Protein Tyrosine Phosphatase 1B Inhibition and Glucose Uptake Potentials of Mulberrofuran G, Albanol B, and Kuwanon G from Root Bark of Morus alba L. in Insulin-Resistant HepG2 Cells: An In Vitro and In Silico Study



Abstract
:1. Introduction
2. Results
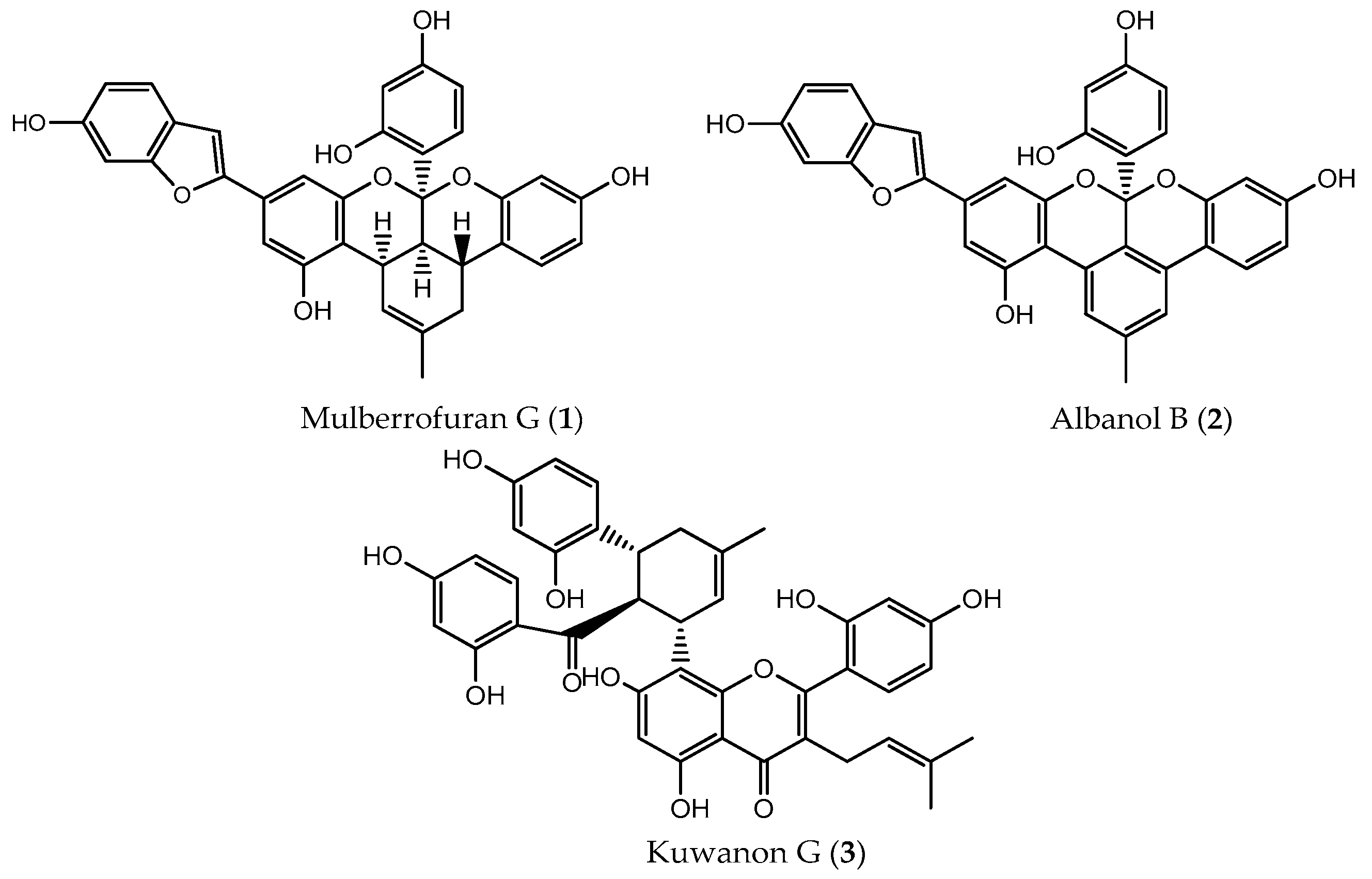
2.1. Inhibitory Activity Against PTP1B and α-Glucosidase
2.2. Enzyme Kinetics of PTP1B and α-Glucosidase Inhibition
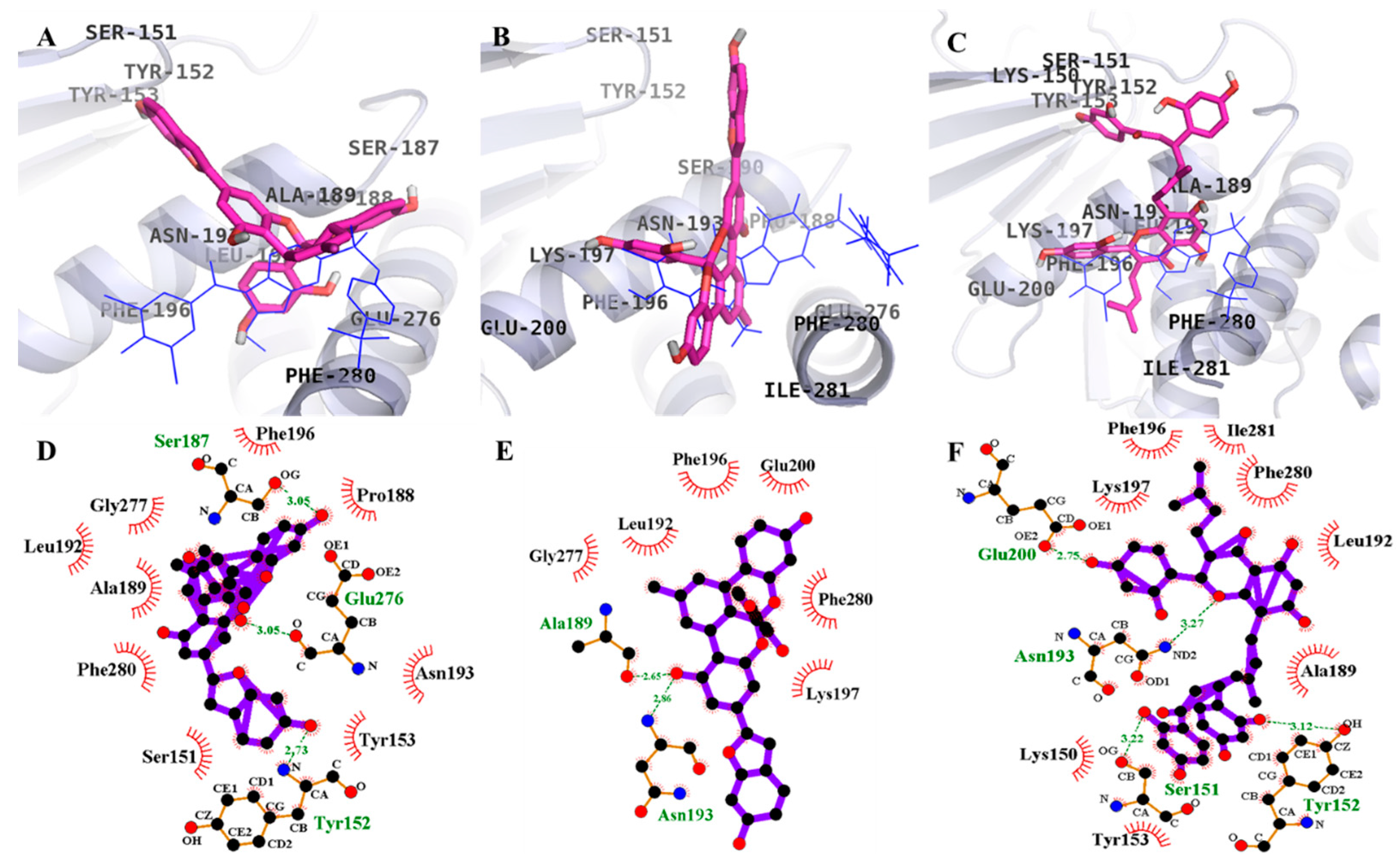
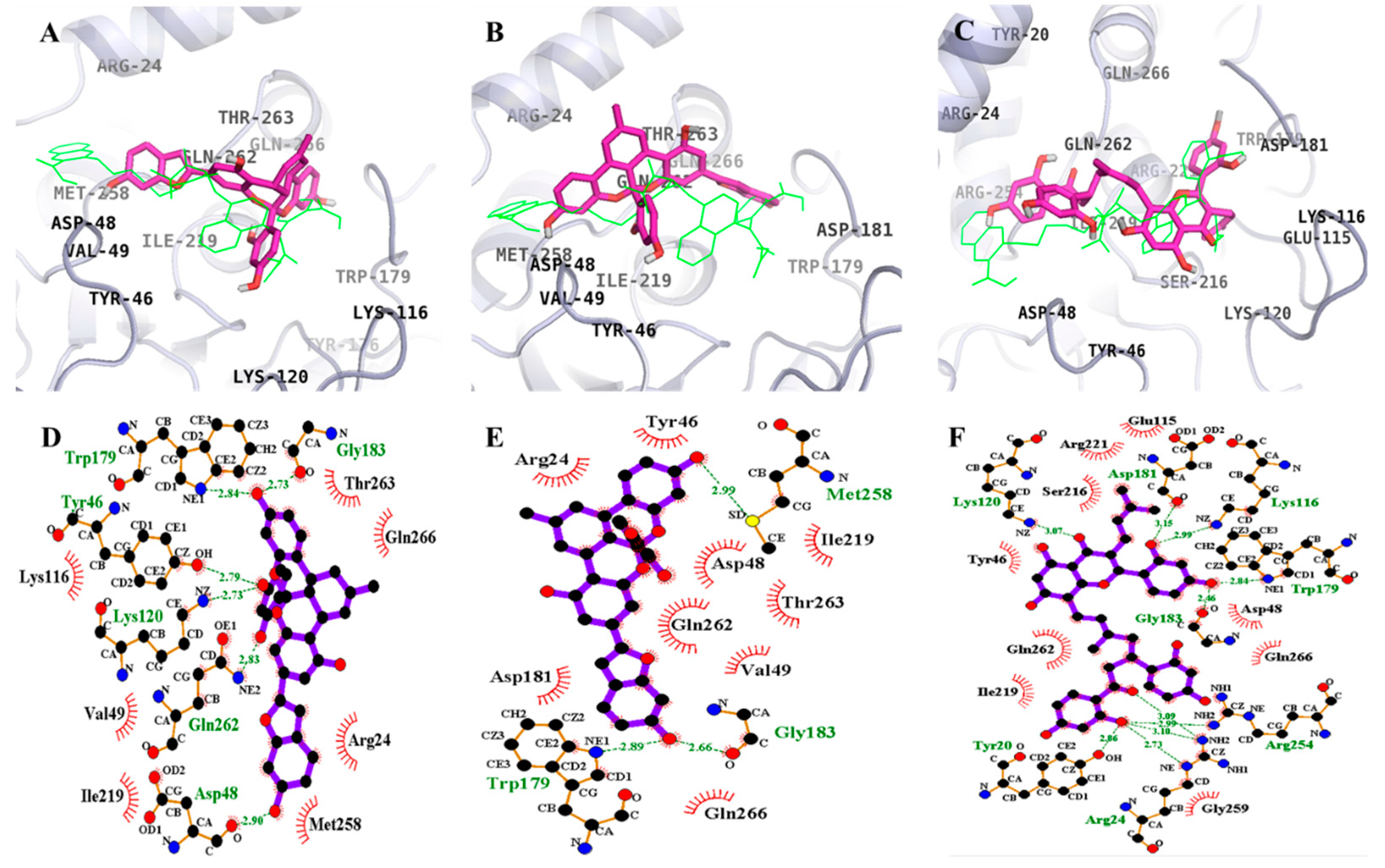
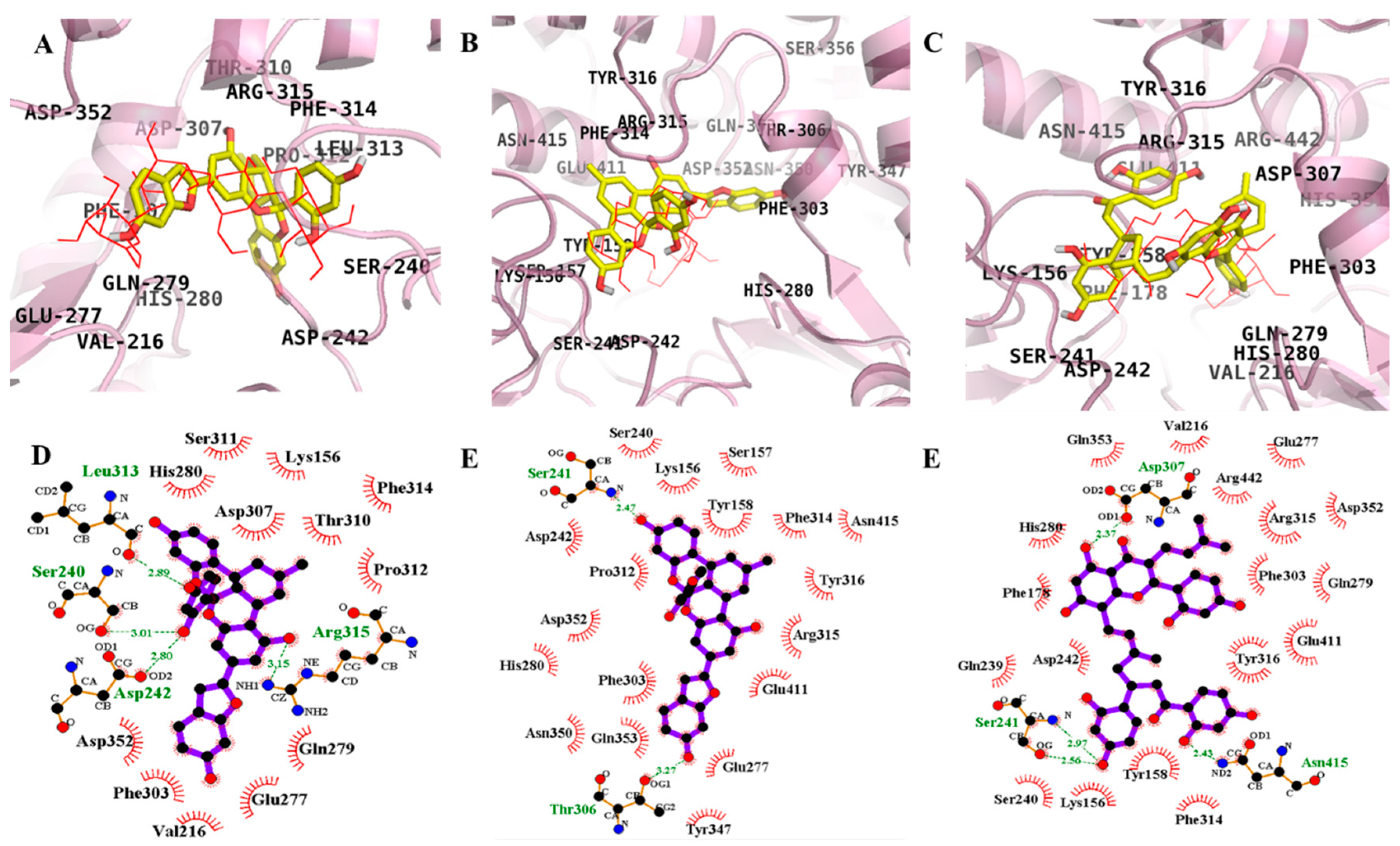
2.3. Molecular Docking Simulation of PTP1B Inhibition
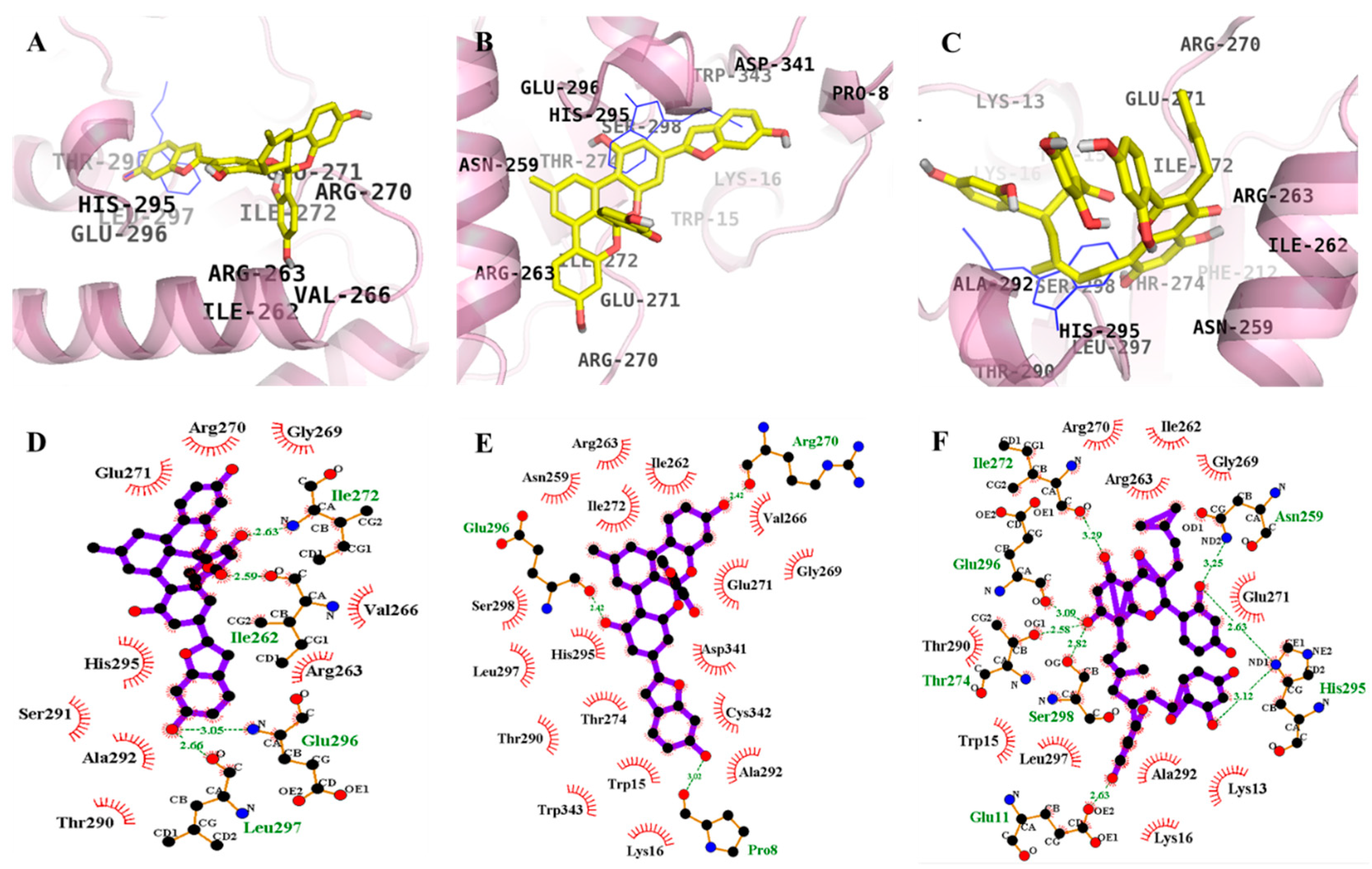
2.4. Molecular Docking Simulation of α-Glucosidase Inhibition
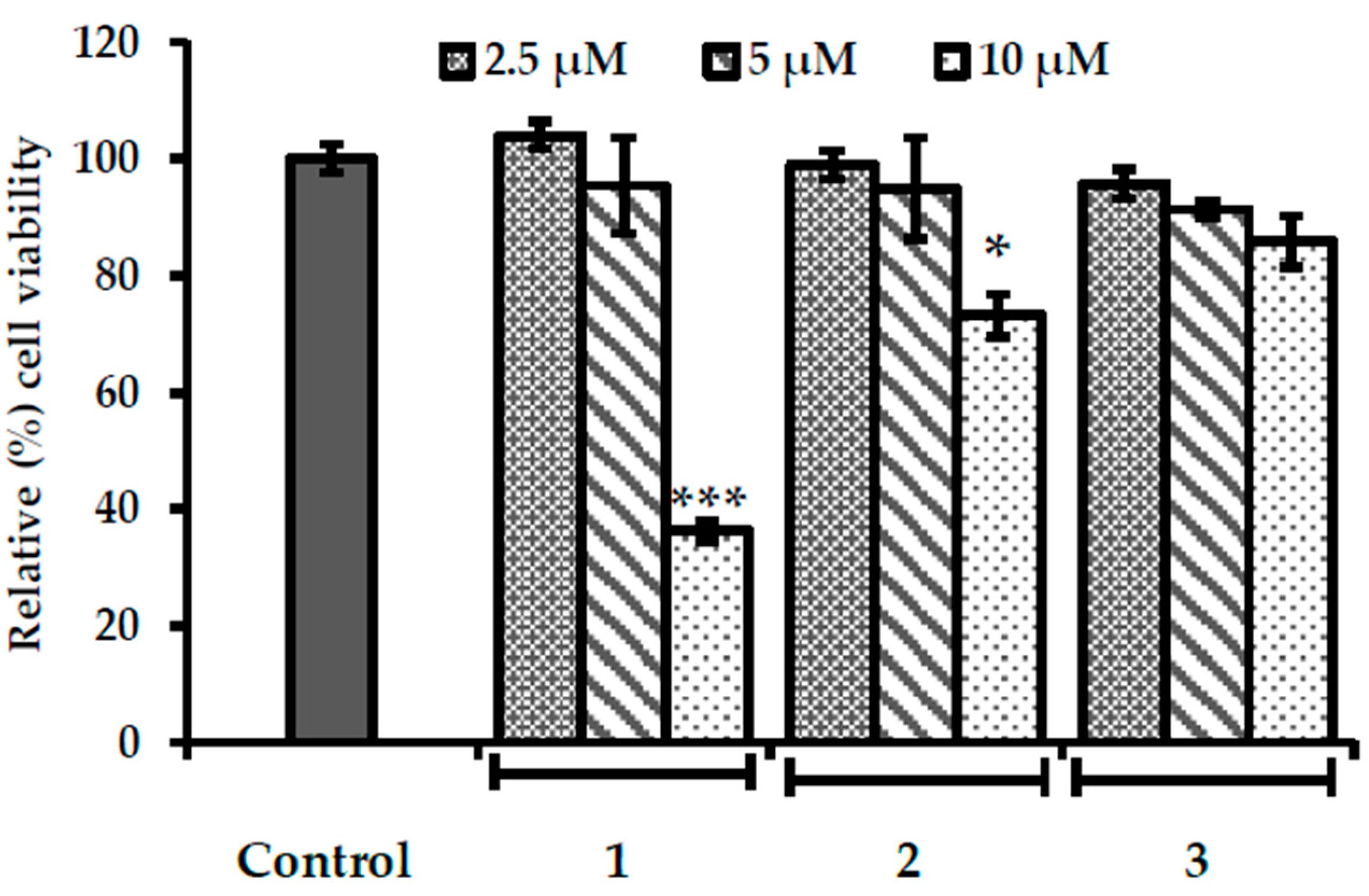
2.5. Evaluation of Cytotoxicity in HepG2 Cells
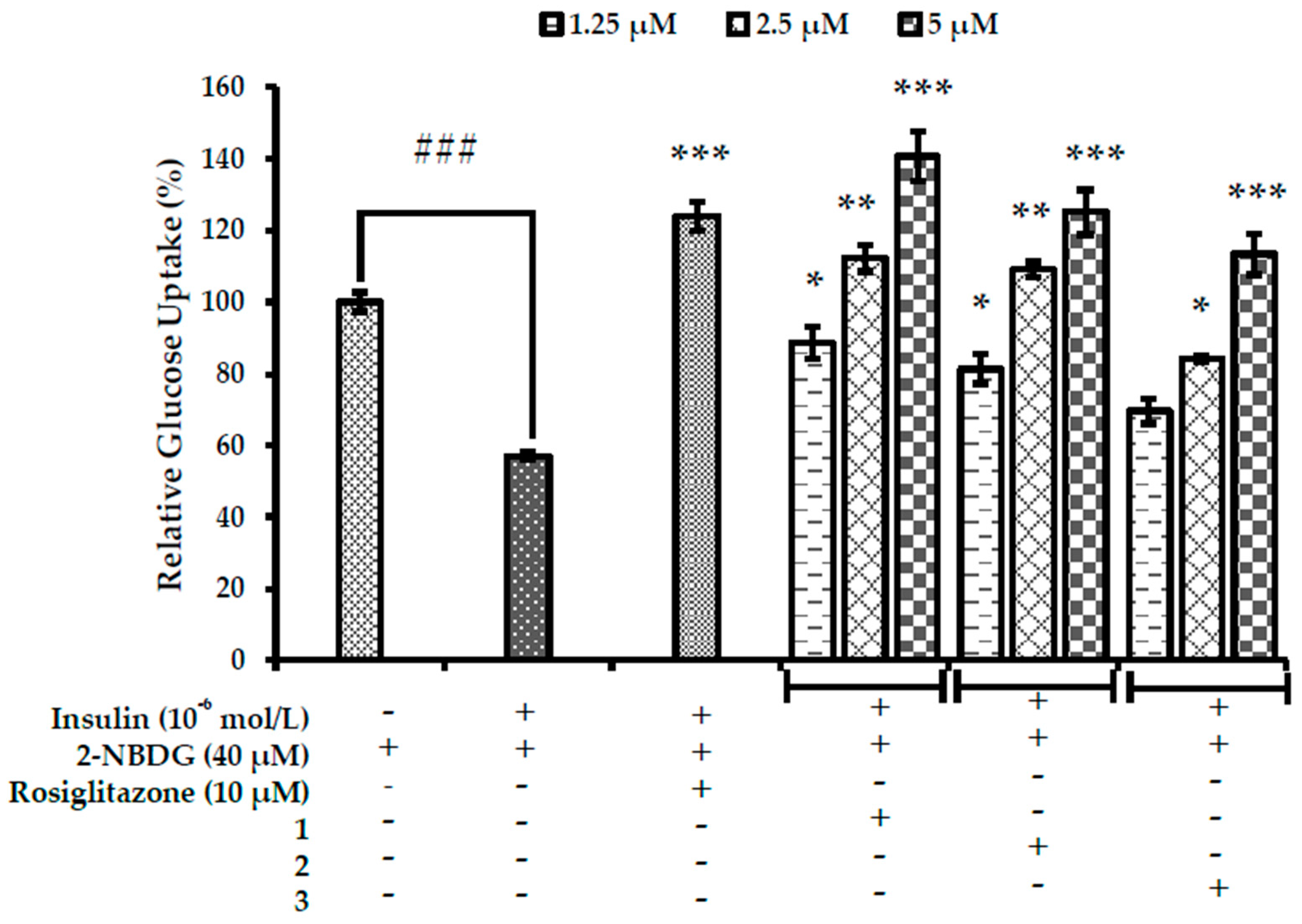
2.6. Effects on Glucose Uptake in Insulin-Resistant HepG2 Cells
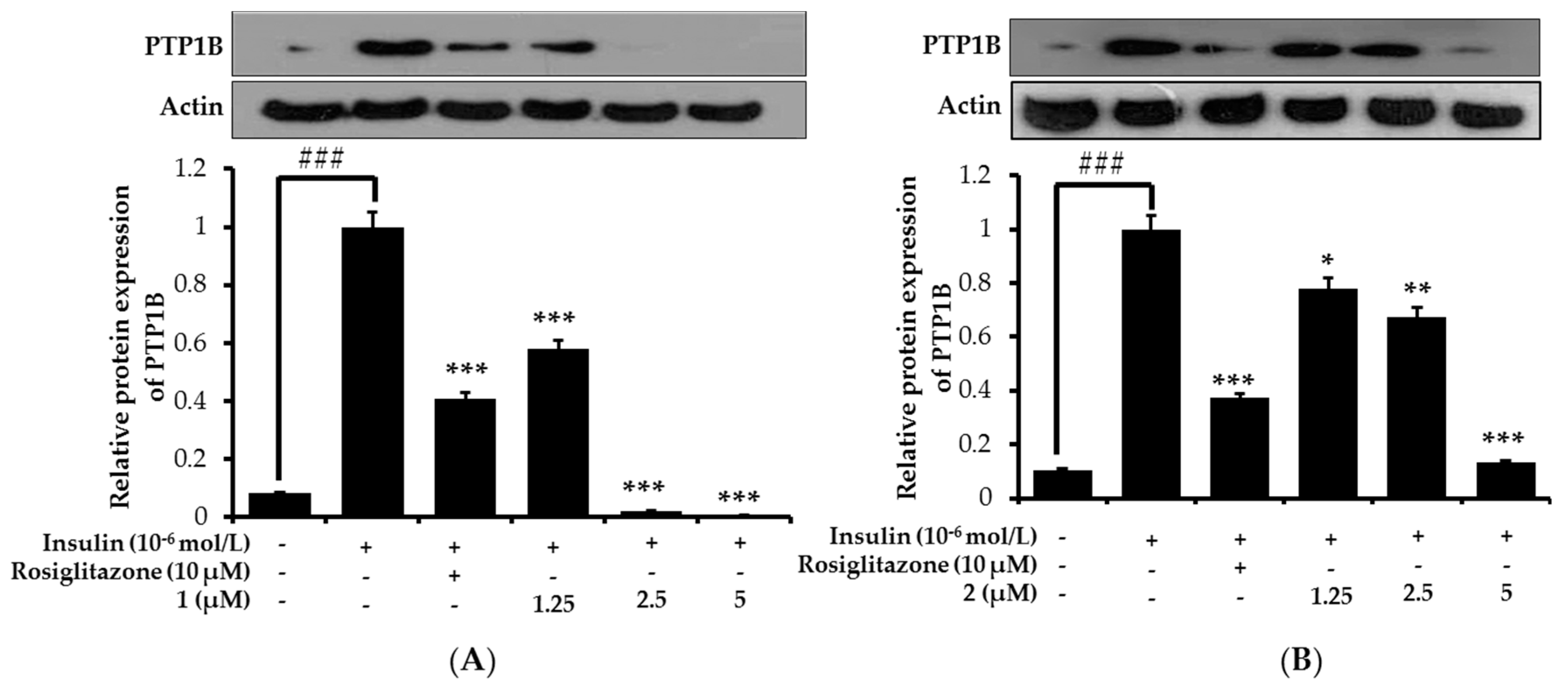
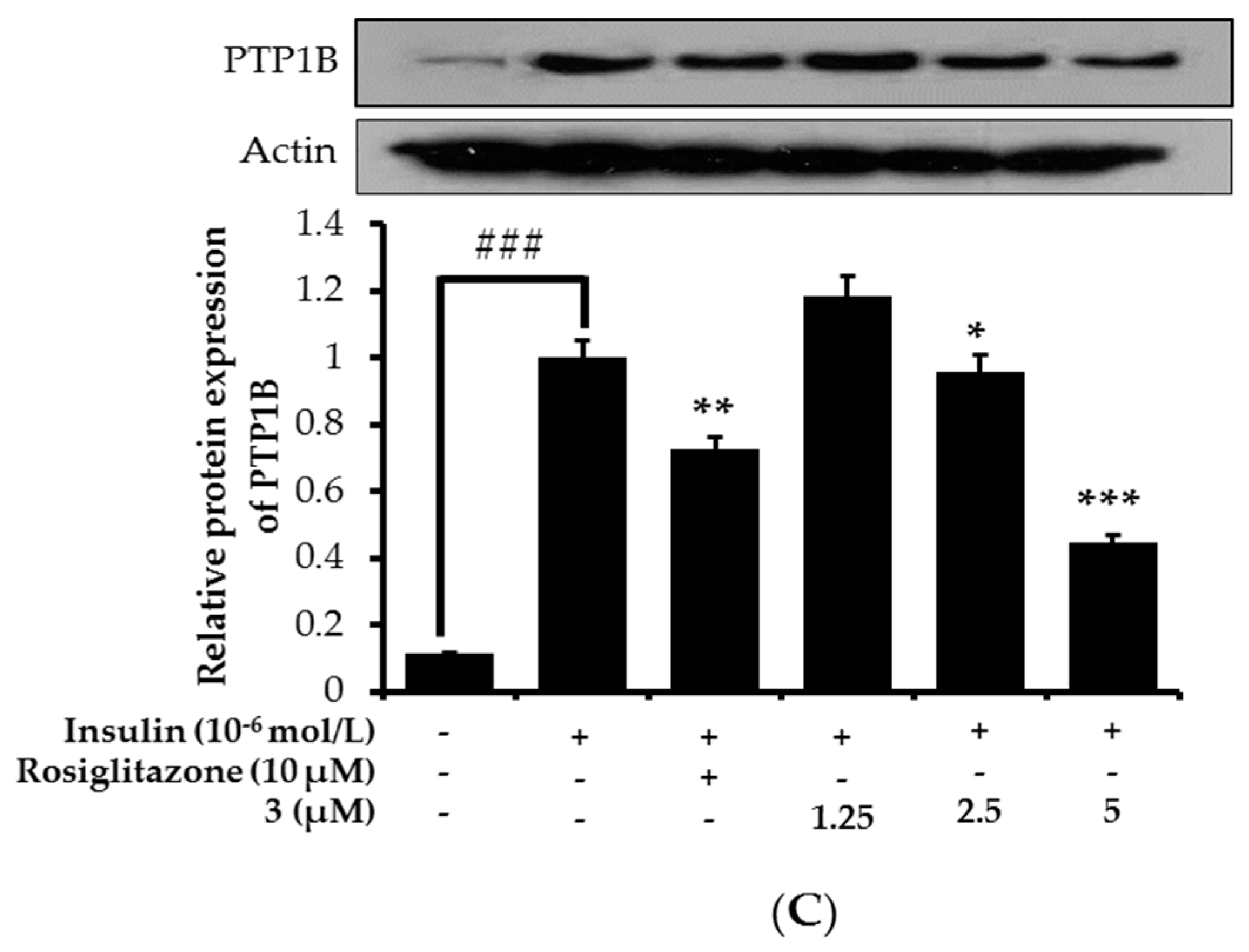
2.7. Effects on PTP1B Expression Level in Insulin-Resistant HepG2 Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Protein Tyrosine Phosphate 1B (PTP1B) Inhibitory Assay
4.3. α-Glucosidase Inhibitory Assay
4.4. Kinetic Study Against PTP1B and α-Glucosidase
4.5. Molecular Docking Simulation of PTP1B and α-Glucosidase Inhibition
4.6. Cell Culture, MTT Assay and Insulin Resistance Induction
4.7. Glucose Uptake Assay
4.8. Preparation of Cell Lysates and Western Blot Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| ADT | AutoDockTools |
| IR | Insulin resistance |
| PTP1B | Protein tyrosine phosphatase 1B |
| TCM | Traditional Chinese Medicine |
| T2DM | Type II diabetes mellitus |
References
- Deshpande, A.D.; Harris-Hayes, M.; Schootman, M. Epidemiology of diabetes and diabetes-related complications. Phys. Ther. 2008, 88, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [Google Scholar] [PubMed]
- Johnson, T.O.; Ermolieff, J.; Jirousek, M.R. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat. Rev. Drug Discov. 2002, 1, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Kawamori, R.; Tajima, N.; Iwamoto, Y.; Kashiwagi, A.; Shimamoto, K.; Kaku, K. Voglibose for prevention of type 2 diabetes mellitus: A randomised, double-blind trial in Japanese individuals with impaired glucose tolerance. Lancet 2009, 373, 1607–1614. [Google Scholar] [CrossRef]
- Singh, R.; Bagachi, A.; Semwal, A.; Kaur, S.; Bharadwaj, A. Traditional uses, phytochemistry and pharmacology of Morus alba Linn.: A review. J. Med. Plant Res. 2013, 7, 461–469. [Google Scholar]
- Kusano, G.; Orihara, S.; Tsukamoto, D.; Shibano, M.; Coskun, M.; Guvenc, A.; Erdurak, C.S. Five new nortropane alkaloids and six new amino acids from the fruit of Morus alba Linne growing in Turkey. Chem. Pharm. Bull. 2002, 50, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.K. Mulberry cultivation and utilization in China. In Mulberry for Animal Production; Sánchez, M.D., Ed.; FAO Animal Production and Health Paper; FAO: Rome, Italy, 2002; Volume 147, pp. 11–43. [Google Scholar]
- Sun, S.G.; Chen, R.Y.; Yu, D.Q. Structures of two new benzofuran derivatives from the bark of mulberry tree (Morus macroura Miq.). J. Asian Nat. Prod. Res. 2001, 3, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Suh, H.J. Antioxidant activities of five different mulberry cultivars in Korea. LWT Food Sci. Technol. 2007, 40, 955–962. [Google Scholar] [CrossRef]
- Chan, E.W.C.; Lye, P.Y.; Wong, S.K. Phytochemistry, pharmacology, and clinical trials of Morus alba. Chin. J. Nat. Med. 2016, 14, 17–30. [Google Scholar] [PubMed]
- Yang, Y.; Tan, Y.X.; Chen, R.Y.; Kang, J. The latest review on the polyphenols and their bioactivities of Chinese Morus plants. J. Asian Nat. Prod. Res. 2014, 16, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Su, S.L.; Duan, J.A.; Ouyang, Z.; Guo, S.; Li, L. Progress on resource chemistry of the medicinal plants in genus Morus L. in China. Mod. Chin. Med. 2012, 14, 1–6. [Google Scholar]
- Gryn-Rynko, A.; Bazylak, G.; Olszewska-Slonina, D. New potential phytotherapeutics obtained from white mulberry (Morus alba L.) leaves. Biomed. Pharmacother. 2016, 84, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Sun, W.; Fan, Y.; Guo, X.; Xu, G.; Xu, T.; Hou, Y.; Zhao, B.; Feng, X.; Liu, T. Effect of mulberry leaf (Folium Mori) on insulin resistance via IRS-1/PI3K/Glut-4 signalling pathway in type 2 diabetes mellitus rats. Pharm. Biol. 2016, 54, 2685–2691. [Google Scholar] [CrossRef] [PubMed]
- Hunyadi, A.; Veres, K.; Danko, B.; Kele, Z.; Weber, E.; Hetenyi, A.; Zupko, I.; Hsieh, T.J. In vitro anti-diabetic activity and chemical characterization of an apolar fraction of Morus alba leaf water extract. Phytother Res. 2013, 27, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Nazari, M.; Hajizadeh, M.R.; Mahmoodi, M.; Mirzaei, M.R.; Hassanshahi, G. The regulatory impacts of Morus alba leaf extract on some enzymes involved in glucose metabolism pathways in diabetic rat liver. Clin. Lab. 2013, 59, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.D.; Islam, M.S. Effects of white mulberry (Morus alba) leaf tea investigated in a type 2 diabetes model of rats. Acta. Pol. Pharm. 2015, 72, 153–160. [Google Scholar] [PubMed]
- Krol, E.; Jeszka-Skowron, M.; Krejpcio, Z.; Flaczyk, E.; Wojciak, R.W. The effects of supplementary mulberry leaf (Morus alba) extracts on the trace element status (Fe, Zn and Cu) in relation to diabetes management and antioxidant indices in diabetic rats. Biol. Trace Elem. Res. 2016, 174, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Nakamura, S.; Omagari, K.; Oku, T. Repeated ingestion of the leaf extract from Morus alba reduces insulin resistance in KK-Ay mice. Nutr. Res. 2011, 31, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.H.; Lee, H.A.; Park, M.H.; Han, J.S. Mulberry (Morus alba L.) fruit extract containing anthocyanins improves glycemic control and insulin sensitivity via activation of AMP-activated protein kinase in diabetic C57BL/Ksj-db/db mice. J. Med. Food. 2016, 19, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Singab, A.N.; El-Beshbishy, H.A.; Yonekawa, M.; Nomura, T.; Fukai, T. Hypoglycemic effect of Egyptian Morus alba root bark extract: Effect on diabetes and lipid peroxidation of streptozotocin-induced diabetic rats. J. Ethnopharmacol. 2005, 100, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, J.; Naik, P.R. Evaluation of hypoglycemic effect of Morus alba in an animal model. Indian J. Pharmacol. 2008, 40, 15–18. [Google Scholar] [PubMed]
- Yamada, H. Screening of α-glucosidase II inhibitor from Chinese herbs and its application on the quality control of mulberry bark. Shoyakugaku Zasshi 1993, 47, 47–55. [Google Scholar]
- Asano, N.; Yamashita, T.; Yasuda, K.; Ikeda, K.; Kizu, H.; Kameda, Y.; Kato, A.; Nash, R.J.; Lee, H.S.; Ryu, K.S. Polyhydroxylated alkaloids isolated from mulberry trees (Morus alba L.) and silkworms (Bombyx mori L.). J. Agric. Food Chem. 2001, 49, 4208–4213. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wu, X.; Wang, X.; Shen, T.; Ren, D. Two novel compounds from the root bark of Morus alba L. Nat. Prod. Res. 2018, 32, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Luo, J.G.; Wan, C.X.; Zhou, Z.B.; Kong, L.Y. Geranylated 2-arylbenzofurans from Morus alba var. tatarica and their α-glucosidase and protein tyrosine phosphatase 1B inhibitory activities. Fitoterapia 2014, 92, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Luo, J.G.; Wan, C.X.; Zhou, Z.B.; Kong, L.Y. Four new flavonoids with α-glucosidase inhibitory activities from Morus alba var. tatarica. Chem. Biodivers. 2015, 12, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Sarikaphuti, A.; Nararatwanchai, T.; Hashiguchi, T.; Ito, T.; Thaworanunta, S.; Kikuchi, K.; Oyama, Y.; Maruyama, I.; Tancharoen, S. Preventive effects of Morus alba L. anthocyanins on diabetes in Zucker diabetic fatty rats. Exp. Ther. Med. 2013, 6, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Hunyadi, A.; Martins, A.; Hsieh, T.J.; Seres, A.; Zupko, I. Chlorogenic acid and rutin play a major role in the in vivo anti-diabetic activity of Morus alba leaf extract on type II diabetic rats. PLoS ONE 2012, 7, e50619. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.H.; Lei, C.; Wang, P.P.; Li, J.Y.; Li, J.; Hou, A.J. Isoprenylated phenolic compounds with PTP1B inhibition from Morus alba. Fitoterapia 2017, 122, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Gershell, L. Type 2 diabetes market. Nat. Rev. Drug Discov. 2005, 4, 367–368. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Fantus, I.G. Inhibition of the protein tyrosine phosphatase PTP1B: Potential therapy for obesity, insulin resistance and type-2 diabetes mellitus. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 621–640. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; Maffioli, P. α-Glucosidase inhibitors and their use in clinical practice. Arch. Med. Sci. 2012, 8, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Kuk, E.B.; Jo, A.R.; Oh, S.I.; Sohn, H.S.; Seong, S.H.; Roy, A.; Choi, J.S.; Jung, H.A. Anti-Alzheimer’s disease activity of compounds from the root bark of Morus alba L. Arch. Pharm. Res. 2017, 40, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Seong, S.H.; Roy, A.; Jung, H.A.; Jung, H.J.; Choi, J.S. Protein tyrosine phosphatase 1B and α-glucosidase inhibitory activities of Pueraria lobata root and its constituents. J. Ethnopharmacol. 2016, 194, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Puius, Y.A.; Zhao, Y.; Sullivan, M.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.Y. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: A paradigm for inhibitor design. Proc. Natl. Acad. Sci. USA 1997, 94, 13420–13425. [Google Scholar] [CrossRef] [PubMed]
- Elchebly, M.; Payette, P.; Michaliszyn, E.; Cromlish, W.; Collins, S.; Loy, A.L.; Normandin, D.; Cheng, A.; Himms-Hagen, J.; Chan, C.C.; et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 1999, 283, 1544–1548. [Google Scholar] [CrossRef] [PubMed]
- Kenner, K.A.; Anyanwu, E.; Olefsky, J.M.; Kusari, J. Protein-tyrosine phosphatase 1B is a negative regulator of insulin-and insulin-like growth factor-I-stimulated signaling. J. Biol. Chem. 1996, 271, 19810–19816. [Google Scholar] [CrossRef] [PubMed]
- Zabolotny, J.M.; Haj, F.G.; Kim, Y.B.; Kim, H.J.; Shulman, G.I.; Kim, J.K.; Neel, B.G.; Kahn, B.B. Transgenic overexpression of protein-tyrosine phosphatase 1B in muscle causes insulin resistance, but overexpression with leukocyte antigen-related phosphatase does not additively impair insulin action. J. Biol. Chem. 2004, 279, 24844–24851. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Kai, K.; Ishii, M.; Ishii, T.; Akagawa, M. Safranal, a novel protein tyrosine phosphatase 1B inhibitor, activates insulin signaling in C2C12 myotubes and improves glucose tolerance in diabetic KK-Ay mice. Mol. Nutr. Food Res. 2014, 58, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Takada, M.; Sumi, M.; Maeda, A.; Watanabe, F.; Kamiya, T.; Ishii, T.; Nakano, M.; Akagawa, M. Pyrroloquinoline quinone, a novel protein tyrosine phosphatase 1B inhibitor, activates insulin signaling in C2C12 myotubes and improves impaired glucose tolerance in diabetic KK-Ay mice. Biochem. Biophys. Res. Commun. 2012, 428, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Q.; Liu, T.; Chen, C.; Li, M.Y.; Wang, Z.Y.; Chen, R.S.; Wei, G.X.; Wang, X.Y.; Luo, D.Q. Fumosorinone, a novel PTP1B inhibitor, activates insulin signaling in insulin-resistance HepG2 cells and shows anti-diabetic effect in diabetic KKAy mice. Toxicol. Appl. Pharmacol. 2015, 285, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Seo, S.Y.; Kim, T.H.; Kim, S.G. Decrease of microRNA-122 causes hepatic insulin resistance by inducing protein tyrosine phosphatase 1B, which is reversed by licorice flavonoid. Hepatology 2012, 56, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Na, M.; Oh, H.; Bae, E.Y.; Jeong, D.G.; Ryu, S.E.; Kim, S.; Kim, B.Y.; Oh, W.K.; Ahn, J.S. Protein tyrosine phosphatase 1B inhibitors from Morus root bark. Bioorg. Med. Chem. Lett. 2006, 16, 1426–1429. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, X.D.; Song, Y.W.; Liu, J.-W. A microplate-based screening method for α-glucosidase inhibitors. Chin. J. Clin. Pharmacol. Ther. 2005, 1128–1134. [Google Scholar]
- Dixon, M. The determination of enzyme inhibitor constants. Biochem. J. 1953, 55, 170–171. [Google Scholar] [CrossRef] [PubMed]
- Cornish-Bowden, A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors (Short Communication). Biochem. J. 1974, 137, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Wiesmann, C.; Barr, K.J.; Kung, J.; Zhu, J.; Erlanson, D.A.; Shen, W.; Fahr, B.J.; Zhong, M.; Taylor, L.; Randal, M.; et al. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 2004, 11, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, G.; Pan, J.; Wang, Y. α-Glucosidase inhibition by luteolin: Kinetics, interaction and molecular docking. Int. J. Biol. Macromol. 2014, 64, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Jung, H.A.; Ali, M.Y.; Bhakta, H.K.; Min, B.S.; Choi, J.S. Prunin is a highly potent flavonoid from Prunus davidiana stems that inhibits protein tyrosine phosphatase 1B and stimulates glucose uptake in insulin-resistant HepG2 cells. Arch. Pharm. Res. 2017, 40, 37–48. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Protein Tyrosine Phosphatase 1B | α-Glucosidase | ||||
|---|---|---|---|---|---|---|
| IC50 (μM) a | Inhibition Type b | Ki (μM) c | IC50 (μM) a | Inhibition Type b | Ki (μM) c | |
| 1 | 0.57 ± 0.04 | Mixed type | 0.70 | 1.67 ± 0.02 | Mixed type | 1.2 |
| 2 | 0.80 ± 0.02 | Mixed type | 1.02 | 1.31 ± 0.01 | Mixed type | 0.9 |
| 3 | 2.26 ± 0.03 | Mixed type | 1.98 | 2.35 ± 0.03 | Mixed type | 2.51 |
| Ursolic acid d | 3.54 ± 0.06 | |||||
| Acarbose e | 119.16 ± 3.25 | |||||
| Compound | Binding Energy a (kcal/mol) | No. of H-Bond b | H-Bond Interacting Residues c | Hydrophobic Interacting Residues d |
|---|---|---|---|---|
| 1 (Allosteric inhibition mode) | –8.24 | 3 | Ser187, Glu276, Tyr152 | Phe196, Pro188, Asn193, Tyr153, Ser151, Phe280, Ala189, Leu192, Gly277 |
| 1 (Catalytic inhibition mode) | –6.85 | 6 | Trp179, Tyr46, Lys120, Gln262, Asp48, Gly183 | Thr263, Gln266, Arg24, Met258, Ile219, Val49, Lys116 |
| 2 (Allosteric inhibition mode) | –7.37 | 2 | Ala189, Asn193 | Gly277, Leu192, Phe196, Glu200, Phe280, Lys197 |
| 2 (Catalytic inhibition mode) | –6.7 | 3 | Met258, Gly183, Trp179 | Arg24, Tyr46, Ile219, Asp48, Thr263, Gln262, Val149, Gln266, Asp181 |
| 3 (Allosteric inhibition mode) | –7.81 | 4 | Glu200, Asn193, Ser151, Tyr152 | Lys197, Phe196, Ile281, Phe280, Leu192, Ala189, Tyr153, Lys150 |
| 3 (Catalytic inhibition mode) | –7.62 | 10 | Asp181, Lys120, Lys116, Trp179, Gly183, Arg254, Tyr20, Arg24 | Glu115, Arg221, Ser216, Tyr46, Gln262, Asp48, Gln266, Gly259, Ile219 |
| Compound 2 (Allosteric inhibitor) | –10.98 | 2 | Asn193, Glu276 | Phe196, Gly277, Phe280, Ile281, Met282, Lys279, Ala189, Leu192 |
| Compound 23 (Catalytic inhibitor) | –11.23 | 11 | Asp48, Tyr46, Arg24, Ser216, Ala217, Arg221, Arg254, Gln262 | Yls116, Phe182, Gln266, Gln262, Ala217, Et258, Gly259, Asp29, Ser28, Val49, Ile219, Tyr46 |
| Compound | Binding Energy a (kcal/mol) | No. of H-Bond b | H-Bond Interacting Residues c | Hydrophobic Interacting Residues d |
|---|---|---|---|---|
| 1 (Allosteric inhibition mode) | −8.65 | 4 | Ile262, Tle272, Glu296, Leu297 | Arg263, Val266, Gly269, Arg270, Glu271, Thr290, Ser291, Ala292, His295 |
| 1 (Catalytic inhibition mode) | −10.43 | 4 | Ser240, Asp242, Leu313, Arg315 | His280, Ser311, Lys156, Asp307, Thr310, Pro312, Gln279, Glu277, Val216, Phe303, Asp352, Phe314 |
| 2 (Allosteric inhibition mode) | −11.71 | 3 | Pro8, Arg270, Glu296 | Trp15, Lys16, Thr274, Thr290, His295, Leu297, Ser298, Trp343, Cys342, Ala292, Asp341, Glu271, Gly269, Val266, Ile262, Ile272, Asn259, Arg263 |
| 2 (Catalytic inhibition mode) | −9.48 | 2 | Ser241, Thr306 | Lys156, Ser157, Tyr158, Glu227, Ser240, Asp242, His280, Phe303, Pro312, Phe314, Arg315, Tyr316, Tyr347 Asn350, Asp352, Gln353, Glu411, Asn415 |
| 3 (Allosteric inhibition mode) | −7.36 | 8 | Ile272, Glu296, Thr274, Glu11, His295, Asn259, Ser298 | Arg270, Ile262, Arg263, Gly269, Glu271, Lys13, Ala292, Lys16, Leu297, Trp15, The290 |
| 3 (Catalytic inhibition mode) | −11.53 | 4 | Ser241, Asp307, Asn415 | Lys156, Tyr158, Phe178, Val216, Gln239, Ser240, Asp242, Glu277, Gln279, His280, Phe303, Phe314, Arg315, Tyr316, Asp351, Gln353, Glu411, Arg442, |
| BIP (Allosteric inhibitor) | −6.85 | 2 | Glu296, His295 | Asp341, Cys342, Ala292, Thr290, Arg294, Leu297, Asn259, Ser291, Ser298, Trp15, Lys16, Trp343 |
| Acarbose (Catalytic inhibitor) | −8.60 | 17 | Tyr158, His112, Gln182, Asp69, Asp215, Arg213, Glu277, Asp352, Arg442, Asp307, His280, Asp242, Ser240 | Lys156, Gln279, Arg315, Phe178, Phe303, Gln353, Tyr72, Val216, His351, Glu411 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paudel, P.; Yu, T.; Seong, S.H.; Kuk, E.B.; Jung, H.A.; Choi, J.S. Protein Tyrosine Phosphatase 1B Inhibition and Glucose Uptake Potentials of Mulberrofuran G, Albanol B, and Kuwanon G from Root Bark of Morus alba L. in Insulin-Resistant HepG2 Cells: An In Vitro and In Silico Study. Int. J. Mol. Sci. 2018, 19, 1542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051542
Paudel P, Yu T, Seong SH, Kuk EB, Jung HA, Choi JS. Protein Tyrosine Phosphatase 1B Inhibition and Glucose Uptake Potentials of Mulberrofuran G, Albanol B, and Kuwanon G from Root Bark of Morus alba L. in Insulin-Resistant HepG2 Cells: An In Vitro and In Silico Study. International Journal of Molecular Sciences. 2018; 19(5):1542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051542
Chicago/Turabian StylePaudel, Pradeep, Ting Yu, Su Hui Seong, Eun Bi Kuk, Hyun Ah Jung, and Jae Sue Choi. 2018. "Protein Tyrosine Phosphatase 1B Inhibition and Glucose Uptake Potentials of Mulberrofuran G, Albanol B, and Kuwanon G from Root Bark of Morus alba L. in Insulin-Resistant HepG2 Cells: An In Vitro and In Silico Study" International Journal of Molecular Sciences 19, no. 5: 1542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051542