The Role of Midkine in Arteriogenesis, Involving Mechanosensing, Endothelial Cell Proliferation, and Vasodilation


Abstract
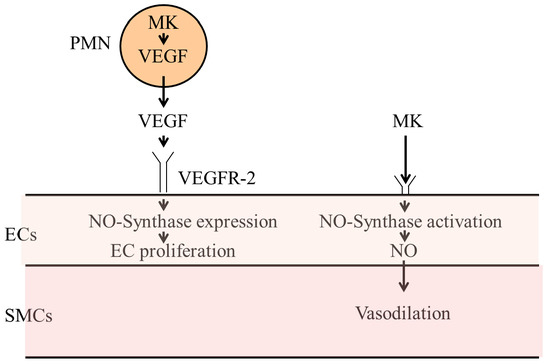
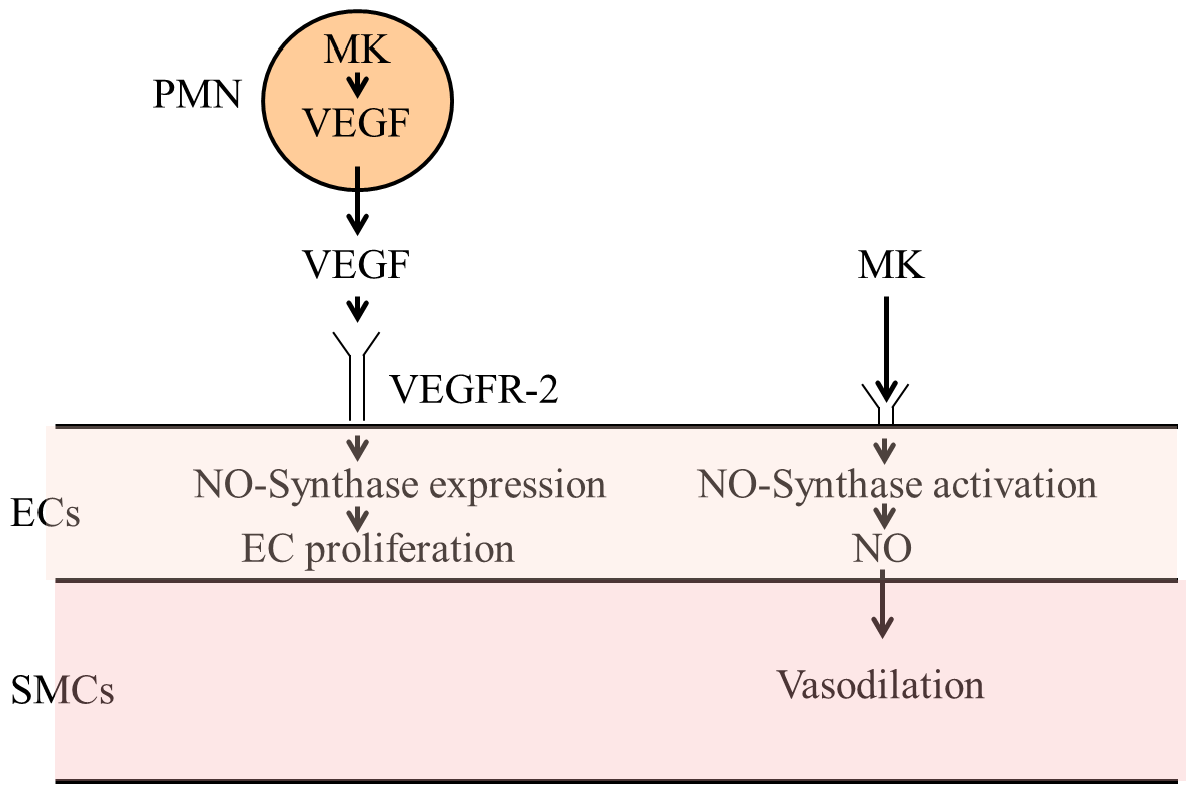
:
1. Introduction
2. Midkine
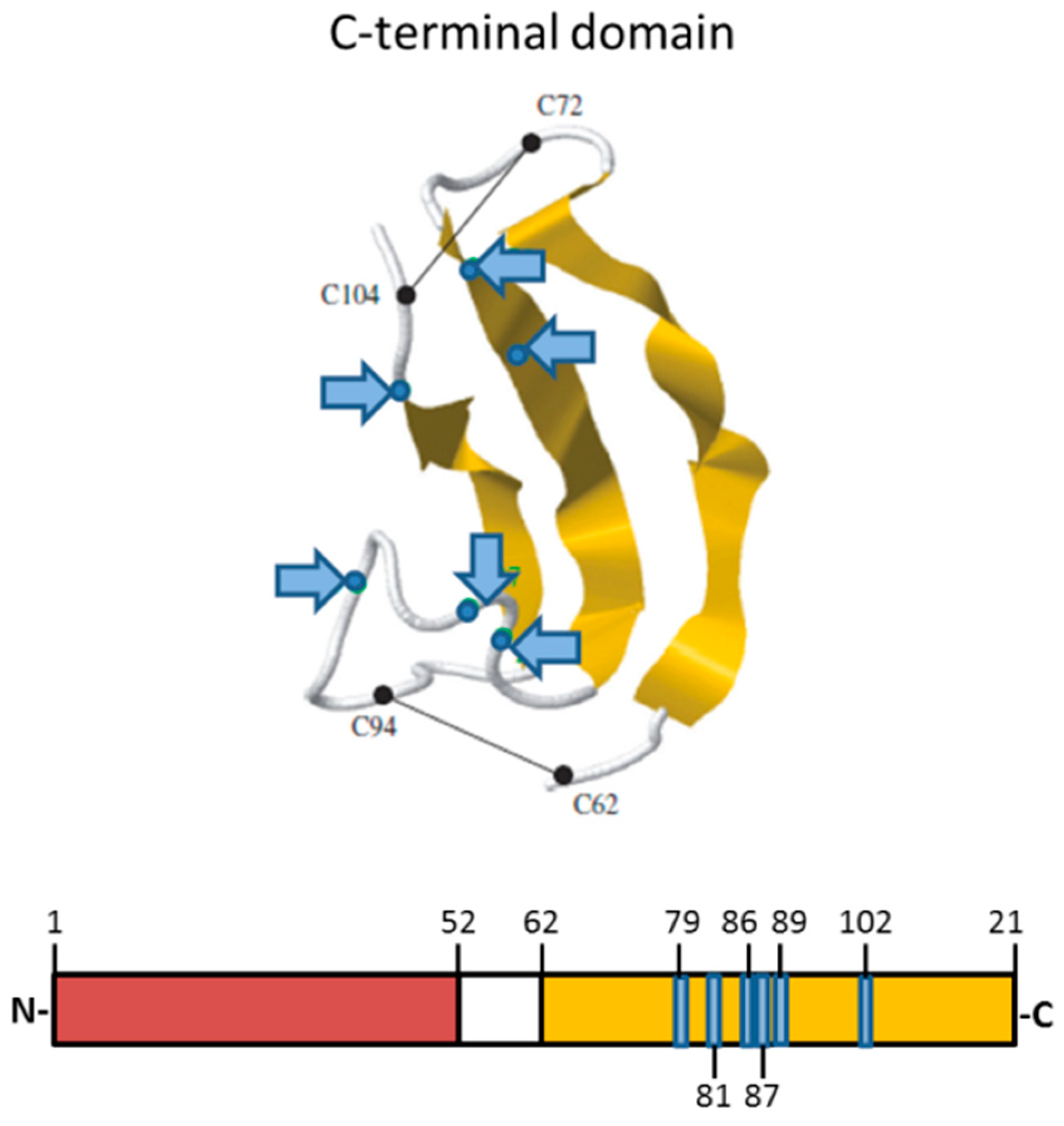
2.1. MK Gene and Protein
2.2. MK Receptors
2.3. Sources of MK and Levels of the Protein in the Vascular System
2.4. MK in Inflammation
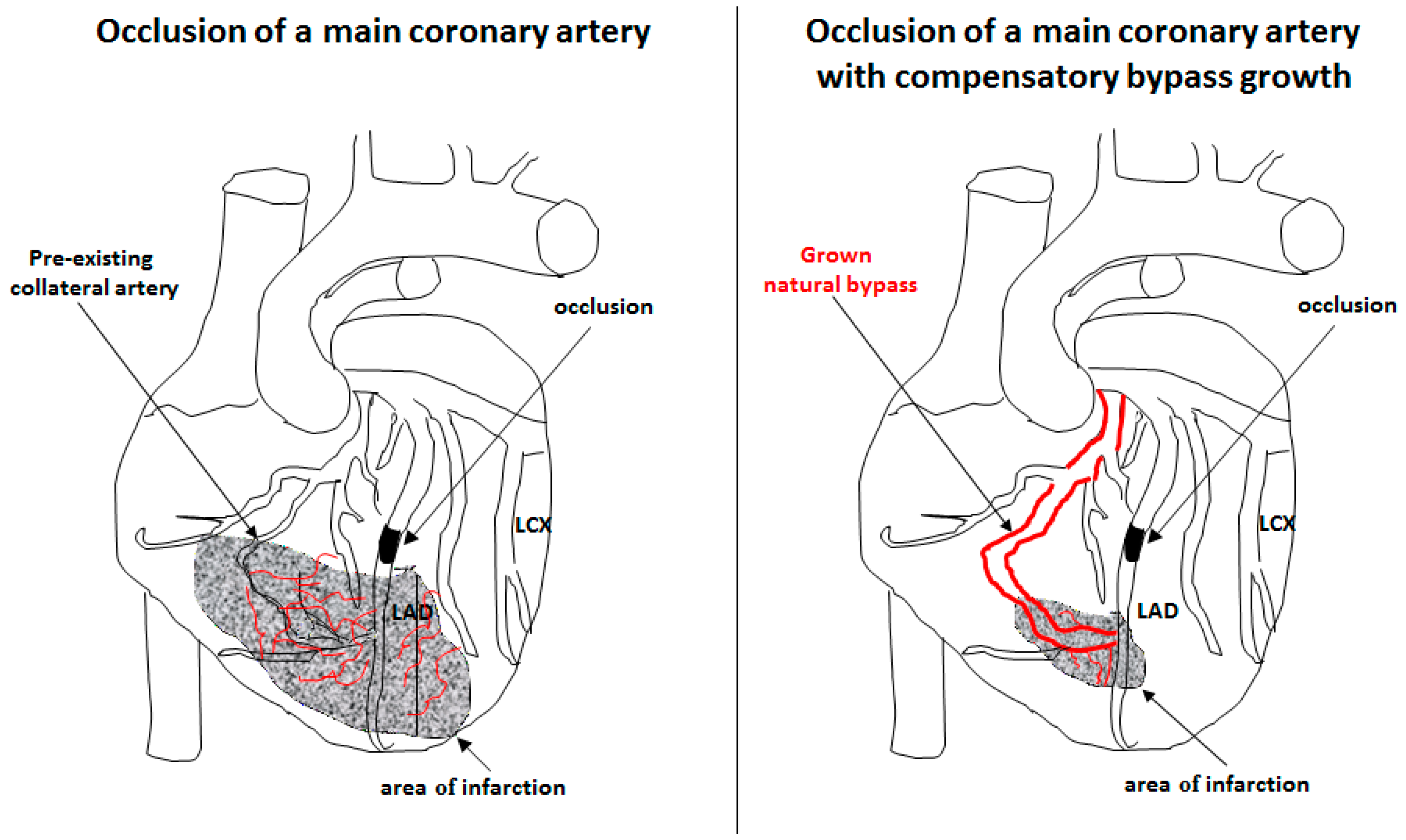
3. Arteriogenesis
3.1. Innate Immunity
3.2. Mechanosensing/Shear Stress
3.3. Vascular Cell Proliferation
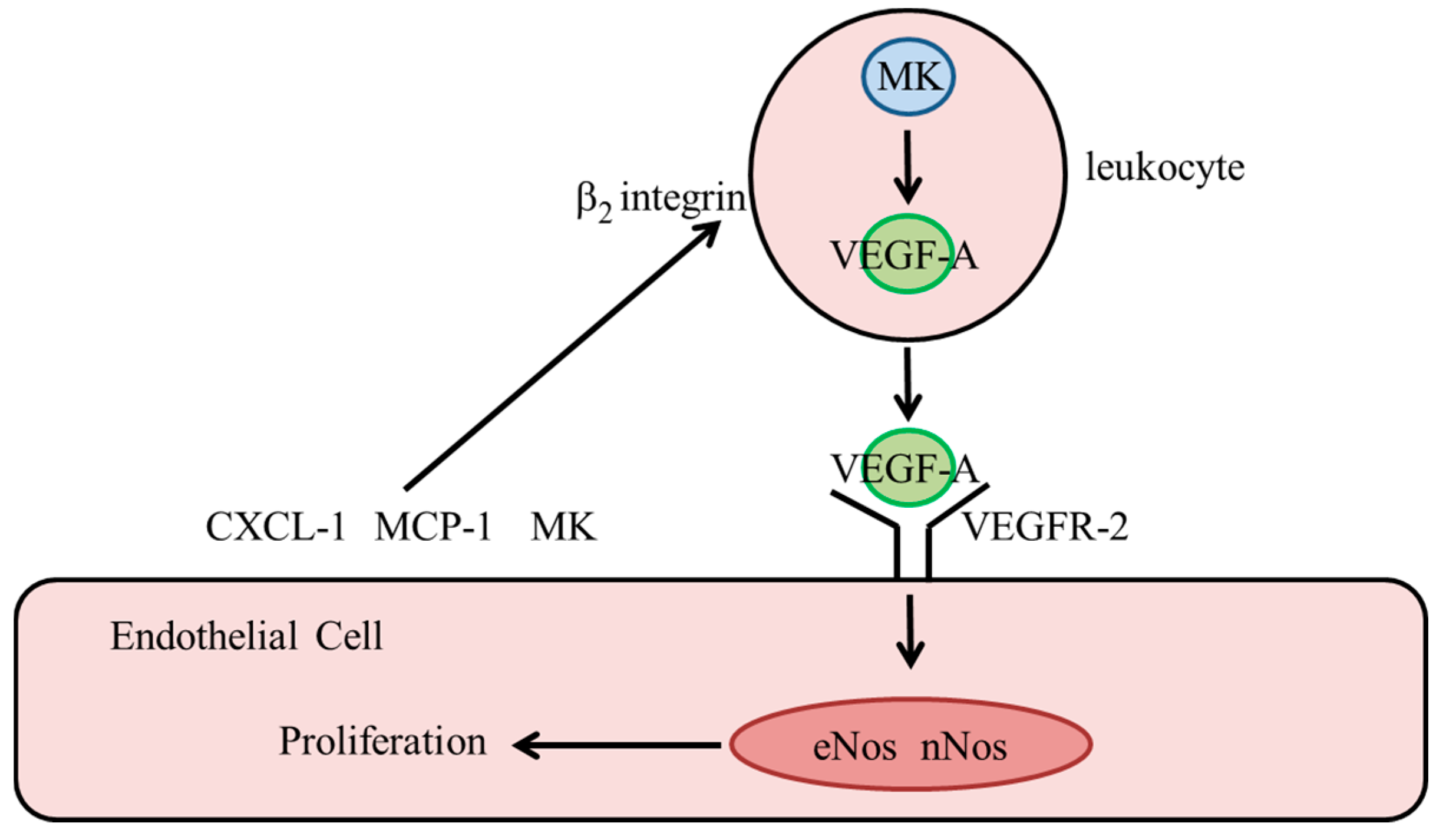
3.3.1. Vascular Endothelial Growth Factor A (VEGF-A)
3.3.2. Midkine
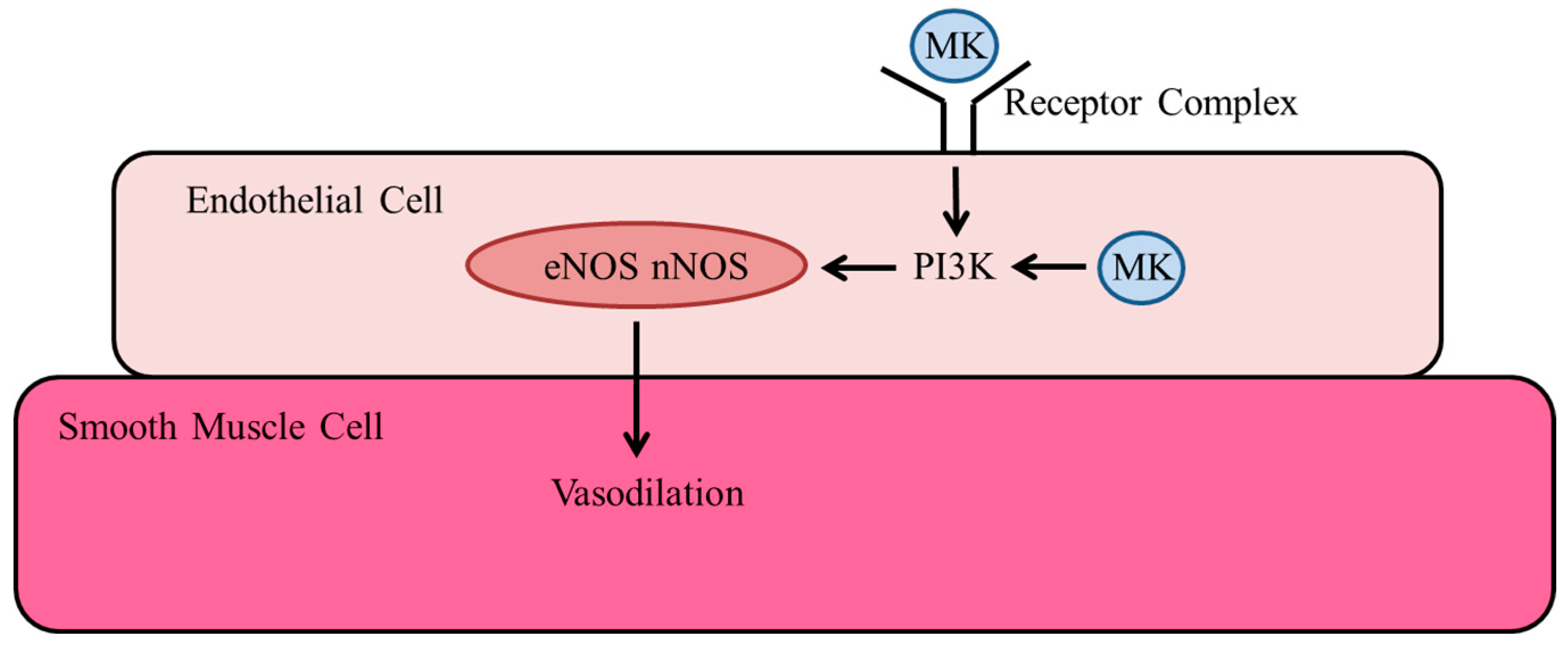
3.3.3. Nitric Oxide Synthases
3.4. Vasodilation
4. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| MK | midkine |
| PTN | pleiotrophin |
| PTPζ | receptor-like protein tyrosine phosphatase β/protein tyrosine phosphatase ζ |
| WT-1 | Wilms tumor suppressor gene |
| PI3K | phosphatidylinositol 3-kinase |
| MAPK | mitogen-activated protein kinase |
| ALK | anaplastic lymphoma kinase |
| LRP | low-density-lipoprotein receptor-related protein |
| ICAM-1 | intercellular adhesion molecule-1 |
| NFκB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| EAE | autoimmune encephalomyelitis |
| CR | cysteine rich complement-like repeats |
| PTA | percutaneous transluminal angioplasty |
| PTCA | percutaneous transluminal coronary angioplasty |
| GPIbα | glycoprotein 1bα |
| PSGL-1 | P-selectin glycoprotein ligand 1 |
| PNA | platelet-neutrophil aggregate |
| Nox2 | neutrophil-NADPH oxidase 2 |
| uPA | urokinase plasminogen activator |
| ROS | reactive oxygen species |
| TNFα | tumor necrosis factor α |
| MCP-1 | monocyte chemoattractant protein-1 |
| LAD | Left Anterior Descending |
| LCX | Left Circumflex |
| NO | nitric oxide |
| PECAM-1 | platelet adhesion molecule-1 |
| VE-Cadherin | vascular endothelial cell cadherin |
| VEGFR2 | vascular endothelial growth factor receptor 2 |
| TRPV-4 | transient receptor potential vanilloid 4 |
| VEGF-A | vascular endothelial growth factor A |
| CXCL-1 | chemokine (C-X-C motif) ligand 1 |
| MIP-2 | macrophage inflammatory protein-2 |
| CXCR2 | chemokine (C-X-C motif) receptor 2 |
| IL-8 | CXCL-8/interleukin-8 |
| NRP1 | Neuropilin 1 |
| vWF | von Willebrand factor |
| Mac-1 | macrophage-1 antigen |
| LFA-1 | lymphocyte function-associated antigen 1 |
| iNOS | inducible NOS |
| nNOS | neuronal NOS |
| CA | cerebral aneurisma |
References
- Kadomatsu, K.; Tomomura, M.; Muramatsu, T. cDNA cloning and sequencing of a new gene intensely expressed in early differentiation stages of embryonal carcinoma cells and in mid-gestation period of mouse embryogenesis. Biochem. Biophys. Res. Commun. 1988, 151, 1312–1318. [Google Scholar] [CrossRef]
- Shaheen, K.Y.; Abdel-Mageed, A.I.; Safwat, E.; AlBreedy, A.M. The value of serum midkine level in diagnosis of hepatocellular carcinoma. Int. J. Hepatol. 2015, 2015, 146389. [Google Scholar] [CrossRef] [PubMed]
- Vu Van, D.; Heberling, U.; Wirth, M.P.; Fuessel, S. Validation of the diagnostic utility of urinary midkine for the detection of bladder cancer. Oncol. Lett. 2016, 12, 3143–3152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Q.; Meng, Z.; Xu, K.; He, X.; Tan, J.; Zhang, G.; Li, X.; Liu, N.; Hu, T.; Zhou, P.; et al. Serum midkine as a surrogate biomarker for metastatic prediction in differentiated thyroid cancer patients with positive thyroglobulin antibody. Sci. Rep. 2017, 7, 43516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Song, X.; Shao, Y.; Wu, C.; Jiang, J. Prognostic value of Midkine expression in patients with solid tumors: A systematic review and meta-analysis. Oncotarget 2018, 9, 24821–24829. [Google Scholar] [CrossRef] [PubMed]
- Weckbach, L.T.; Muramatsu, T.; Walzog, B. Midkine in inflammation. Sci. World J. 2011, 11, 2491–2505. [Google Scholar] [CrossRef] [PubMed]
- Kadomatsu, K.; Huang, R.P.; Suganuma, T.; Murata, F.; Muramatsu, T. A retinoic acid responsive gene MK found in the teratocarcinoma system is expressed in spatially and temporally controlled manner during mouse embryogenesis. J. Cell Biol. 1990, 110, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muramatsu, T. Structure and function of midkine as the basis of its pharmacological effects. Br. J. Pharmacol. 2014, 171, 814–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaname, T.; Kuwano, A.; Murano, I.; Uehara, K.; Muramatsu, T.; Kajii, T. Midkine gene (MDK), a gene for prenatal differentiation and neuroregulation, maps to band 11p11.2 by fluorescence in situ hybridization. Genomics 1993, 17, 514–515. [Google Scholar] [CrossRef] [PubMed]
- Simon-Chazottes, D.; Matsubara, S.; Miyauchi, T.; Muramatsu, T.; Guenet, J.L. Chromosomal localization of two cell surface-associated molecules of potential importance in development: Midkine (Mdk) and basigin (Bsg). Mamm. Genome 1992, 2, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.R.; Mucenski, M.L.; Le Cras, T.D.; Nichols, W.C.; Whitsett, J.A. Midkine is regulated by hypoxia and causes pulmonary vascular remodeling. J. Biol. Chem. 2004, 279, 37124–37132. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, S.; Take, M.; Pedraza, C.; Muramatsu, T. Mapping and characterization of a retinoic acid-responsive enhancer of midkine, a novel heparin-binding growth/differentiation factor with neurotrophic activity. J. Biochem. 1994, 115, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Matsubara, S.; Pedraza, C.; Ozawa, M.; Tsutsui, J.; Takamatsu, H.; Noguchi, H.; Akiyama, T.; Muramatsu, T. Midkine as a novel target gene for the Wilms’ tumor suppressor gene (WT1). Oncogene 1996, 13, 2197–2203. [Google Scholar] [PubMed]
- Iwasaki, W.; Nagata, K.; Hatanaka, H.; Inui, T.; Kimura, T.; Muramatsu, T.; Yoshida, K.; Tasumi, M.; Inagaki, F. Solution structure of midkine, a new heparin-binding growth factor. EMBO J. 1997, 16, 6936–6946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, T.; Katsumi, A.; Yamazaki, T.; Muramatsu, T.; Nagasaka, T.; Ohsumi, K.; Saito, H. Human ryudocan from endothelium-like cells binds basic fibroblast growth factor, midkine, and tissue factor pathway inhibitor. J. Biol. Chem. 1996, 271, 5914–5920. [Google Scholar] [CrossRef] [PubMed]
- Novotny, W.F.; Maffi, T.; Mehta, R.L.; Milner, P.G. Identification of novel heparin-releasable proteins, as well as the cytokines midkine and pleiotrophin, in human postheparin plasma. Arterioscler. Thromb. 1993, 13, 1798–1805. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, T. Midkine and pleiotrophin: Two related proteins involved in development, survival, inflammation and tumorigenesis. J. Biochem. 2002, 132, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Fabri, L.; Maruta, H.; Muramatsu, H.; Muramatsu, T.; Simpson, R.J.; Burgess, A.W.; Nice, E.C. Structural characterisation of native and recombinant forms of the neurotrophic cytokine MK. J. Chromatogr. 1993, 646, 213–225. [Google Scholar] [CrossRef]
- Asai, T.; Watanabe, K.; Ichihara-Tanaka, K.; Kaneda, N.; Kojima, S.; Iguchi, A.; Inagaki, F.; Muramatsu, T. Identification of heparin-binding sites in midkine and their role in neurite-promotion. Biochem. Biophys. Res. Commun. 1997, 236, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, J.; Uehara, K.; Kadomatsu, K.; Matsubara, S.; Muramatsu, T. A new family of heparin-binding factors: Strong conservation of midkine (MK) sequences between the human and the mouse. Biochem. Biophys. Res. Commun. 1991, 176, 792–797. [Google Scholar] [CrossRef]
- Winkler, C.; Schafer, M.; Duschl, J.; Schartl, M.; Volff, J.N. Functional divergence of two zebrafish midkine growth factors following fish-specific gene duplication. Genome Res. 2003, 13, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Englund, C.; Birve, A.; Falileeva, L.; Grabbe, C.; Palmer, R.H. Miple1 and miple2 encode a family of MK/PTN homologues in Drosophila melanogaster. Dev. Genes Evol. 2006, 216, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Maeda, N.; Ichihara-Tanaka, K.; Kimura, T.; Kadomatsu, K.; Muramatsu, T.; Noda, M. A receptor-like protein-tyrosine phosphatase PTPzeta/RPTPbeta binds a heparin-binding growth factor midkine. Involvement of arginine 78 of midkine in the high affinity binding to PTPzeta. J. Biol. Chem. 1999, 274, 12474–12479. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Ikematsu, S.; Maeda, N.; Ichihara-Tanaka, K.; Sakuma, S.; Noda, M.; Muramatsu, T.; Kadomatsu, K. Haptotactic migration induced by midkine. Involvement of protein-tyrosine phosphatase zeta. Mitogen-activated protein kinase, and phosphatidylinositol 3-kinase. J. Biol. Chem. 2001, 276, 15868–15875. [Google Scholar] [CrossRef] [PubMed]
- Stoica, G.E.; Kuo, A.; Powers, C.; Bowden, E.T.; Sale, E.B.; Riegel, A.T.; Wellstein, A. Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell types. J. Biol. Chem. 2002, 277, 35990–35998. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, H.; Zou, K.; Sakaguchi, N.; Ikematsu, S.; Sakuma, S.; Muramatsu, T. LDL receptor-related protein as a component of the midkine receptor. Biochem. Biophys. Res. Commun. 2000, 270, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Weckbach, L.T.; Gola, A.; Winkelmann, M.; Jakob, S.M.; Groesser, L.; Borgolte, J.; Pogoda, F.; Pick, R.; Pruenster, M.; Muller-Hocker, J.; et al. The cytokine midkine supports neutrophil trafficking during acute inflammation by promoting adhesion via beta2 integrins (CD11/CD18). Blood 2014, 123, 1887–1896. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, H.; Zou, P.; Suzuki, H.; Oda, Y.; Chen, G.Y.; Sakaguchi, N.; Sakuma, S.; Maeda, N.; Noda, M.; Takada, Y.; et al. alpha4beta1- and alpha6beta1-integrins are functional receptors for midkine, a heparin-binding growth factor. J. Cell Sci. 2004, 117, 5405–5415. [Google Scholar] [CrossRef] [PubMed]
- Ueoka, C.; Kaneda, N.; Okazaki, I.; Nadanaka, S.; Muramatsu, T.; Sugahara, K. Neuronal cell adhesion, mediated by the heparin-binding neuroregulatory factor midkine, is specifically inhibited by chondroitin sulfate E. Structural ans functional implications of the over-sulfated chondroitin sulfate. J. Biol. Chem. 2000, 275, 37407–37413. [Google Scholar] [CrossRef] [PubMed]
- Ichihara-Tanaka, K.; Oohira, A.; Rumsby, M.; Muramatsu, T. Neuroglycan C is a novel midkine receptor involved in process elongation of oligodendroglial precursor-like cells. J. Biol. Chem. 2006, 281, 30857–30864. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, N.; Chen, G.Y.; Kadomatsu, K.; Ikematsu, S.; Sakuma, S.; Muramatsu, T. Glypican-2 binds to midkine: The role of glypican-2 in neuronal cell adhesion and neurite outgrowth. Glycoconj. J. 2001, 18, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Kadomatsu, K.; Okamoto, T.; Ichihara-Tanaka, K.; Kojima, T.; Saito, H.; Tomoda, Y.; Muramatsu, T. Expression of syndecan-1 and -3 during embryogenesis of the central nervous system in relation to binding with midkine. J. Biochem. 1997, 121, 197–205. [Google Scholar] [PubMed]
- Gungor, C.; Zander, H.; Effenberger, K.E.; Vashist, Y.K.; Kalinina, T.; Izbicki, J.R.; Yekebas, E.; Bockhorn, M. Notch signaling activated by replication stress-induced expression of midkine drives epithelial-mesenchymal transition and chemoresistance in pancreatic cancer. Cancer Res. 2011, 71, 5009–5019. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.W.; Sanders, J.M.; Bevard, M.; Coleman, E.; Sarembock, I.J.; Schwartz, M.A. The subendothelial extracellular matrix modulates NF-kappaB activation by flow: A potential role in atherosclerosis. J. Cell Biol. 2005, 169, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hoque, M.O.; Wu, F.; Trink, B.; Sidransky, D.; Ratovitski, E.A. Midkine induces epithelial-mesenchymal transition through Notch2/Jak2-Stat3 signaling in human keratinocytes. Cell Cycle 2008, 7, 1613–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, V.K.; Anstey, C.M.; Gately, R.P.; Comeau, D.C.; Clark, C.J.; Noble, E.P.; Mahadevan, K.; Hollett, P.R.; Pollock, A.J.; Hall, S.T.; et al. Urine and serum midkine levels in an Australian chronic kidney disease clinic population: An observational study. BMJ Open 2017, 7, e014615. [Google Scholar] [CrossRef] [PubMed]
- Weckbach, L.T.; Groesser, L.; Borgolte, J.; Pagel, J.I.; Pogoda, F.; Schymeinsky, J.; Muller-Hocker, J.; Shakibaei, M.; Muramatsu, T.; Deindl, E.; et al. Midkine acts as proangiogenic cytokine in hypoxia-induced angiogenesis. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Takada, T.; Toriyama, K.; Muramatsu, H.; Song, X.J.; Torii, S.; Muramatsu, T. Midkine, a retinoic acid-inducible heparin-binding cytokine in inflammatory responses: Chemotactic activity to neutrophils and association with inflammatory synovitis. J. Biochem. 1997, 122, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, T.; Yuzawa, Y.; Sato, W.; Arata-Kawai, H.; Suzuki, N.; Kato, N.; Matsuo, S.; Kadomatsu, K. Midkine is involved in tubulointerstitial inflammation associated with diabetic nephropathy. Lab. Investig. 2007, 87, 903–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banno, H.; Takei, Y.; Muramatsu, T.; Komori, K.; Kadomatsu, K. Controlled release of small interfering RNA targeting midkine attenuates intimal hyperplasia in vein grafts. J. Vasc. Surg. 2006, 44, 633–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Mashour, G.A.; Webster, H.F.; Kurtz, A. Basic FGF and FGF receptor 1 are expressed in microglia during experimental autoimmune encephalomyelitis: Temporally distinct expression of midkine and pleiotrophin. Glia 1998, 24, 390–397. [Google Scholar] [CrossRef]
- Wang, J.; Takeuchi, H.; Sonobe, Y.; Jin, S.; Mizuno, T.; Miyakawa, S.; Fujiwara, M.; Nakamura, Y.; Kato, T.; Muramatsu, H.; et al. Inhibition of midkine alleviates experimental autoimmune encephalomyelitis through the expansion of regulatory T cell population. Proc. Natl. Acad. Sci. USA 2008, 105, 3915–3920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruyama, K.; Muramatsu, H.; Ishiguro, N.; Muramatsu, T. Midkine, a heparin-binding growth factor, is fundamentally involved in the pathogenesis of rheumatoid arthritis. Arthritis Rheum. 2004, 50, 1420–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiba, M.; Kadomatsu, K.; Nakamura, E.; Muramatsu, H.; Ikematsu, S.; Sakuma, S.; Hayashi, K.; Yuzawa, Y.; Matsuo, S.; Kuzuya, M.; et al. Neointima formation in a restenosis model is suppressed in midkine-deficient mice. J. Clin. Investig. 2000, 105, 489–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walzog, B.; Scharffetter-Kochanek, K.; Gaehtgens, P. Impairment of neutrophil emigration in CD18-null mice. Am. J. Physiol. 1999, 276, 1125–1130. [Google Scholar] [CrossRef]
- Lillis, A.P.; Mikhailenko, I.; Strickland, D.K. Beyond endocytosis: LRP function in cell migration, proliferation and vascular permeability. J. Thromb. Haemost. JTH 2005, 3, 1884–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lillis, A.P.; Van Duyn, L.B.; Murphy-Ullrich, J.E.; Strickland, D.K. LDL receptor-related protein 1: Unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 2008, 88, 887–918. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Bu, G.; Takei, Y.; Sakamoto, K.; Ikematsu, S.; Muramatsu, T.; Kadomatsu, K. Midkine and LDL-receptor-related protein 1 contribute to the anchorage-independent cell growth of cancer cells. J. Cell Sci. 2007, 120, 4009–4015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranganathan, S.; Cao, C.; Catania, J.; Migliorini, M.; Zhang, L.; Strickland, D.K. Molecular basis for the interaction of low density lipoprotein receptor-related protein 1 (LRP1) with integrin alphaMbeta2: Identification of binding sites within alphaMbeta2 for LRP1. J. Biol. Chem. 2011, 286, 30535–30541. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Maeda, K.; Sato, W.; Kosugi, T.; Sato, Y.; Kojima, H.; Kato, N.; Ishimoto, T.; Tsuboi, N.; Uchimura, K.; et al. Growth Factor Midkine Promotes T-Cell Activation through Nuclear Factor of Activated T Cells Signaling and Th1 Cell Differentiation in Lupus Nephritis. Am. J. Pathol. 2017, 187, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Sonobe, Y.; Li, H.; Jin, S.; Kishida, S.; Kadomatsu, K.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Midkine inhibits inducible regulatory T cell differentiation by suppressing the development of tolerogenic dendritic cells. J. Immunol. 2012, 188, 2602–2611. [Google Scholar] [CrossRef] [PubMed]
- Svensson, S.L.; Pasupuleti, M.; Walse, B.; Malmsten, M.; Morgelin, M.; Sjogren, C.; Olin, A.I.; Collin, M.; Schmidtchen, A.; Palmer, R.; et al. Midkine and pleiotrophin have bactericidal properties: Preserved antibacterial activity in a family of heparin-binding growth factors during evolution. J. Biol. Chem. 2010, 285, 16105–16115. [Google Scholar] [CrossRef] [PubMed]
- Krzystek-Korpacka, M.; Mierzchala, M.; Neubauer, K.; Durek, G.; Gamian, A. Midkine, a multifunctional cytokine, in patients with severe sepsis and septic shock: A pilot study. Shock 2011, 35, 471–477. [Google Scholar] [CrossRef] [PubMed]
- World-Health-Organisation. WHO Fact Sheet N317: Cardiovascular Diseases; World-Health-Organisation: Geneva, Switzerland, 2013. [Google Scholar]
- Deindl, E.; Schaper, W. The art of arteriogenesis. Cell Biochem. Biophys. 2005, 43, 1–15. [Google Scholar] [CrossRef]
- Faber, J.E.; Chilian, W.M.; Deindl, E.; van Royen, N.; Simons, M. A brief etymology of the collateral circulation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1854–1859. [Google Scholar] [CrossRef] [PubMed]
- Seiler, C.; Stoller, M.; Pitt, B.; Meier, P. The human coronary collateral circulation: Development and clinical importance. Eur. Heart J. 2013, 34, 2674–2682. [Google Scholar] [CrossRef] [PubMed]
- Pipp, F.; Boehm, S.; Cai, W.J.; Adili, F.; Ziegler, B.; Karanovic, G.; Ritter, R.; Balzer, J.; Scheler, C.; Schaper, W.; et al. Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expression in the pig hind limb. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Chandraratne, S.; von Bruehl, M.L.; Pagel, J.I.; Stark, K.; Kleinert, E.; Konrad, I.; Farschtschi, S.; Coletti, R.; Gartner, F.; Chillo, O.; et al. Critical role of platelet glycoprotein ibalpha in arterial remodeling. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Ichikawa, N.; Lee, M.; Goto, M.; Sakai, H.; Kim, J.J.; Yoshida, M.; Handa, M.; Ikeda, Y.; Handa, S. Platelet surface P-selectin molecules increased after exposing platelet to a high shear flow. Int. Angiol. 2000, 19, 147–151. [Google Scholar] [PubMed]
- Chillo, O.; Kleinert, E.C.; Lautz, T.; Lasch, M.; Pagel, J.I.; Heun, Y.; Troidl, K.; Fischer, S.; Caballero-Martinez, A.; Mauer, A.; et al. Perivascular Mast Cells Govern Shear Stress-Induced Arteriogenesis by Orchestrating Leukocyte Function. Cell Rep. 2016, 16, 2197–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arras, M.; Ito, W.D.; Scholz, D.; Winkler, B.; Schaper, J.; Schaper, W. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J. Clin. Investig. 1998, 101, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Deindl, E.; Zaruba, M.M.; Brunner, S.; Huber, B.; Mehl, U.; Assmann, G.; Hoefer, I.E.; Mueller-Hoecker, J.; Franz, W.M. G-CSF administration after myocardial infarction in mice attenuates late ischemic cardiomyopathy by enhanced arteriogenesis. FASEB J. 2006, 20, 956–958. [Google Scholar] [CrossRef] [PubMed]
- Lucitti, J.L.; Jones, E.A.; Huang, C.; Chen, J.; Fraser, S.E.; Dickinson, M.E. Vascular remodeling of the mouse yolk sac requires hemodynamic force. Development 2007, 134, 3317–3326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malota, Z.; Glowacki, J.; Sadowski, W.; Kostur, M. Numerical analysis of the impact of flow rate, heart rate, vessel geometry, and degree of stenosis on coronary hemodynamic indices. BMC Cardiovasc. Disord. 2018, 18, 132. [Google Scholar] [CrossRef] [PubMed]
- Givens, C.; Tzima, E. Endothelial Mechanosignaling: Does One Sensor Fit All? Antioxid. Redox Signal. 2016, 25, 373–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tkachenko, E.; Rhodes, J.M.; Simons, M. Syndecans: New kids on the signaling block. Circ. Res. 2005, 96, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Weinbaum, S.; Zhang, X.; Han, Y.; Vink, H.; Cowin, S.C. Mechanotransduction and flow across the endothelial glycocalyx. Proc. Natl. Acad. Sci. USA 2003, 100, 7988–7995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Sweet, D.T.; Irani-Tehrani, M.; Maeda, N.; Tzima, E. Shc coordinates signals from intercellular junctions and integrins to regulate flow-induced inflammation. J. Cell Biol. 2008, 182, 185–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voyvodic, P.L.; Min, D.; Liu, R.; Williams, E.; Chitalia, V.; Dunn, A.K.; Baker, A.B. Loss of syndecan-1 induces a pro-inflammatory phenotype in endothelial cells with a dysregulated response to atheroprotective flow. J. Biol. Chem. 2014, 289, 9547–9559. [Google Scholar] [CrossRef] [PubMed]
- Cabral, P.D.; Garvin, J.L. TRPV4 activation mediates flow-induced nitric oxide production in the rat thick ascending limb. Am. J. Physiol. Renal Physiol. 2014, 307, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Nauli, S.M.; Kawanabe, Y.; Kaminski, J.J.; Pearce, W.J.; Ingber, D.E.; Zhou, J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation 2008, 117, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- dela Paz, N.G.; Melchior, B.; Shayo, F.Y.; Frangos, J.A. Heparan sulfates mediate the interaction between platelet endothelial cell adhesion molecule-1 (PECAM-1) and the Galphaq/11 subunits of heterotrimeric G proteins. J. Biol. Chem. 2014, 289, 7413–7424. [Google Scholar] [CrossRef] [PubMed]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.B.; Dejana, E.; Schultz, D.A.; Engelhardt, B.; Cao, G.; DeLisser, H.; Schwartz, M.A. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 2005, 437, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Jazwa, A.; Florczyk, U.; Grochot-Przeczek, A.; Krist, B.; Loboda, A.; Jozkowicz, A.; Dulak, J. Limb ischemia and vessel regeneration: Is there a role for VEGF? Vasc. Pharmacol. 2016, 86, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Lautz, T.; Lasch, M.; Borgolte, J.; Troidl, K.; Pagel, J.I.; Caballero-Martinez, A.; Kleinert, E.C.; Walzog, B.; Deindl, E. Midkine Controls Arteriogenesis by Regulating the Bioavailability of Vascular Endothelial Growth Factor A and the Expression of Nitric Oxide Synthase 1 and 3. EBioMedicine 2018, 27, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Deindl, E.; Buschmann, I.; Hoefer, I.E.; Podzuweit, T.; Boengler, K.; Vogel, S.; van Royen, N.; Fernandez, B.; Schaper, W. Role of ischemia and hypoxia-inducible genes in arteriogenesis after femoral artery occlusion in the rabbit. Circ. Res. 2001, 89, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Scapini, P.; Morini, M.; Tecchio, C.; Minghelli, S.; Di Carlo, E.; Tanghetti, E.; Albini, A.; Lowell, C.; Berton, G.; Noonan, D.M.; et al. CXCL1/macrophage inflammatory protein-2-induced angiogenesis in vivo is mediated by neutrophil-derived vascular endothelial growth factor-A. J. Immunol. 2004, 172, 5034–5040. [Google Scholar] [CrossRef] [PubMed]
- Vries, M.H.; Wagenaar, A.; Verbruggen, S.E.; Molin, D.G.; Dijkgraaf, I.; Hackeng, T.H.; Post, M.J. Erratum to: CXCL1 promotes arteriogenesis through enhanced monocyte recruitment into the peri-collateral space. Angiogenesis 2015, 18, 173. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, H.; Mitsumata, M.; Yamane, T.; Jin, X.; Yoshida, Y. Laminar shear stress-induced GRO mRNA and protein expression in endothelial cells. Circulation 1998, 98, 2584–2590. [Google Scholar] [CrossRef] [PubMed]
- Ito, W.D.; Arras, M.; Winkler, B.; Scholz, D.; Schaper, J.; Schaper, W. Monocyte chemotactic protein-1 increases collateral and peripheral conductance after femoral artery occlusion. Circ. Res. 1997, 80, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.R.; Yarovinsky, T.O.; Young, B.D.; Moraes, F.; Ross, T.D.; Ceneri, N.; Zhang, J.; Zhuang, Z.W.; Sinusas, A.J.; Pardi, R.; et al. Chemokine-coupled beta2 integrin-induced macrophage Rac2-Myosin IIA interaction regulates VEGF-A mRNA stability and arteriogenesis. J. Exp. Med. 2014, 211, 1957–1968. [Google Scholar] [CrossRef] [PubMed]
- Lanahan, A.; Zhang, X.; Fantin, A.; Zhuang, Z.; Rivera-Molina, F.; Speichinger, K.; Prahst, C.; Zhang, J.; Wang, Y.; Davis, G.; et al. The neuropilin 1 cytoplasmic domain is required for VEGF-A-dependent arteriogenesis. Dev. Cell 2013, 25, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Nishio, M.; Peters, S.C.; Tschernatsch, M.; Walberer, M.; Weidemann, S.; Heidenreich, R.; Couraud, P.O.; Weksler, B.B.; Romero, I.A.; et al. Signaling mechanism of extracellular RNA in endothelial cells. FASEB J. 2009, 23, 2100–2109. [Google Scholar] [CrossRef] [PubMed]
- Mulvany, M.J. Vascular remodelling of resistance vessels: Can we define this? Cardiovasc. Res. 1999, 41, 9–13. [Google Scholar] [CrossRef]
- Narita, H.; Chen, S.; Komori, K.; Kadomatsu, K. Midkine is expressed by infiltrating macrophages in in-stent restenosis in hypercholesterolemic rabbits. J. Vasc. Surg. 2008, 47, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.L.; Zhang, Y.J.; Li, M.H.; Zhang, X.L.; Chen, S.L. Mesenchymal stem cells with overexpression of midkine enhance cell survival and attenuate cardiac dysfunction in a rat model of myocardial infarction. Stem Cell Res. Ther. 2014, 5, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gahete, M.D.; Rincon-Fernandez, D.; Duran-Prado, M.; Hergueta-Redondo, M.; Ibanez-Costa, A.; Rojo-Sebastian, A.; Gracia-Navarro, F.; Culler, M.D.; Casanovas, O.; Moreno-Bueno, G.; et al. The truncated somatostatin receptor sst5TMD4 stimulates the angiogenic process and is associated to lymphatic metastasis and disease-free survival in breast cancer patients. Oncotarget 2016, 7, 60110–60122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Abram, C.L.; Lowell, C.A. The ins and outs of leukocyte integrin signaling. Annu. Rev. Immunol. 2009, 27, 339–362. [Google Scholar] [CrossRef] [PubMed]
- Jakob, S.M.; Pick, R.; Brechtefeld, D.; Nussbaum, C.; Kiefer, F.; Sperandio, M.; Walzog, B. Hematopoietic progenitor kinase 1 (HPK1) is required for LFA-1-mediated neutrophil recruitment during the acute inflammatory response. Blood 2013, 121, 4184–4194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, C.T.; Rossaint, J.; Moser, M.; Petrich, B.G.; Zarbock, A.; Monkley, S.J.; Critchley, D.R.; Ginsberg, M.H.; Fassler, R.; Ley, K. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood 2012, 119, 4275–4282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentzen, E.R.; Neelamegham, S.; Kansas, G.S.; Benanti, J.A.; McIntire, L.V.; Smith, C.W.; Simon, S.I. Sequential binding of CD11a/CD18 and CD11b/CD18 defines neutrophil capture and stable adhesion to intercellular adhesion molecule-1. Blood 2000, 95, 911–920. [Google Scholar] [PubMed]
- Pick, R.; Brechtefeld, D.; Walzog, B. Intraluminal crawling versus interstitial neutrophil migration during inflammation. Mol. Immunol. 2013, 55, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Pagel, J.I.; Borgolte, J.; Hoefer, I.; Fernandez, B.; Schaper, W.; Deindl, E. Involvement of neuronal NO synthase in collateral artery growth. Indian J. Biochem. Biophys. 2011, 48, 270–274. [Google Scholar] [PubMed]
- Troidl, K.; Tribulova, S.; Cai, W.J.; Ruding, I.; Apfelbeck, H.; Schierling, W.; Troidl, C.; Schmitz-Rixen, T.; Schaper, W. Effects of endogenous nitric oxide and of DETA NONOate in arteriogenesis. J. Cardiovasc. Pharmacol. 2010, 55, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Jiang, W.G.; Ahmed, A.; Boulton, M. Vascular endothelial growth factor-induced endothelial cell proliferation is regulated by interaction between VEGFR-2, SH-PTP1 and eNOS. Microvasc. Res. 2006, 71, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Gohongi, T.; Kadambi, A.; Izumi, Y.; Ang, J.; Yun, C.O.; Buerk, D.G.; Huang, P.L.; Jain, R.K. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. USA 2001, 98, 2604–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.C.; Salyapongse, A.N.; Bragdon, G.A.; Shears, L.L., 2nd; Watkins, S.C.; Edington, H.D.; Billiar, T.R. Impaired wound healing and angiogenesis in eNOS-deficient mice. Am. J. Physiol. 1999, 277, 1600–1608. [Google Scholar] [CrossRef]
- Mees, B.; Wagner, S.; Ninci, E.; Tribulova, S.; Martin, S.; van Haperen, R.; Kostin, S.; Heil, M.; de Crom, R.; Schaper, W. Endothelial nitric oxide synthase activity is essential for vasodilation during blood flow recovery but not for arteriogenesis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1926–1933. [Google Scholar] [CrossRef] [PubMed]
- Capettini, L.S.; Cortes, S.F.; Lemos, V.S. Relative contribution of eNOS and nNOS to endothelium-dependent vasodilation in the mouse aorta. Eur. J. Pharmacol. 2010, 643, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.D.; Rezende, B.A.; Cortes, S.F.; Lemos, V.S. Neuronal Nitric Oxide Synthase in Vascular Physiology and Diseases. Front. Physiol. 2016, 7, 206. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wallerath, T.; Munzel, T.; Forstermann, U. Regulation of endothelial-type NO synthase expression in pathophysiology and in response to drugs. Nitric Oxide 2002, 7, 149–164. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.J.; Zhang, S.X. NADPH oxidase 4-derived H2O2 promotes aberrant retinal neovascularization via activation of VEGF receptor 2 pathway in oxygen-induced retinopathy. J. Diabetes Res. 2015, 2015, 963289. [Google Scholar] [CrossRef] [PubMed]
- Oshikawa, J.; Urao, N.; Kim, H.W.; Kaplan, N.; Razvi, M.; McKinney, R.; Poole, L.B.; Fukai, T.; Ushio-Fukai, M. Extracellular SOD-derived H2O2 promotes VEGF signaling in caveolae/lipid rafts and post-ischemic angiogenesis in mice. PLoS ONE 2010, 5, e10189. [Google Scholar] [CrossRef] [PubMed]
- Son, H.; Hawkins, R.D.; Martin, K.; Kiebler, M.; Huang, P.L.; Fishman, M.C.; Kandel, E.R. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell 1996, 87, 1015–1023. [Google Scholar] [CrossRef]
- Kimura, H.; Esumi, H. Reciprocal regulation between nitric oxide and vascular endothelial growth factor in angiogenesis. Acta Biochim. Pol. 2003, 50, 49–59. [Google Scholar] [PubMed]
- Kroll, J.; Waltenberger, J. VEGF-A induces expression of eNOS and iNOS in endothelial cells via VEGF receptor-2 (KDR). Biochem. Biophys. Res. Commun. 1998, 252, 743–746. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, H.; Horiba, M.; Ishiguro, H.; Sumida, A.; Hojo, M.; Usui, A.; Akita, T.; Sakuma, S.; Ueda, Y.; Kodama, I.; et al. Midkine prevents ventricular remodeling and improves long-term survival after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Fujibayashi, T.; Kurauchi, Y.; Hisatsune, A.; Seki, T.; Shudo, K.; Katsuki, H. Mitogen-activated protein kinases regulate expression of neuronal nitric oxide synthase and neurite outgrowth via non-classical retinoic acid receptor signaling in human neuroblastoma SH-SY5Y cells. J. Pharmacol. Sci. 2015, 129, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.T.; Ren, C.Z.; Yang, Y.H.; Zhang, R.W.; Sun, J.C.; Wang, Y.K.; Su, D.F.; Wang, W.Z. The PI3K signaling-mediated nitric oxide contributes to cardiovascular effects of angiotensin-(1-7) in the nucleus tractus solitarii of rats. Nitric Oxide 2016, 52, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Fisslthaler, B.; Dimmeler, S.; Hermann, C.; Busse, R.; Fleming, I. Phosphorylation and activation of the endothelial nitric oxide synthase by fluid shear stress. Acta Physiol. Scand. 2000, 168, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Nagl, F.; Schonhofer, K.; Seidler, B.; Mages, J.; Allescher, H.D.; Schmid, R.M.; Schneider, G.; Saur, D. Retinoic acid-induced nNOS expression depends on a novel PI3K/Akt/DAX1 pathway in human TGW-nu-I neuroblastoma cells. Am. J. Physiol. Cell Physiol. 2009, 297, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Binder, L.; Pantakani, D.V.K.; Asif, A.R. MPA Modulates Tight Junctions’ Permeability via Midkine/PI3K Pathway in Caco-2 Cells: A Possible Mechanism of Leak-Flux Diarrhea in Organ Transplanted Patients. Front. Physiol. 2017, 8, 438. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Horiguchi, K.; Maliza, R.; Tofrizal, A.; Batchuluun, K.; Ramadhani, D.; Syaidah, R.; Tsukada, T.; Azuma, M.; Kikuchi, M.; et al. Expression of the heparin-binding growth factor midkine and its receptor, Ptprz1, in adult rat pituitary. Cell Tissue Res. 2015, 359, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Caicedo, D.; Diaz, O.; Devesa, P.; Devesa, J. Growth Hormone (GH) and Cardiovascular System. Int. J. Mol. Sci. 2018, 19, 290. [Google Scholar] [CrossRef] [PubMed]
- Rubinek, T.; Modan-Moses, D. Klotho and the Growth Hormone/Insulin-Like Growth Factor 1 Axis: Novel Insights into Complex Interactions. Vitam. Horm. 2016, 101, 85–118. [Google Scholar] [PubMed]
- Caicedo, D.; Devesa, P.; Arce, V.M.; Requena, J.; Devesa, J. Chronic limb-threatening ischemia could benefit from growth hormone therapy for wound healing and limb salvage. Ther. Adv. Cardiovasc. Dis. 2018, 12, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.M.; Davies, J.S.; Anderson, R.A.; Ellis, G.R.; Jackson, S.K.; Lewis, M.J.; Frenneaux, M.P.; Rees, A.; Scanlon, M.F. The effect of GH replacement therapy on endothelial function and oxidative stress in adult growth hormone deficiency. Eur. J. Endocrinol. 2000, 142, 254–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setola, E.; Monti, L.D.; Lanzi, R.; Lucotti, P.; Losa, M.; Gatti, E.; Galluccio, E.; Oldani, M.; Fermo, I.; Giovannelli, M.; et al. Effects of growth hormone treatment on arginine to asymmetric dimethylarginine ratio and endothelial function in patients with growth hormone deficiency. Metabolism 2008, 57, 1685–1690. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor Family | Receptor | Reference |
|---|---|---|
| Protein tyrosine phosphatase (PTP) | PTPζ | Maeda et al., 1999 [23] |
| Qi et al., 2001 [24] | ||
| LDL-receptor-related protein (LRP) | LRP-1 | Muramatsu et al., 2000 [26] |
| megalin/brushin | Muramatsu et al., 2000 [26] | |
| LRP-6 | Muramatsu et al., 2004 [28] | |
| apoE receptor-2 | Muramatsu et al., 2000 [26] | |
| Integrins | α4β1 | Muramatsu et al., 2004 [28] |
| α6β1 | Muramatsu et al., 2004 [28] | |
| Notch | notch2 | Huang et al., 2008 [35] |
| Receptor tyrosine kinase | ALK | Stoica et al., 2007 [25] |
| Glycosaminoglycans | Heparan sulfate trisulfated units | Ueoka et al., 2000 [29] |
| Chondroitin sulfate E units | Ueoka et al., 2000 [29] | |
| Syndecan-1 | Nakanishi et al., 1997 [32] | |
| Syndecan-3 | Nakanishi et al., 1997 [32] | |
| Glypican-2 | Kurosawa et al., 2001 [31] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weckbach, L.T.; Preissner, K.T.; Deindl, E. The Role of Midkine in Arteriogenesis, Involving Mechanosensing, Endothelial Cell Proliferation, and Vasodilation. Int. J. Mol. Sci. 2018, 19, 2559. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092559
Weckbach LT, Preissner KT, Deindl E. The Role of Midkine in Arteriogenesis, Involving Mechanosensing, Endothelial Cell Proliferation, and Vasodilation. International Journal of Molecular Sciences. 2018; 19(9):2559. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092559
Chicago/Turabian StyleWeckbach, Ludwig T., Klaus T. Preissner, and Elisabeth Deindl. 2018. "The Role of Midkine in Arteriogenesis, Involving Mechanosensing, Endothelial Cell Proliferation, and Vasodilation" International Journal of Molecular Sciences 19, no. 9: 2559. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092559