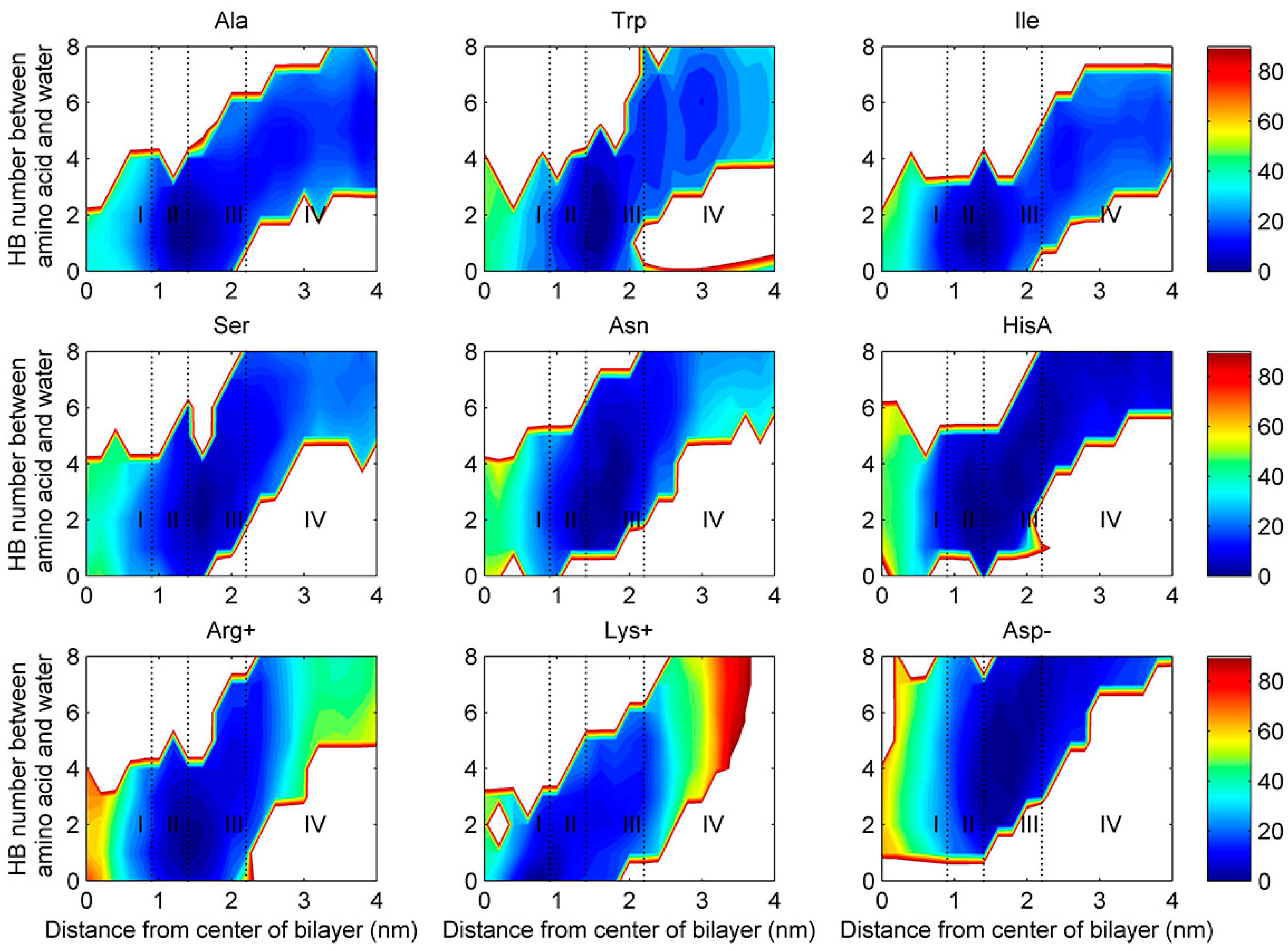
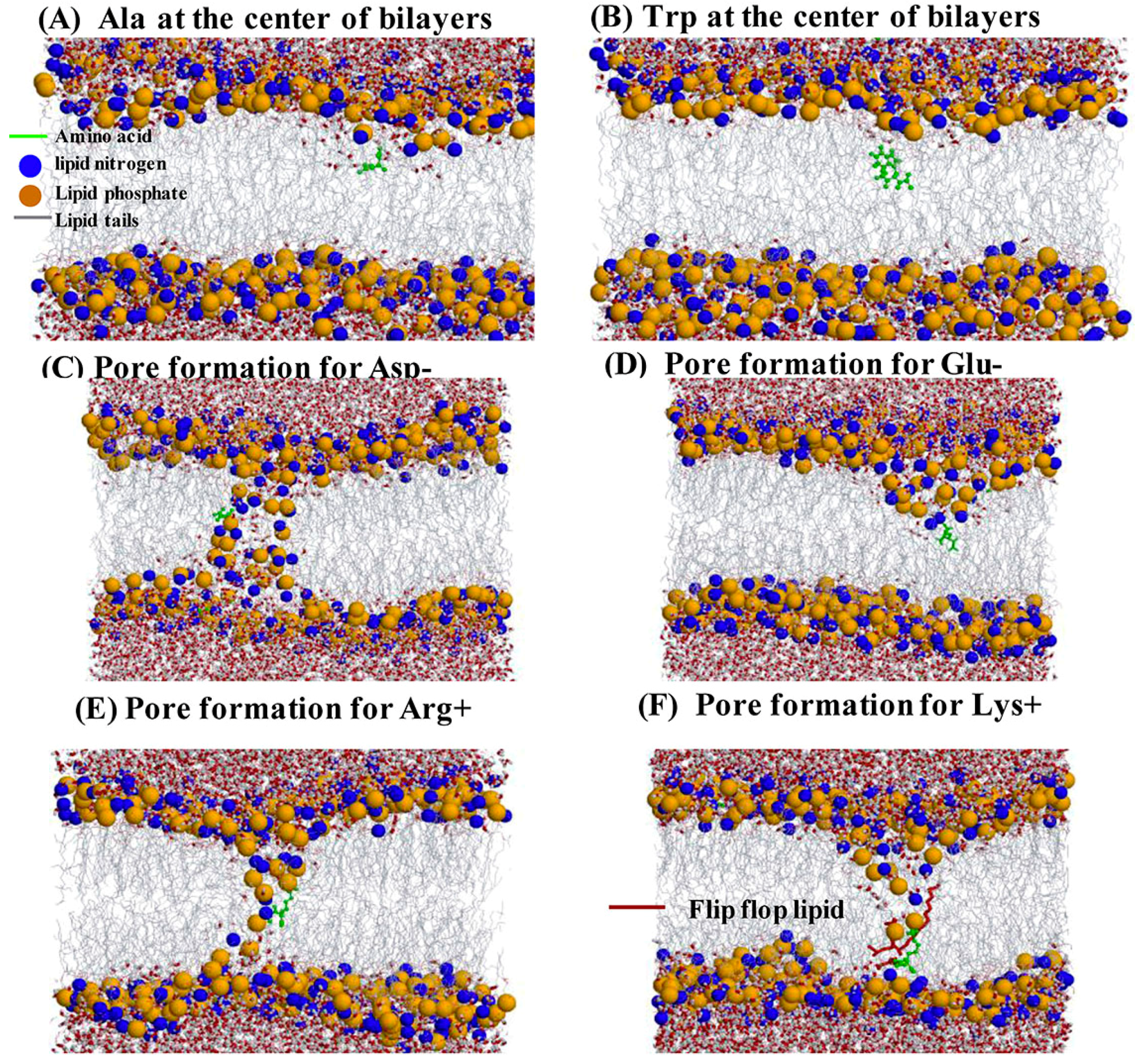
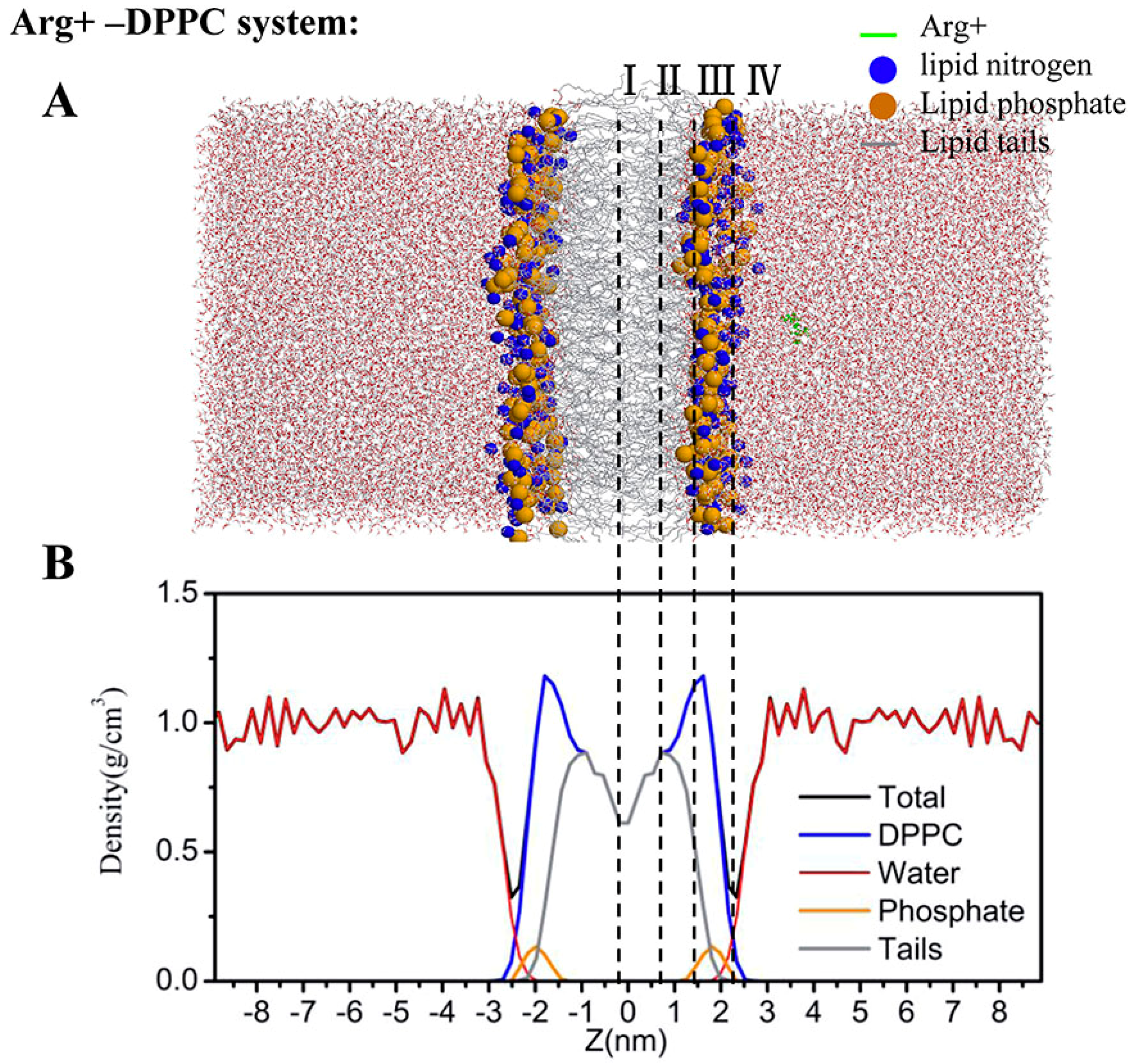
2.4. Water Defects and Pore Formation
3. Discussion
4. Materials and Methods
4.1. MD Simulation
4.2. Amino Acid-Lipid Bilayer Systems
4.3. Bias-Exchange Metadynamics
4.4. Multidimensional Free Energy Profiles
4.5. Regions of Bilayer Simulations
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Engelman, D.M. Membranes are more mosaic than fluid. Nature 2005, 438, 578–580. [Google Scholar] [CrossRef] [PubMed]
- Almen, M.S.; Nordstrom, K.J.; Fredriksson, R.; Schioth, H.B. Mapping the human membrane proteome: A majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Bhalla, S.; Usmani, S.S.; Singh, S.; Chaudhary, K.; Raghava, G.P.; Gautam, A. CPPsite 2.0: A repository of experimentally validated cell-penetrating peptides. Nucleic Acids Res. 2016, 44, D1098–D1103. [Google Scholar] [CrossRef] [PubMed]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.; Levchenko, T.S.; Torchilin, V.P. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv. Drug Deliv. Rev. 2005, 57, 637–651. [Google Scholar] [CrossRef] [PubMed]
- White, S.H.; Wimley, W.C. Membrane protein folding and stability: Physical principles. Annu. Rev. Biophys. Biomol. 1999, 28, 319–365. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Hessa, T.; Kim, H.; Bihlmaier, K.; Lundin, C.; Boekel, J.; Andersson, H.; Nilsson, I.; White, S.H.; von Heijne, G. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 2005, 433, 377–381. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.K.; Fleming, K.G. Aromatic Side Chain Water-to-Lipid Transfer Free Energies Show a Depth Dependence across the Membrane Normal. J. Am. Chem. Soc. 2016, 138, 7946–7950. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.C. Permeability of Membranes to Amino-Acids and Modified Amino-Acids—Mechanisms Involved in Translocation. Amino Acids 1994, 6, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhou, Y. Quantifying the effect of burial of amino acid residues on protein stability. Proteins 2004, 54, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.Y.; Zhou, Y.Q. Predicting the topology of transmembrane helical proteins using mean burial propensity and a hidden-Markov-model-based method. Protein Sci. 2003, 12, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xu, Y. Energetics and stability of transmembrane helix packing: A replica-exchange simulation with a knowledge-based membrane potential. Proteins 2006, 62, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Aliste, M.P.; Tieleman, D.P. Computer simulation of partitioning of ten pentapeptides Ace-WLXLL at the cyclohexane/water and phospholipid/water interfaces. BMC Biochem. 2005, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- MacCallum, J.L.; Tieleman, D.P. Calculation of the water-cyclohexane transfer free energies of neutral amino acid side-chain analogs using the OPLS all-atom force field. J. Comput. Chem. 2003, 24, 1930–1935. [Google Scholar] [CrossRef] [PubMed]
- MacCallum, J.L.; Bennett, W.F.; Tieleman, D.P. Distribution of amino acids in a lipid bilayer from computer simulations. Biophys. J. 2008, 94, 3393–3404. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, A.E.; Shrestha, R.; Webb, L.J.; Elber, R. Membrane Permeation of a Peptide: It Is Better to be Positive. J. Phys. Chem. B 2015, 119, 6412–6420. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Cui, Q. Does arginine remain protonated in the lipid membrane? Insights from microscopic pKa calculations. Biophys. J. 2008, 94, L61–L63. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ou, S.; Patel, S. Free energetics of arginine permeation into model DMPC lipid bilayers: Coupling of effective counterion concentration and lateral bilayer dimensions. J. Phys. Chem. 2013, 117, 11641–11653. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Vorobyov, I.; Allen, T.W. Potential of mean force and pKa profile calculation for a lipid membrane-exposed arginine side chain. J. Phys. Chem. B 2008, 112, 9574–9587. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, F.; Pietrucci, F.; Laio, A.; Piana, S. A kinetic model of trp-cage folding from multiple biased molecular dynamics simulations. PLoS Comput. Biol. 2009, 5, e1000452. [Google Scholar] [CrossRef] [PubMed]
- Granata, D.; Camilloni, C.; Vendruscolo, M.; Laio, A. Characterization of the free-energy landscapes of proteins by NMR-guided metadynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 6817–6822. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Jambeck, J.P.M.; Lyubartsev, A.P. Exploring the Free Energy Landscape of Solutes Embedded in Lipid Bilayers. J. Phys. Chem. Lett. 2013, 4, 1781–1787. [Google Scholar] [CrossRef] [PubMed]
- Elvati, P.; Violi, A. Free energy calculation of permeant-membrane interactions using molecular dynamics simulations. Methods Mol. Biol. 2012, 926, 189–202. [Google Scholar] [PubMed]
- Minozzi, M.; Lattanzi, G.; Benz, R.; Costi, M.P.; Venturelli, A.; Carloni, P. Permeation through the Cell Membrane of a Boron-Based beta-Lactamase Inhibitor. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaemi, Z.; Minozzi, M.; Carloni, P.; Laio, A. A Novel Approach to the Investigation of Passive Molecular Permeation through Lipid Bilayers from Atomistic Simulations. J. Phys. Chem. B 2012, 116, 8714–8721. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef]
- Laio, A.; Gervasio, F.L. Metadynamics: A method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008, 71, 291–312. [Google Scholar] [CrossRef]
- Kawashima, S.; Pokarowski, P.; Pokarowska, M.; Kolinski, A.; Katayama, T.; Kanehisa, M. AAindex: Amino acid index database, progress report 2008. Nucleic Acids Res. 2008, 36, D202–D205. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Kanehisa, M. AAindex: Amino acid index database. Nucleic Acids Res. 2000, 28, 374. [Google Scholar] [CrossRef] [PubMed]
- Chong, I.G.; Jun, C.H. Performance of some variable selection methods when multicollinearity is present. Chemom. Intell. Lab. Syst. 2005, 78, 103–112. [Google Scholar] [CrossRef]
- Biou, V.; Gibrat, J.F.; Levin, J.M.; Robson, B.; Garnier, J. Secondary structure prediction: Combination of three different methods. Protein Eng. 1988, 2, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Naderi-Manesh, H.; Sadeghi, M.; Arab, S.; Moosavi Movahedi, A.A. Prediction of protein surface accessibility with information theory. Proteins 2001, 42, 452–459. [Google Scholar] [CrossRef]
- Hu, H.; Elstner, M.; Hermans, J. Comparison of a QM/MM force field and molecular mechanics force fields in simulations of alanine and glycine “dipeptides” (Ace-Ala-Nme and Ace-Gly-Nme) in water in relation to the problem of modeling the unfolded peptide backbone in solution. Proteins 2003, 50, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Giorgino, T.; Laio, A.; Rodriguez, A. METAGUI 3: A graphical user interface for choosing the collective variables in molecular dynamics simulations. Comput. Phys. Commun. 2017, 217, 204–209. [Google Scholar] [CrossRef]
- Sengupta, D.; Leontiadou, H.; Mark, A.E.; Marrink, S.J. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Biophys. Acta 2008, 1778, 2308–2317. [Google Scholar] [CrossRef] [PubMed]
- Naoi, M.; Naoi, M.; Shimizu, T.; Malviya, A.N.; Yagi, K. Permeability of amino acids into liposomes. Biochim. Biophys. Acta 1977, 471, 305–310. [Google Scholar] [CrossRef]
- Chakrabarti, A.C.; Clark-Lewis, I.; Harrigan, P.R.; Cullis, P.R. Uptake of basic amino acids and peptides into liposomes in response to transmembrane pH gradients. Biophys. J. 1992, 61, 228–234. [Google Scholar] [CrossRef]
- Berger, O.; Edholm, O.; Jahnig, F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997, 72, 2002–2013. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Herce, H.D.; Garcia, A.E. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 20805–20810. [Google Scholar] [CrossRef] [PubMed]
- Yesylevskyy, S.; Marrink, S.J.; Mark, A.E. Alternative mechanisms for the interaction of the cell-penetrating peptides penetratin and the TAT peptide with lipid bilayers. Biophys. J. 2009, 97, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Forsman, J.; Lund, M.; Woodward, C.E. Effect of arginine-rich cell penetrating peptides on membrane pore formation and life-times: A molecular simulation study. Phys. Chem. Chem. Phys. 2014, 16, 20785–20795. [Google Scholar] [CrossRef] [PubMed]
- Nagle, J.F. Area/lipid of bilayers from NMR. Biophys. J. 1993, 64, 1476–1481. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single-Crystals—A New Molecular-Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Piana, S.; Laio, A. A bias-exchange approach to protein folding. J. Phys. Chem. 2007, 111, 4553–4559. [Google Scholar] [CrossRef] [PubMed]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| From Water | From Minimum | Expt. (Hessa et al.) | |
|---|---|---|---|
| ILE | 24 | 35 | −2.5 |
| LEU | 25 | 35 | −2.3 |
| PHE | 14 | 44 | −1.3 |
| VAL | 18 | 40 | −1.3 |
| TRP | 18 | 40 | 1.3 |
| ALA | 28 | 38 | 0.5 |
| MET | 32 | 44 | −0.4 |
| PRO | 22 | 28 | 9.3 |
| CYS | 19 | 42 | −0.5 |
| GLY | 21 | 34 | 3.1 |
| THR | 19 | 36 | 2.2 |
| TYR | 41 | 49 | 2.8 |
| HISA | 36 | 42 | 8.6 |
| SER | 18 | 35 | 3.5 |
| ASN | 22 | 46 | 8.6 |
| GLN | 30 | 50 | 9.9 |
| ARG+ | 23 | 60 | 10.8 |
| LYS+ | 29 | 101 | 11.3 |
| GLU- | 45 | 60 | 11.2 |
| ASP- | 42 | 56 | 14.6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Z.; Bian, Y.; Hu, G.; Zhao, L.; Kong, Z.; Yang, Y.; Wang, J.; Zhou, Y. Bias-Exchange Metadynamics Simulation of Membrane Permeation of 20 Amino Acids. Int. J. Mol. Sci. 2018, 19, 885. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19030885
Cao Z, Bian Y, Hu G, Zhao L, Kong Z, Yang Y, Wang J, Zhou Y. Bias-Exchange Metadynamics Simulation of Membrane Permeation of 20 Amino Acids. International Journal of Molecular Sciences. 2018; 19(3):885. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19030885
Chicago/Turabian StyleCao, Zanxia, Yunqiang Bian, Guodong Hu, Liling Zhao, Zhenzhen Kong, Yuedong Yang, Jihua Wang, and Yaoqi Zhou. 2018. "Bias-Exchange Metadynamics Simulation of Membrane Permeation of 20 Amino Acids" International Journal of Molecular Sciences 19, no. 3: 885. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19030885