High-Risk Human Papillomaviral Oncogenes E6 and E7 Target Key Cellular Pathways to Achieve Oncogenesis


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. HPV in Cancers
1.2. HPV Genome
1.3. HPV Oncogenes
2. Mechanism of E6 and E7 Regulating Cellular Pathways
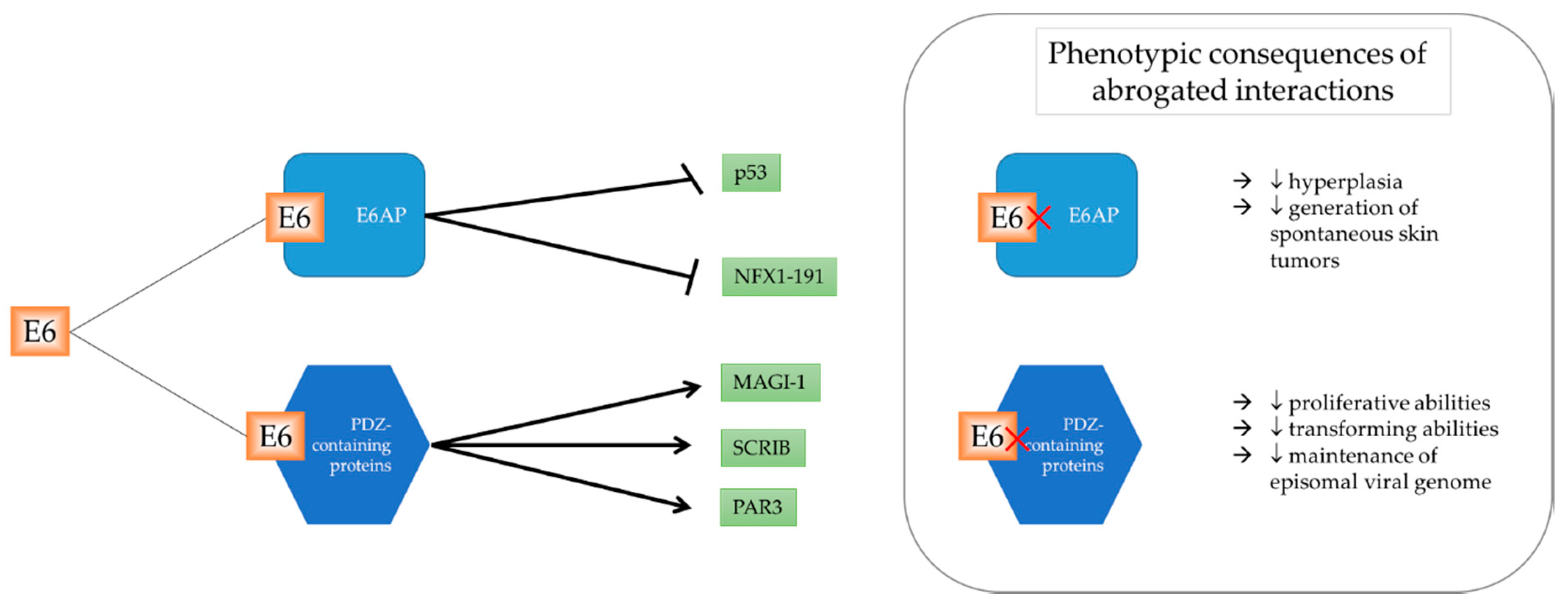
2.1. Protein Targets Dependent on E6AP
2.2. PDZ-Domain Family of E6 Interactors
2.3. Other Protein Targets
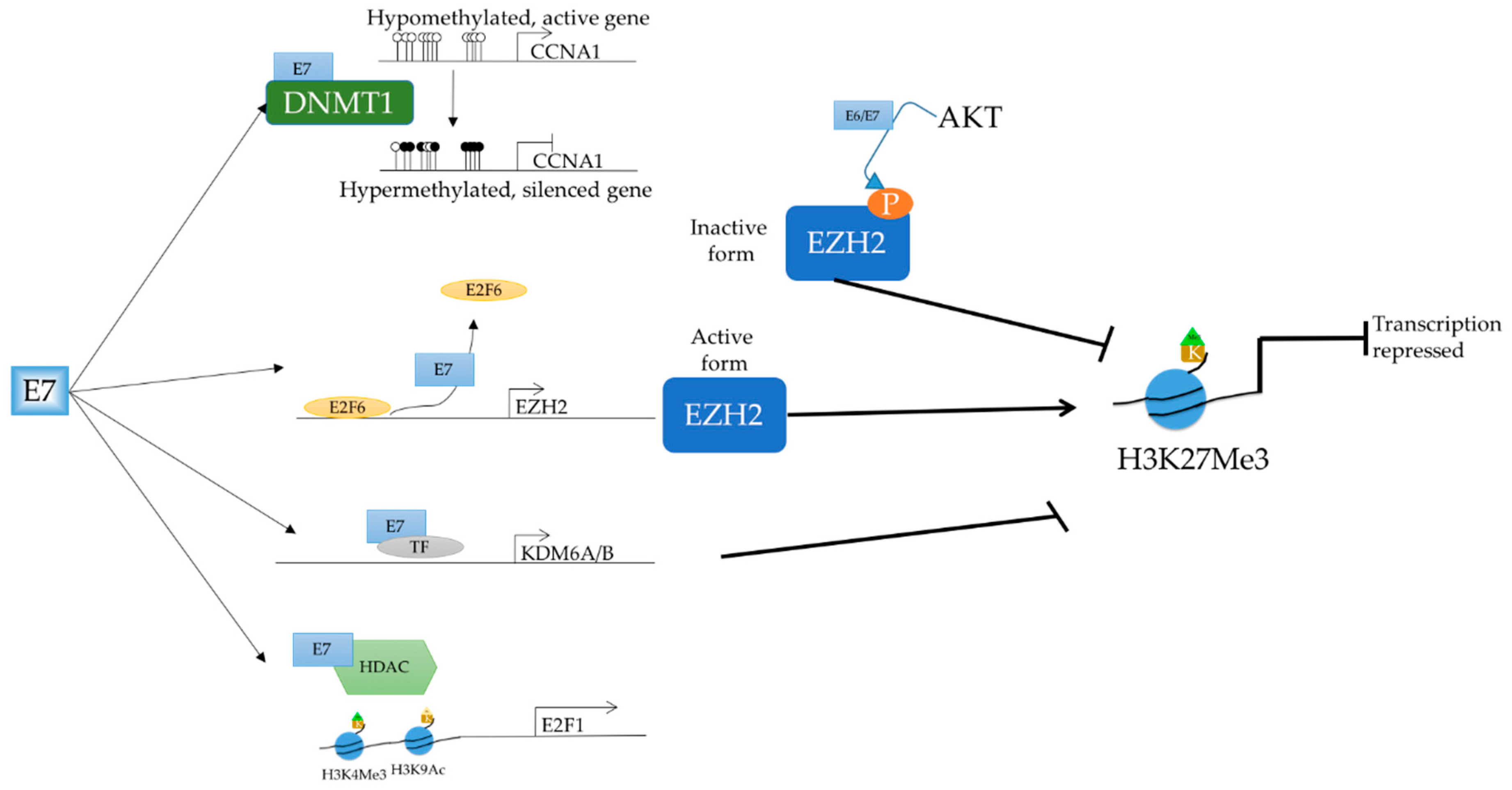
2.4. Epigenetic Targets
E6/E7 Regulating Methylation Status
2.5. E6/E7 Targeting Epiegnetic Modifiers
2.6. RNA Targets of HPV
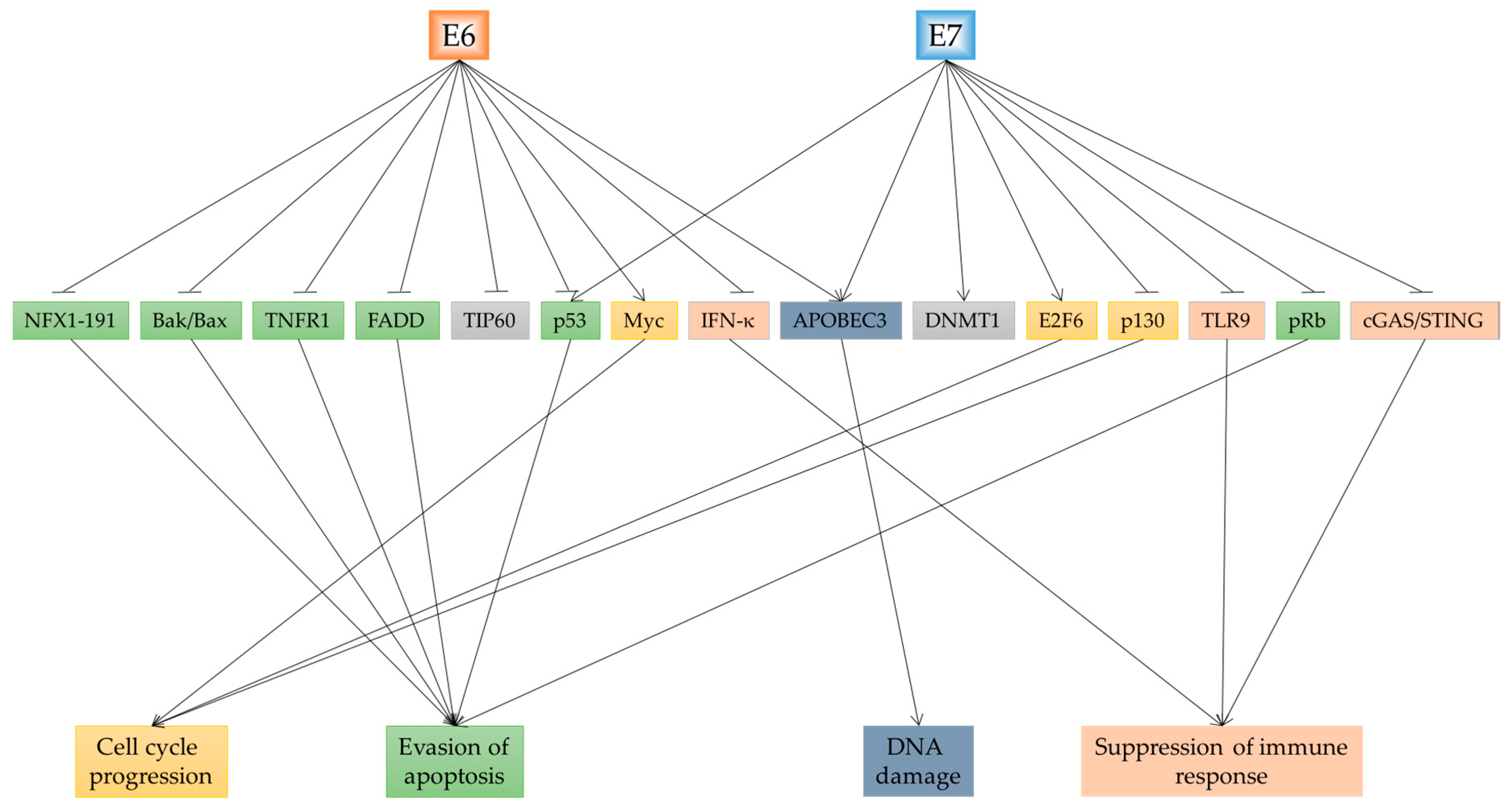
3. Cellular Consequences Affected by E6 and E7
3.1. Cell Cycle Changes and Growth Promotion
3.2. Evading Apoptosis
3.3. Immune Response
3.4. DNA Damage
4. Therapeutics against HPV
4.1. Vaccinations
4.2. Treatment
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AS | Angelman syndrome |
| APOBEC3 | apolipoprotein B mRNA editing enzyme catalytic-polypeptide-like proteins 3 |
| BGS | bisulfite genome sequencing |
| BTB | BR-C, ttk and bab |
| CARM1 | co-activator associated arginine methyltransferase 1 |
| CDEs | cycle-dependent elements |
| CDK | cyclin-dependent kinase |
| CHRs | cell cycle genes homology regions |
| CLE | CHR-like elements |
| CUL2 | cullin 2 |
| DNA-FISH | DNA breakage detection-fluorescence in situ hybridization |
| DREAM | DP, RB-like, E2F4, and MuvB |
| DUBs | deubiquitinating enzymes |
| E6AP | E6-associated protein |
| EBV | Epstein–Barr virus |
| FADD | FAS-associated protein with death domain |
| FDA | Food and Drug Administration |
| GPCA | Gaussia princeps luciferase protein complementation assay |
| HAT | histone acetyltransferase |
| HBV | hepatitis B virus |
| HC2 | hybrid capture 2 |
| HCV | hepatitis C virus |
| HDAC | histone deacetylases |
| HECT | homologous to E6AP carboxyl-terminus |
| HFKs | human foreskin keratinocytes |
| HMTs | histone methyltransferases |
| HNCs | head and neck cancers |
| HNSCC | head and neck squamous cell carcinoma |
| HPV | human papillomavirus |
| HSV | herpes simplex virus |
| HTLV-1 | human T-lymphotropic virus-1 |
| KSHV | Kaposi’s sarcoma-associated herpesvirus |
| LCR | long control region |
| MCV | Merkel cell polyomavirus |
| miRNAs | microRNAs |
| MSDK | methylation-specific digital karyotyping |
| MVP | methylation variable positions |
| NIKs | normal immortalized human keratinocytes |
| NLS3 | nuclear localization signal 3 |
| P/CAF | p300/CBP-associated factor |
| PBM | PDZ-binding motif |
| PKA | protein kinase A |
| pRb | retinoblastoma protein |
| PRC | polycomb repressive complex |
| PRMT1 | protein arginine methyltransferase 1 |
| PRRs | pathogen recognition receptors |
| RNAi | RNA interference |
| SCC | squamous cell carcinoma |
| SET7 | SET domain containing lysine methyltransferase 7 |
| SR | splicing regulatory |
| SSA | single-strand annealing |
| TALEN | transcription activator-like effector nuclease |
| TNFR1 | TNF receptor 1 |
| TRAIL | TNF-related apoptosis-inducing ligand |
| TRAM | transcriptional adapter motif |
| UPS | ubiquitin–proteasome system |
| VLP | virus-like particles |
References
- Rous, P. A Sarcoma of the Fowl Transmissible by an Agent Separable from the Tumor Cells. J. Exp. Med. 1911, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Pagano, J.S.; Blaser, M.; Buendia, M.A.; Damania, B.; Khalili, K.; Raab-Traub, N.; Roizman, B. Infectious agents and cancer: Criteria for a causal relation. Semin. Cancer Biol. 2004, 14, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Doorbar, J. The low-risk papillomaviruses. Virus Res. 2017, 231, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Carifi, M.; Napolitano, D.; Morandi, M.; Dall’Olio, D. Recurrent respiratory papillomatosis: Current and future perspectives. Ther. Clin. Risk Manag. 2015, 11, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Taberna, M.; Mena, M.; Pavon, M.A.; Alemany, L.; Gillison, M.L.; Mesia, R. Human papillomavirus-related oropharyngeal cancer. Ann. Oncol. 2017, 28, 2386–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durst, M.; Gissmann, L.; Ikenberg, H.; zur Hausen, H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Natl. Acad. Sci. USA 1983, 80, 3812–3815. [Google Scholar] [CrossRef] [PubMed]
- Society, A.A.C. Global Cancer Facts & Figures, 3rd ed.; American Cancer Society: Atlanta, GA, USA, 2015. [Google Scholar]
- Arbyn, M.; Castellsague, X.; de Sanjose, S.; Bruni, L.; Saraiya, M.; Bray, F.; Ferlay, J. Worldwide burden of cervical cancer in 2008. Ann. Oncol. 2011, 22, 2675–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch, F.X.; Manos, M.M.; Munoz, N.; Sherman, M.; Jansen, A.M.; Peto, J.; Schiffman, M.H.; Moreno, V.; Kurman, R.; Shah, K.V.; et al. Prevalence of human papillomavirus in cervical cancer: A worldwide perspective. J. Natl. Cancer Inst. 1995, 87, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Walboomers, J.M.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.F.; Peto, J.; Meijer, C.J.L.M.; Munoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Walboomers, J.; Meijer, C. Do HPV-negative cervical carcinomas exist? J. Pathol. 1997, 181, 253–254. [Google Scholar] [CrossRef]
- Tanaka, T.I.; Alawi, F. Human Papillomavirus and Oropharyngeal Cancer. Dent. Clin. N. Am. 2018, 62, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Spence, T.; Bruce, J.; Yip, K.W.; Liu, F.F. HPV Associated Head and Neck Cancer. Cancers 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Heng, B.; Glenn, W.K.; Ye, Y.; Tran, B.; Delprado, W.; Lutze-Mann, L.; Whitaker, N.J.; Lawson, J.S. Human papilloma virus is associated with breast cancer. Br. J. Cancer 2009, 101, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Gannon, O.M.; Antonsson, A.; Bennett, I.C.; Saunders, N.A. Viral infections and breast cancer—A current perspective. Cancer Lett. 2018, 420, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Lawson, J.S.; Salmons, B.; Glenn, W.K. Oncogenic Viruses and Breast Cancer: Mouse Mammary Tumor Virus (MMTV), Bovine Leukemia Virus (BLV), Human Papilloma Virus (HPV), and Epstein-Barr Virus (EBV). Front. Oncol. 2018, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Chaturvedi, A.K.; Lowy, D.R. HPV prophylactic vaccines and the potential prevention of noncervical cancers in both men and women. Cancer 2008, 113, 3036–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glenn, W.K.; Heng, B.; Delprado, W.; Iacopetta, B.; Whitaker, N.J.; Lawson, J.S. Epstein-Barr virus, human papillomavirus and mouse mammary tumour virus as multiple viruses in breast cancer. PLoS ONE 2012, 7, e48788. [Google Scholar] [CrossRef] [PubMed]
- Salman, N.A.; Davies, G.; Majidy, F.; Shakir, F.; Akinrinade, H.; Perumal, D.; Ashrafi, G.H. Association of High Risk Human Papillomavirus and Breast cancer: A UK based Study. Sci. Rep. 2017, 7, 43591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ElAmrani, A.; Gheit, T.; Benhessou, M.; McKay-Chopin, S.; Attaleb, M.; Sahraoui, S.; El Mzibri, M.; Corbex, M.; Tommasino, M.; Khyatti, M. Prevalence of mucosal and cutaneous human papillomavirus in Moroccan breast cancer. Papillomavirus Res. 2018, 5, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.W.; Zhang, K.; Zhao, S.; Lv, Y.; Zhu, J.; Liu, H.; Feng, J.; Liang, W.; Ma, R.; Wang, J. HPV Status and Its Correlation with BCL2, p21, p53, Rb, and Survivin Expression in Breast Cancer in a Chinese Population. BioMed Res. Int. 2017, 2017, 6315392. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zeng, X.; Li, W.; Zhu, H.; Wang, G.; Liu, X.; Lv, Y.; Wu, J.; Zhuang, X.; Zhang, J.; et al. Detection and analysis of human papillomavirus (HPV) DNA in breast cancer patients by an effective method of HPV capture. PLoS ONE 2014, 9, e90343. [Google Scholar] [CrossRef] [PubMed]
- Ghittoni, R.; Accardi, R.; Hasan, U.; Gheit, T.; Sylla, B.; Tommasino, M. The biological properties of E6 and E7 oncoproteins from human papillomaviruses. Virus Genes 2010, 40, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomavirus infections, a major cause of human cancers. Biochim. Biophys. Acta Rev. Cancer 1996, 1288, F55–F78. [Google Scholar] [CrossRef]
- Shukla, S.; Mahata, S.; Shishodia, G.; Pande, S.; Verma, G.; Hedau, S.; Bhambhani, S.; Kumari, A.; Batra, S.; Basir, S.F.; et al. Physical state & copy number of high risk human papillomavirus type 16 DNA in progression of cervical cancer. Indian J. Med. Res. 2014, 139, 531–543. [Google Scholar] [PubMed]
- Thierry, F.; Yaniv, M. The BPV1-E2 trans-acting protein can be either an activator or a repressor of the HPV18 regulatory region. EMBO J. 1987, 6, 3391–3397. [Google Scholar] [PubMed]
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Werness, B.A.; Dyson, N.; Phelps, W.C.; Harlow, E.; Howley, P.M. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989, 8, 4099–4105. [Google Scholar] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997. [Google Scholar] [PubMed]
- Cancer Genome Atlas Research Network; Albert Einstein College of Medicine; Analytical Biological Services; Barretos Cancer Hospital; Baylor College of Medicine; Beckman Research Institute of City of Hope; Buck Institute for Research on Aging; Canada’s Michael Smith Genome Sciences Centre; Harvard Medical School; Helen, F.; et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef]
- Chaiwongkot, A.; Vinokurova, S.; Pientong, C.; Ekalaksananan, T.; Kongyingyoes, B.; Kleebkaow, P.; Chumworathayi, B.; Patarapadungkit, N.; Reuschenbach, M.; von Knebel Doeberitz, M. Differential methylation of E2 binding sites in episomal and integrated HPV 16 genomes in preinvasive and invasive cervical lesions. Int. J. Cancer 2013, 132, 2087–2094. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.P.; Stubenrauch, F.; Beyer-Finkler, E.; Pfister, H. Prevalence of deletions of YY1-binding sites in episomal HPV 16 DNA from cervical cancers. Int. J. Cancer 1994, 58, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, B.; Sengupta, S. CpG methylation of HPV 16 LCR at E2 binding site proximal to P97 is associated with cervical cancer in presence of intact E2. Virology 2006, 354, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Werness, B.A.; Levine, A.J.; Howley, P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990, 248, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E.; Brasiskyte, D.; Van der Haegen, B.A. E6 of human papillomavirus type 16 can overcome the M1 stage of immortalization in human mammary epithelial cells but not in human fibroblasts. Oncogene 1993, 8, 1407–1413. [Google Scholar] [PubMed]
- Richard, C.; Lanner, C.; Naryzhny, S.N.; Sherman, L.; Lee, H.; Lambert, P.F.; Zehbe, I. The immortalizing and transforming ability of two common human papillomavirus 16 E6 variants with different prevalences in cervical cancer. Oncogene 2010, 29, 3435–3445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasson, C.W.; Morgan, E.L.; Muller, M.; Ross, R.L.; Hartley, M.; Roberts, S.; Macdonald, A. Human papillomavirus type 18 E5 oncogene supports cell cycle progression and impairs epithelial differentiation by modulating growth factor receptor signalling during the virus life cycle. Oncotarget 2017, 8, 103581–103600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straight, S.W.; Hinkle, P.M.; Jewers, R.J.; McCance, D.J. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J. Virol. 1993, 67, 4521–4532. [Google Scholar] [PubMed]
- Pim, D.; Collins, M.; Banks, L. Human papillomavirus type 16 E5 gene stimulates the transforming activity of the epidermal growth factor receptor. Oncogene 1992, 7, 27–32. [Google Scholar] [PubMed]
- Genther Williams, S.M.; Disbrow, G.L.; Schlegel, R.; Lee, D.; Threadgill, D.W.; Lambert, P.F. Requirement of epidermal growth factor receptor for hyperplasia induced by E5, a high-risk human papillomavirus oncogene. Cancer Res. 2005, 65, 6534–6542. [Google Scholar] [CrossRef] [PubMed]
- Stoppler, M.C.; Straight, S.W.; Tsao, G.; Schlegel, R.; McCance, D.J. The E5 gene of HPV-16 enhances keratinocyte immortalization by full-length DNA. Virology 1996, 223, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Kishino, T.; Lalande, M.; Wagstaff, J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997, 15, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991, 10, 4129–4135. [Google Scholar] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Martinez-Zapien, D.; Ruiz, F.X.; Poirson, J.; Mitschler, A.; Ramirez, J.; Forster, A.; Cousido-Siah, A.; Masson, M.; Vande Pol, S.; Podjarny, A.; et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 2016, 529, 541–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, M.; Song, S.; Liem, A.; Androphy, E.; Liu, Y.; Lambert, P.F. A mutant of human papillomavirus type 16 E6 deficient in binding alpha-helix partners displays reduced oncogenic potential in vivo. J. Virol. 2002, 76, 13039–13048. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.L.; Keiger, K.E.; Lee, C.J.; Huibregtse, J.M. The global transcriptional effects of the human papillomavirus E6 protein in cervical carcinoma cell lines are mediated by the E6AP ubiquitin ligase. J. Virol. 2005, 79, 3737–3747. [Google Scholar] [CrossRef] [PubMed]
- Songyang, Z.; Fanning, A.S.; Fu, C.; Xu, J.; Marfatia, S.M.; Chishti, A.H.; Crompton, A.; Chan, A.C.; Anderson, J.M.; Cantley, L.C. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science 1997, 275, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Delury, C.P.; Marsh, E.K.; James, C.D.; Boon, S.S.; Banks, L.; Knight, G.L.; Roberts, S. The role of protein kinase A regulation of the E6 PDZ-binding domain during the differentiation-dependent life cycle of human papillomavirus type 18. J. Virol. 2013, 87, 9463–9472. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.L.; Nguyen, M.M.; Lee, D.; Griep, A.E.; Lambert, P.F. The PDZ ligand domain of the human papillomavirus type 16 E6 protein is required for E6’s induction of epithelial hyperplasia in vivo. J. Virol. 2003, 77, 6957–6964. [Google Scholar] [CrossRef] [PubMed]
- Kranjec, C.; Banks, L. A systematic analysis of human papillomavirus (HPV) E6 PDZ substrates identifies MAGI-1 as a major target of HPV type 16 (HPV-16) and HPV-18 whose loss accompanies disruption of tight junctions. J. Virol. 2011, 85, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Glaunsinger, B.A.; Lee, S.S.; Thomas, M.; Banks, L.; Javier, R. Interactions of the PDZ-protein MAGI-1 with adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins. Oncogene 2000, 19, 5270–5280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, M.; Kojima, T.; Yamamoto, T.; Go, M.; Takano, K.; Chiba, H.; Tokino, T.; Sawada, N. Tight junction protein MAGI-1 is up-regulated by transfection with connexin 32 in an immortalized mouse hepatic cell line: cDNA microarray analysis. Cell Tissue Res. 2005, 319, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Laimins, L.A. Role of the PDZ domain-binding motif of the oncoprotein E6 in the pathogenesis of human papillomavirus type 31. J. Virol. 2004, 78, 12366–12377. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.A.; Thomas, M.; Banks, L.; Roberts, S. Activity of the human papillomavirus E6 PDZ-binding motif correlates with an enhanced morphological transformation of immortalized human keratinocytes. J. Cell Sci. 2003, 116, 4925–4934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyono, T.; Hiraiwa, A.; Fujita, M.; Hayashi, Y.; Akiyama, T.; Ishibashi, M. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 11612–11616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimatsu, Y.; Nakahara, T.; Tanaka, K.; Inagawa, Y.; Narisawa-Saito, M.; Yugawa, T.; Ohno, S.I.; Fujita, M.; Nakagama, H.; Kiyono, T. Roles of the PDZ-binding motif of HPV 16 E6 protein in oncogenic transformation of human cervical keratinocytes. Cancer Sci. 2017, 108, 1303–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougherty, M.K.; Morrison, D.K. Unlocking the code of 14-3-3. J. Cell Sci. 2004, 117, 1875–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boon, S.S.; Banks, L. High-risk human papillomavirus E6 oncoproteins interact with 14-3-3zeta in a PDZ binding motif-dependent manner. J. Virol. 2013, 87, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Huibregtse, J.M. Human scribble (Vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6AP ubiquitin-protein ligase. Mol. Cell. Biol. 2000, 20, 8244–8253. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Thomas, M.; Javier, R.; Gardiol, D.; Banks, L. HPV E6 targeted degradation of the discs large protein: Evidence for the involvement of a novel ubiquitin ligase. Oncogene 2000, 19, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Grm, H.S.; Banks, L. Degradation of hDlg and MAGIs by human papillomavirus E6 is E6-AP-independent. J. Gen. Virol. 2004, 85, 2815–2819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poirson, J.; Biquand, E.; Straub, M.L.; Cassonnet, P.; Nomine, Y.; Jones, L.; van der Werf, S.; Trave, G.; Zanier, K.; Jacob, Y.; et al. Mapping the interactome of HPV E6 and E7 oncoproteins with the Ubiquitin-Proteasome System. FEBS J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sartor, M.A.; Dolinoy, D.C.; Jones, T.R.; Colacino, J.A.; Prince, M.E.; Carey, T.E.; Rozek, L.S. Genome-wide methylation and expression differences in HPV(+) and HPV(−) squamous cell carcinoma cell lines are consistent with divergent mechanisms of carcinogenesis. Epigenetics 2011, 6, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Fenton, T.; West, J.; Wilson, G.; Feber, A.; Henderson, S.; Thirlwell, C.; Dibra, H.K.; Jay, A.; Butcher, L.; et al. Identification and functional validation of HPV-mediated hypermethylation in head and neck squamous cell carcinoma. Genome Med 2013, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Steenbergen, R.D.; Ongenaert, M.; Snellenberg, S.; Trooskens, G.; van der Meide, W.F.; Pandey, D.; Bloushtain-Qimron, N.; Polyak, K.; Meijer, C.J.; Snijders, P.J.; et al. Methylation-specific digital karyotyping of HPV16E6E7-expressing human keratinocytes identifies novel methylation events in cervical carcinogenesis. J. Pathol. 2013, 231, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Au Yeung, C.L.; Tsang, W.P.; Tsang, T.Y.; Co, N.N.; Yau, P.L.; Kwok, T.T. HPV-16 E6 upregulation of DNMT1 through repression of tumor suppressor p53. Oncol. Rep. 2010, 24, 1599–1604. [Google Scholar] [PubMed]
- Giannini, S.L.; Hubert, P.; Doyen, J.; Boniver, J.; Delvenne, P. Influence of the mucosal epithelium microenvironment on Langerhans cells: Implications for the development of squamous intraepithelial lesions of the cervix. Int. J. Cancer 2002, 97, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denk, C.; Hulsken, J.; Schwarz, E. Reduced gene expression of E-cadherin and associated catenins in human cervical carcinoma cell lines. Cancer Lett. 1997, 120, 185–193. [Google Scholar] [CrossRef]
- Caberg, J.H.; Hubert, P.; Herman, L.; Herfs, M.; Roncarati, P.; Boniver, J.; Delvenne, P. Increased migration of Langerhans cells in response to HPV16 E6 and E7 oncogene silencing: Role of CCL20. Cancer Immunol. Immunother. 2009, 58, 39–47. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, Z.J.; Jolly, C.; Androphy, E.J.; Mercer, A.; Matthews, C.M.; Hibma, M.H. Transcriptional repression of E-cadherin by human papillomavirus type 16 E6. PLoS ONE 2012, 7, e48954. [Google Scholar] [CrossRef] [PubMed]
- Burgers, W.A.; Blanchon, L.; Pradhan, S.; de Launoit, Y.; Kouzarides, T.; Fuks, F. Viral oncoproteins target the DNA methyltransferases. Oncogene 2007, 26, 1650–1655. [Google Scholar] [CrossRef] [PubMed]
- Laurson, J.; Khan, S.; Chung, R.; Cross, K.; Raj, K. Epigenetic repression of E-cadherin by human papillomavirus 16 E7 protein. Carcinogenesis 2010, 31, 918–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalertpet, K.; Pakdeechaidan, W.; Patel, V.; Mutirangura, A.; Yanatatsaneejit, P. Human papillomavirus type 16 E7 oncoprotein mediates CCNA1 promoter methylation. Cancer Sci. 2015, 106, 1333–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chujan, S.; Kitkumthorn, N.; Siriangkul, S.; Mutirangura, A. CCNA1 promoter methylation: A potential marker for grading Papanicolaou smear cervical squamous intraepithelial lesions. Asian Pac. J. Cancer Prev. 2014, 15, 7971–7975. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Nijhuis, E.R.; Volders, H.H.; Eijsink, J.J.; Lendvai, A.; Zhang, B.; Hollema, H.; Schuuring, E.; Wisman, G.B.; van der Zee, A.G. Gene promoter methylation patterns throughout the process of cervical carcinogenesis. Cell. Oncol. 2010, 32, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Cicchini, L.; Westrich, J.A.; Xu, T.; Vermeer, D.W.; Berger, J.N.; Clambey, E.T.; Lee, D.; Song, J.I.; Lambert, P.F.; Greer, R.O.; et al. Suppression of Antitumor Immune Responses by Human Papillomavirus through Epigenetic Downregulation of CXCL14. MBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.F.; Wang, N.; Bi, X.N.; Yu, X.; Xu, X.H.; Wang, Y.L.; Zhao, C.Q.; Luo, B.; Wang, Y.K. Serine/threonine kinases 31(STK31) may be a novel cellular target gene for the HPV16 oncogene E7 with potential as a DNA hypomethylation biomarker in cervical cancer. Virol. J. 2016, 13, 60. [Google Scholar] [CrossRef] [PubMed]
- Fok, K.L.; Chung, C.M.; Yi, S.Q.; Jiang, X.; Sun, X.; Chen, H.; Chen, Y.C.; Kung, H.F.; Tao, Q.; Diao, R.; et al. STK31 maintains the undifferentiated state of colon cancer cells. Carcinogenesis 2012, 33, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Missaoui, N.; Hmissa, S.; Dante, R.; Frappart, L. Global DNA methylation in precancerous and cancerous lesions of the uterine cervix. Asian Pac. J. Cancer Prev. 2010, 11, 1741–1744. [Google Scholar] [PubMed]
- McLaughlin-Drubin, M.E.; Huh, K.W.; Munger, K. Human papillomavirus type 16 E7 oncoprotein associates with E2F6. J. Virol. 2008, 82, 8695–8705. [Google Scholar] [CrossRef] [PubMed]
- Holland, D.; Hoppe-Seyler, K.; Schuller, B.; Lohrey, C.; Maroldt, J.; Durst, M.; Hoppe-Seyler, F. Activation of the enhancer of zeste homologue 2 gene by the human papillomavirus E7 oncoprotein. Cancer Res. 2008, 68, 9964–9972. [Google Scholar] [CrossRef] [PubMed]
- Hyland, P.L.; McDade, S.S.; McCloskey, R.; Dickson, G.J.; Arthur, K.; McCance, D.J.; Patel, D. Evidence for alteration of EZH2, BMI1, and KDM6A and epigenetic reprogramming in human papillomavirus type 16 E6/E7-expressing keratinocytes. J. Virol. 2011, 85, 10999–11006. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Crum, C.P.; Munger, K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc. Natl. Acad. Sci. USA 2011, 108, 2130–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, T.L.; Zhou, B.P.; Xia, W.; Wu, Y.; Yang, C.C.; Chen, C.T.; Ping, B.; Otte, A.P.; Hung, M.C. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 2005, 310, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Menges, C.W.; Baglia, L.A.; Lapoint, R.; McCance, D.J. Human papillomavirus type 16 E7 up-regulates AKT activity through the retinoblastoma protein. Cancer Res. 2006, 66, 5555–5559. [Google Scholar] [CrossRef] [PubMed]
- Spangle, J.M.; Munger, K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J. Virol. 2010, 84, 9398–9407. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Vande Pol, S.; Banerjee, N.S.; Dutta, A.B.; Chow, L.T.; Dutta, A. Destabilization of TIP60 by human papillomavirus E6 results in attenuation of TIP60-dependent transcriptional regulation and apoptotic pathway. Mol. Cell 2010, 38, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Lee, A.Y.; Hou, S.Y.; Kemper, J.K.; Erdjument-Bromage, H.; Tempst, P.; Chiang, C.M. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006, 20, 2383–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamaki, J.I.; Wilkinson, S.; Hahn, M.; Tasdemir, N.; O’Prey, J.; Clark, W.; Hedley, A.; Nixon, C.; Long, J.S.; New, M.; et al. Bromodomain Protein BRD4 Is a Transcriptional Repressor of Autophagy and Lysosomal Function. Mol. Cell 2017, 66, 517–532.e9. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Degenkolbe, R.; Bernard, H.U.; O’Connor, M.J. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J. Virol. 1999, 73, 6209–6219. [Google Scholar] [PubMed]
- O’Connor, M.J.; Zimmermann, H.; Nielsen, S.; Bernard, H.U.; Kouzarides, T. Characterization of an E1A-CBP interaction defines a novel transcriptional adapter motif (TRAM) in CBP/p300. J. Virol. 1999, 73, 3574–3581. [Google Scholar] [PubMed]
- Subbaiah, V.K.; Zhang, Y.; Rajagopalan, D.; Abdullah, L.N.; Yeo-Teh, N.S.; Tomaic, V.; Banks, L.; Myers, M.P.; Chow, E.K.; Jha, S. E3 ligase EDD1/UBR5 is utilized by the HPV E6 oncogene to destabilize tumor suppressor TIP60. Oncogene 2015, 35, 2062–2074. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H.; Peng, K.L.; Jhang, H.C.; Lin, C.H.; Wu, S.Y.; Chiang, C.M.; Lee, S.C.; Yu, W.C.; Juan, L.J. The HPV E6 oncoprotein targets histone methyltransferases for modulating specific gene transcription. Oncogene 2012, 31, 2335–2349. [Google Scholar] [CrossRef] [PubMed]
- Brehm, A.; Nielsen, S.J.; Miska, E.A.; McCance, D.J.; Reid, J.L.; Bannister, A.J.; Kouzarides, T. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 1999, 18, 2449–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longworth, M.S.; Laimins, L.A. The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. J. Virol. 2004, 78, 3533–3541. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Cam, H.; Takahashi, Y.; Volkert, T.; Terragni, J.; Young, R.A.; Dynlacht, B.D. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 2002, 16, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Longworth, M.S.; Wilson, R.; Laimins, L.A. HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with HDACs. EMBO J. 2005, 24, 1821–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Laribee, R.N.; Klemsz, M.J.; Roman, A. Human papillomavirus type 16 E7 protein increases acetylation of histone H3 in human foreskin keratinocytes. Virology 2004, 329, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, K.; Chuikov, S.; Sarma, K.; Erdjument-Bromage, H.; Allis, C.D.; Tempst, P.; Reinberg, D. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002, 16, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodaghi, S.; Jia, R.; Zheng, Z.M. Human papillomavirus type 16 E2 and E6 are RNA-binding proteins and inhibit in vitro splicing of pre-mRNAs with suboptimal splice sites. Virology 2009, 386, 32–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Fang, Y.; Qin, J.; Chen, X.; Liang, X.; Xie, X.; Lu, W. A transcriptomic landscape of human papillomavirus 16 E6-regulated gene expression and splicing events. FEBS Lett. 2016, 590, 4594–4605. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.; Pietila, T.; Ronty, M.; Michon, F.; Frilander, M.J.; Ritari, J.; Tarkkanen, J.; Paulin, L.; Auvinen, P.; Auvinen, E. Identification and validation of human papillomavirus encoded microRNAs. PLoS ONE 2013, 8, e70202. [Google Scholar] [CrossRef] [PubMed]
- MacLean, G.; Abu-Abed, S.; Dolle, P.; Tahayato, A.; Chambon, P.; Petkovich, M. Cloning of a novel retinoic-acid metabolizing cytochrome P450, Cyp26B1, and comparative expression analysis with Cyp26A1 during early murine development. Mech. Dev. 2001, 107, 195–201. [Google Scholar] [CrossRef]
- Borutinskaite, V.V.; Navakauskiene, R.; Magnusson, K.E. Retinoic acid and histone deacetylase inhibitor BML-210 inhibit proliferation of human cervical cancer HeLa cells. Ann. N. Y. Acad. Sci. 2006, 1091, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; An, J.; Ye, P.; Zhao, K.N.; Antonsson, A. Prediction of conserved microRNAs from skin and mucosal human papillomaviruses. Arch. Virol. 2011, 156, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Honegger, A.; Schilling, D.; Bastian, S.; Sponagel, J.; Kuryshev, V.; Sultmann, H.; Scheffner, M.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Dependence of intracellular and exosomal microRNAs on viral E6/E7 oncogene expression in HPV-positive tumor cells. PLoS Pathog. 2015, 11, e1004712. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Yu, L.; Zhu, D.; Ding, W.; Wang, X.; Zhang, C.; Wang, L.; Jiang, X.; Shen, H.; He, D.; et al. Disruption of HPV16-E7 by CRISPR/Cas system induces apoptosis and growth inhibition in HPV16 positive human cervical cancer cells. BioMed Res. Int. 2014, 2014, 612823. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ding, W.; Zhu, D.; Yu, L.; Jiang, X.; Wang, X.; Zhang, C.; Wang, L.; Ji, T.; Liu, D.; et al. TALEN-mediated targeting of HPV oncogenes ameliorates HPV-related cervical malignancy. J. Clin. Investig. 2015, 125, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Bonetta, A.C.; Mailly, L.; Robinet, E.; Trave, G.; Masson, M.; Deryckere, F. Artificial microRNAs against the viral E6 protein provoke apoptosis in HPV positive cancer cells. Biochem. Biophys. Res. Commun. 2015, 465, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Lea, J.S.; Sunaga, N.; Sato, M.; Kalahasti, G.; Miller, D.S.; Minna, J.D.; Muller, C.Y. Silencing of HPV 18 oncoproteins With RNA interference causes growth inhibition of cervical cancer cells. Reprod. Sci. 2007, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.H.; Alexander, K.A. RNA interference of human papillomavirus type 18 E6 and E7 induces senescence in HeLa cells. J. Virol. 2003, 77, 6066–6069. [Google Scholar] [CrossRef] [PubMed]
- Conesa-Zamora, P.; Domenech-Peris, A.; Orantes-Casado, F.J.; Ortiz-Reina, S.; Sahuquillo-Frias, L.; Acosta-Ortega, J.; Garcia-Solano, J.; Perez-Guillermo, M. Effect of human papillomavirus on cell cycle-related proteins p16, Ki-67, Cyclin D1, p53, and ProEx C in precursor lesions of cervical carcinoma: A tissue microarray study. Am. J. Clin. Pathol. 2009, 132, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Incassati, A.; Wang, N.; McCance, D.J. Human papillomavirus type 16 E6 and E7 cause polyploidy in human keratinocytes and up-regulation of G2-M-phase proteins. Cancer Res. 2004, 64, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Glover, D.M.; Hagan, I.M.; Tavares, A.A. Polo-like kinases: A team that plays throughout mitosis. Genes Dev. 1998, 12, 3777–3787. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A. Polo-like kinases: Positive regulators of cell division from start to finish. Curr. Opin. Cell Biol. 1998, 10, 776–783. [Google Scholar] [CrossRef]
- Peters, J.M. The anaphase-promoting complex: Proteolysis in mitosis and beyond. Mol. Cell 2002, 9, 931–943. [Google Scholar] [CrossRef]
- Knoepfler, P.S. Myc goes global: New tricks for an old oncogene. Cancer Res. 2007, 67, 5061–5063. [Google Scholar] [CrossRef] [PubMed]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Abba, M.C.; Laguens, R.M.; Dulout, F.N.; Golijow, C.D. The c-myc activation in cervical carcinomas and HPV 16 infections. Mutat. Res. 2004, 557, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Couturier, J.; Sastre-Garau, X.; Schneider-Maunoury, S.; Labib, A.; Orth, G. Integration of papillomavirus DNA near myc genes in genital carcinomas and its consequences for proto-oncogene expression. J. Virol. 1991, 65, 4534–4538. [Google Scholar] [PubMed]
- Wang, Y.W.; Chang, H.S.; Lin, C.H.; Yu, W.C. HPV-18 E7 conjugates to c-Myc and mediates its transcriptional activity. Int. J. Biochem. Cell Biol. 2007, 39, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dakic, A.; Chen, R.; Dai, Y.; Schlegel, R.; Liu, X. Direct HPV E6/Myc interactions induce histone modifications, Pol II phosphorylation, and hTERT promoter activation. Oncotarget 2017, 8, 96323–96339. [Google Scholar] [CrossRef] [PubMed]
- Tworkowski, K.A.; Salghetti, S.E.; Tansey, W.P. Stable and unstable pools of Myc protein exist in human cells. Oncogene 2002, 21, 8515–8520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldman, T.; Liu, X.; Yuan, H.; Schlegel, R. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc. Natl. Acad. Sci. USA 2003, 100, 8211–8216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, S.B. MYC and the control of apoptosis. Cold Spring Harb. Perspect. Med. 2014, 4, a014407. [Google Scholar] [CrossRef] [PubMed]
- Gross-Mesilaty, S.; Reinstein, E.; Bercovich, B.; Tobias, K.E.; Schwartz, A.L.; Kahana, C.; Ciechanover, A. Basal and human papillomavirus E6 oncoprotein-induced degradation of Myc proteins by the ubiquitin pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 8058–8063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.T.; Kyo, S.; Laimins, L.A. Telomerase Activation by Human Papillomavirus Type 16 E6 Protein: Induction of Human Telomerase Reverse Transcriptase Expression through Myc and GC-Rich Sp1 Binding Sites. J. Virol. 2001, 75, 5559–5566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldman, T.; Horikawa, I.; Barrett, J.C.; Schlegel, R. Transcriptional activation of the telomerase hTERT gene by human papillomavirus type 16 E6 oncoprotein. J. Virol. 2001, 75, 4467–4472. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, D.; Pandey, A.K.; Xiuzhen, M.C.; Lee, K.K.; Hora, S.; Zhang, Y.; Chua, B.H.; Kwok, H.S.; Bhatia, S.S.; Deng, L.W.; et al. TIP60 represses telomerase expression by inhibiting Sp1 binding to the TERT promoter. PLoS Pathog. 2017, 13, e1006681. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Dakic, A.; Zhang, Y.; Dai, Y.; Chen, R.; Schlegel, R. HPV E6 protein interacts physically and functionally with the cellular telomerase complex. Proc. Natl. Acad. Sci. USA 2009, 106, 18780–18785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gewin, L.; Myers, H.; Kiyono, T.; Galloway, D.A. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes Dev. 2004, 18, 2269–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, H.R.; McCance, D.J. Human papillomavirus type 16 E6 activates TERT gene transcription through induction of c-Myc and release of USF-mediated repression. J. Virol. 2003, 77, 9852–9861. [Google Scholar] [CrossRef] [PubMed]
- Westphal, D.; Dewson, G.; Czabotar, P.E.; Kluck, R.M. Molecular biology of Bax and Bak activation and action. Biochim. Biophys. Acta 2011, 1813, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Banks, L. Human papillomavirus (HPV) E6 interactions with Bak are conserved amongst E6 proteins from high and low risk HPV types. J. Gen. Virol. 1999, 80, 1513–1517. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.; Butz, K.; Dymalla, S.; Semzow, J.; Hoppe-Seyler, F. Inhibition of Bax activity is crucial for the antiapoptotic function of the human papillomavirus E6 oncoprotein. Oncogene 2006, 25, 4009–4015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basile, J.R.; Zacny, V.; Munger, K. The cytokines tumor necrosis factor-alpha (TNF-alpha) and TNF-related apoptosis-inducing ligand differentially modulate proliferation and apoptotic pathways in human keratinocytes expressing the human papillomavirus-16 E7 oncoprotein. J. Biol. Chem. 2001, 276, 22522–22528. [Google Scholar] [CrossRef] [PubMed]
- Demers, G.W.; Halbert, C.L.; Galloway, D.A. Elevated wild-type p53 protein levels in human epithelial cell lines immortalized by the human papillomavirus type 16 E7 gene. Virology 1994, 198, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Thompson, D.A.; Munger, K. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology 1997, 239, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Seavey, S.E.; Holubar, M.; Saucedo, L.J.; Perry, M.E. The E7 oncoprotein of human papillomavirus type 16 stabilizes p53 through a mechanism independent of p19(ARF). J. Virol. 1999, 73, 7590–7598. [Google Scholar] [PubMed]
- Nor Rashid, N.; Yusof, R.; Watson, R.J. Disruption of repressive p130-DREAM complexes by human papillomavirus 16 E6/E7 oncoproteins is required for cell-cycle progression in cervical cancer cells. J. Gen. Virol. 2011, 92, 2620–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nor Rashid, N.; Yusof, R.; Watson, R.J. Disruption of pocket protein dream complexes by E7 proteins of different types of human papillomaviruses. Acta Virol. 2013, 57, 447–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeCaprio, J.A. Human papillomavirus type 16 E7 perturbs DREAM to promote cellular proliferation and mitotic gene expression. Oncogene 2014, 33, 4036–4038. [Google Scholar] [CrossRef] [PubMed]
- Mannefeld, M.; Klassen, E.; Gaubatz, S. B-MYB is required for recovery from the DNA damage-induced G2 checkpoint in p53 mutant cells. Cancer Res. 2009, 69, 4073–4080. [Google Scholar] [CrossRef] [PubMed]
- Quaas, M.; Muller, G.A.; Engeland, K. p53 can repress transcription of cell cycle genes through a p21(WAF1/CIP1)-dependent switch from MMB to DREAM protein complex binding at CHR promoter elements. Cell Cycle 2012, 11, 4661–4672. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chen, W.; Roman, A. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Quaas, M.; Wintsche, A.; Muller, G.A.; Engeland, K. Polo-like kinase 4 transcription is activated via CRE and NRF1 elements, repressed by DREAM through CDE/CHR sites and deregulated by HPV E7 protein. Nucl. Acids Res. 2014, 42, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Uxa, S.; Stanko, C.; Magin, T.M.; Engeland, K. Human papilloma virus E7 oncoprotein abrogates the p53-p21-DREAM pathway. Sci. Rep. 2017, 7, 2603. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Grossmann, P.; Padi, M.; DeCaprio, J.A. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucl. Acids Res. 2016, 44, 6070–6086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, M. p21 governs p53’s repressive side. Cell Cycle 2016, 15, 2852–2853. [Google Scholar] [CrossRef] [PubMed]
- Filippova, M.; Song, H.; Connolly, J.L.; Dermody, T.S.; Duerksen-Hughes, P.J. The human papillomavirus 16 E6 protein binds to tumor necrosis factor (TNF) R1 and protects cells from TNF-induced apoptosis. J. Biol. Chem. 2002, 277, 21730–21739. [Google Scholar] [CrossRef] [PubMed]
- Filippova, M.; Parkhurst, L.; Duerksen-Hughes, P.J. The human papillomavirus 16 E6 protein binds to Fas-associated death domain and protects cells from Fas-triggered apoptosis. J. Biol. Chem. 2004, 279, 25729–25744. [Google Scholar] [CrossRef] [PubMed]
- Garnett, T.O.; Filippova, M.; Duerksen-Hughes, P.J. Accelerated degradation of FADD and procaspase 8 in cells expressing human papilloma virus 16 E6 impairs TRAIL-mediated apoptosis. Cell Death Differ. 2006, 13, 1915–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Hasan, U.A.; Bates, E.; Takeshita, F.; Biliato, A.; Accardi, R.; Bouvard, V.; Mansour, M.; Vincent, I.; Gissmann, L.; Iftner, T.; et al. TLR9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16. J. Immunol. 2007, 178, 3186–3197. [Google Scholar] [CrossRef] [PubMed]
- Fiola, S.; Gosselin, D.; Takada, K.; Gosselin, J. TLR9 contributes to the recognition of EBV by primary monocytes and plasmacytoid dendritic cells. J. Immunol. 2010, 185, 3620–3631. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.; Sato, A.; Akira, S.; Medzhitov, R.; Iwasaki, A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 2003, 198, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Ewald, S.E.; Engel, A.; Lee, J.; Wang, M.; Bogyo, M.; Barton, G.M. Nucleic acid recognition by Toll-like receptors is coupled to stepwise processing by cathepsins and asparagine endopeptidase. J. Exp. Med. 2011, 208, 643–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, U.A.; Zannetti, C.; Parroche, P.; Goutagny, N.; Malfroy, M.; Roblot, G.; Carreira, C.; Hussain, I.; Muller, M.; Taylor-Papadimitriou, J.; et al. The human papillomavirus type 16 E7 oncoprotein induces a transcriptional repressor complex on the Toll-like receptor 9 promoter. J. Exp. Med. 2013, 210, 1369–1387. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karim, R.; Meyers, C.; Backendorf, C.; Ludigs, K.; Offringa, R.; van Ommen, G.J.; Melief, C.J.; van der Burg, S.H.; Boer, J.M. Human papillomavirus deregulates the response of a cellular network comprising of chemotactic and proinflammatory genes. PLoS ONE 2011, 6, e17848. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Hurst, J.; Voges, M.; Krauss, P.; Munch, P.; Iftner, T.; Stubenrauch, F. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J. Virol. 2011, 85, 11372–11380. [Google Scholar] [CrossRef] [PubMed]
- Rincon-Orozco, B.; Halec, G.; Rosenberger, S.; Muschik, D.; Nindl, I.; Bachmann, A.; Ritter, T.M.; Dondog, B.; Ly, R.; Bosch, F.X.; et al. Epigenetic silencing of interferon-kappa in human papillomavirus type 16-positive cells. Cancer Res. 2009, 69, 8718–8725. [Google Scholar] [CrossRef] [PubMed]
- Cortes Gutierrez, E.I.; Garcia-Vielma, C.; Aguilar-Lemarroy, A.; Vallejo-Ruiz, V.; Pina-Sanchez, P.; Zapata-Benavides, P.; Gosalvez, J. Expression of the HPV18/E6 oncoprotein induces DNA damage. Eur. J. Histochem. 2017, 61, 2773. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Pihan, G.A.; Purohit, A.; Wallace, J.; Knecht, H.; Woda, B.; Quesenberry, P.; Doxsey, S.J. Centrosome defects and genetic instability in malignant tumors. Cancer Res. 1998, 58, 3974–3985. [Google Scholar] [PubMed]
- Duensing, S.; Duensing, A.; Crum, C.P.; Munger, K. Human papillomavirus type 16 E7 oncoprotein-induced abnormal centrosome synthesis is an early event in the evolving malignant phenotype. Cancer Res. 2001, 61, 2356–2360. [Google Scholar] [PubMed]
- Duensing, S.; Lee, L.Y.; Duensing, A.; Basile, J.; Piboonniyom, S.; Gonzalez, S.; Crum, C.P.; Munger, K. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc. Natl. Acad. Sci. USA 2000, 97, 10002–10007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duensing, S.; Munger, K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002, 62, 7075–7082. [Google Scholar] [PubMed]
- Warren, C.J.; Xu, T.; Guo, K.; Griffin, L.M.; Westrich, J.A.; Lee, D.; Lambert, P.F.; Santiago, M.L.; Pyeon, D. APOBEC3A functions as a restriction factor of human papillomavirus. J. Virol. 2015, 89, 688–702. [Google Scholar] [CrossRef] [PubMed]
- Vieira, V.C.; Leonard, B.; White, E.A.; Starrett, G.J.; Temiz, N.A.; Lorenz, L.D.; Lee, D.; Soares, M.A.; Lambert, P.F.; Howley, P.M.; et al. Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. MBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Westrich, J.A.; Doorslaer, K.V.; Pyeon, D. Roles of APOBEC3A and APOBEC3B in Human Papillomavirus Infection and Disease Progression. Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.H.; Irwin, D.; Kurosu, T.; Tokunaga, K.; Sata, T.; Peterlin, B.M. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J. Virol. 2004, 78, 6073–6076. [Google Scholar] [CrossRef] [PubMed]
- Hultquist, J.F.; Lengyel, J.A.; Refsland, E.W.; LaRue, R.S.; Lackey, L.; Brown, W.L.; Harris, R.S. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 2011, 85, 11220–11234. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Takeuchi, T.; Ishii, Y.; Yugawa, T.; Kiyono, T.; Nishina, H.; Kukimoto, I. Human Papillomavirus 16 E6 Upregulates APOBEC3B via the TEAD Transcription Factor. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Westrich, J.A.; Warren, C.J.; Klausner, M.J.; Guo, K.; Liu, C.W.; Santiago, M.L.; Pyeon, D. Human Papillomavirus 16 E7 Stabilizes APOBEC3A Protein by Inhibiting Cullin 2-Dependent Protein Degradation. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Van Doorslaer, K.; Pandey, A.; Espinosa, J.M.; Pyeon, D. Role of the host restriction factor APOBEC3 on papillomavirus evolution. Virus Evol. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Koneva, L.A.; Virani, S.; Arthur, A.E.; Virani, A.; Hall, P.B.; Warden, C.D.; Carey, T.E.; Chepeha, D.B.; Prince, M.E.; et al. Subtypes of HPV-Positive Head and Neck Cancers Are Associated with HPV Characteristics, Copy Number Alterations, PIK3CA Mutation, and Pathway Signatures. Clin. Cancer Res. 2016, 22, 4735–4745. [Google Scholar] [CrossRef] [PubMed]
- Nichols, A.C.; Palma, D.A.; Chow, W.; Tan, S.; Rajakumar, C.; Rizzo, G.; Fung, K.; Kwan, K.; Wehrli, B.; Winquist, E.; et al. High frequency of activating PIK3CA mutations in human papillomavirus-positive oropharyngeal cancer. JAMA Otolaryngol. Head Neck Surg. 2013, 139, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.; Perry, C.M. Human papillomavirus quadrivalent (types 6, 11, 16, 18) recombinant vaccine (Gardasil). Drugs 2006, 66, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Donovan, B.; Franklin, N.; Guy, R.; Grulich, A.E.; Regan, D.G.; Ali, H.; Wand, H.; Fairley, C.K. Quadrivalent human papillomavirus vaccination and trends in genital warts in Australia: Analysis of national sentinel surveillance data. Lancet Infect. Dis. 2011, 11, 39–44. [Google Scholar] [CrossRef]
- Korostil, I.A.; Ali, H.; Guy, R.J.; Donovan, B.; Law, M.G.; Regan, D.G. Near elimination of genital warts in Australia predicted with extension of human papillomavirus vaccination to males. Sex Trans. Dis. 2013, 40, 833–835. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.T.; Castellsague, X.; Garland, S.M. A review of clinical trials of human papillomavirus prophylactic vaccines. Vaccine 2012, 30 (Suppl. 5), F123–F138. [Google Scholar] [CrossRef] [PubMed]
- Joura, E.A.; Giuliano, A.R.; Iversen, O.E.; Bouchard, C.; Mao, C.; Mehlsen, J.; Moreira, E.D., Jr.; Ngan, Y.; Petersen, L.K.; Lazcano-Ponce, E.; et al. A 9-valent HPV vaccine against infection and intraepithelial neoplasia in women. N. Engl. J. Med. 2015, 372, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Rajasekaran, N.; Ju, W.; Shin, Y.K. Human Papillomavirus: Current and Future RNAi Therapeutic Strategies for Cervical Cancer. J. Clin. Med. 2015, 4, 1126–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.; Bharti, A.C.; Hussain, S.; Mahata, S.; Hedau, S.; Kailash, U.; Kashyap, V.; Bhambhani, S.; Roy, M.; Batra, S.; et al. Elimination of high-risk human papillomavirus type HPV16 infection by ‘Praneem’ polyherbal tablet in women with early cervical intraepithelial lesions. J. Cancer Res. Clin. Oncol. 2009, 135, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Hillan, K.J.; Novotny, W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem. Biophys. Res. Commun. 2005, 333, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Tewari, K.S.; Sill, M.W.; Penson, R.T.; Huang, H.; Ramondetta, L.M.; Landrum, L.M.; Oaknin, A.; Reid, T.J.; Leitao, M.M.; Michael, H.E.; et al. Bevacizumab for advanced cervical cancer: Final overall survival and adverse event analysis of a randomised, controlled, open-label, phase 3 trial (Gynecologic Oncology Group 240). Lancet 2017, 390, 1654–1663. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeo-Teh, N.S.L.; Ito, Y.; Jha, S. High-Risk Human Papillomaviral Oncogenes E6 and E7 Target Key Cellular Pathways to Achieve Oncogenesis. Int. J. Mol. Sci. 2018, 19, 1706. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061706
Yeo-Teh NSL, Ito Y, Jha S. High-Risk Human Papillomaviral Oncogenes E6 and E7 Target Key Cellular Pathways to Achieve Oncogenesis. International Journal of Molecular Sciences. 2018; 19(6):1706. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061706
Chicago/Turabian StyleYeo-Teh, Nicole S. L., Yoshiaki Ito, and Sudhakar Jha. 2018. "High-Risk Human Papillomaviral Oncogenes E6 and E7 Target Key Cellular Pathways to Achieve Oncogenesis" International Journal of Molecular Sciences 19, no. 6: 1706. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061706