A Peptide Inhibitor of NADPH Oxidase (NOX2) Activation Markedly Decreases Mouse Lung Injury and Mortality Following Administration of Lipopolysaccharide (LPS)

Abstract
:
1. Introduction
2. Results
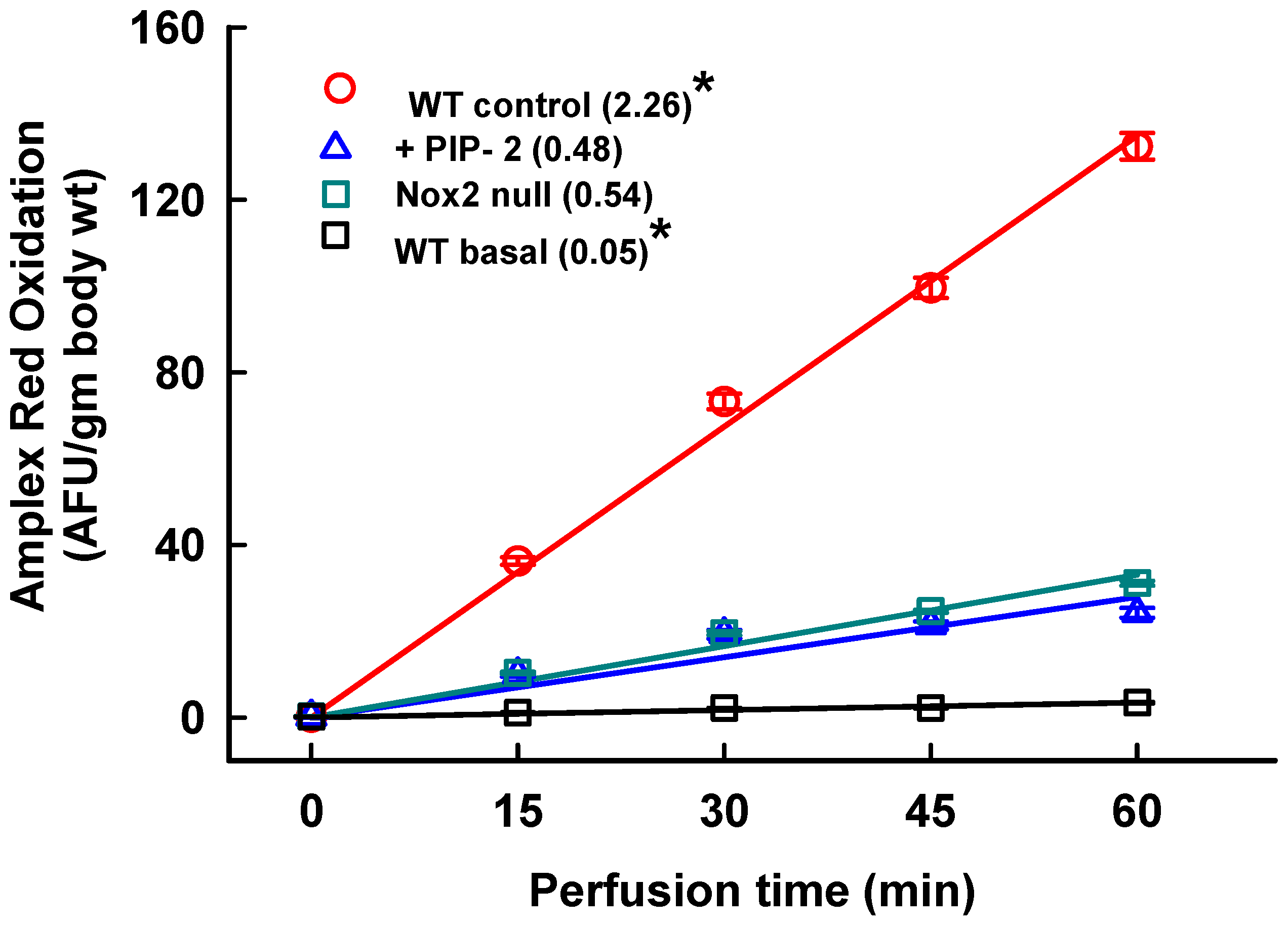
2.1. Inhibition of Lung ROS Production by PIP
2.2. Time Course for LPS-Mediated Lung Injury
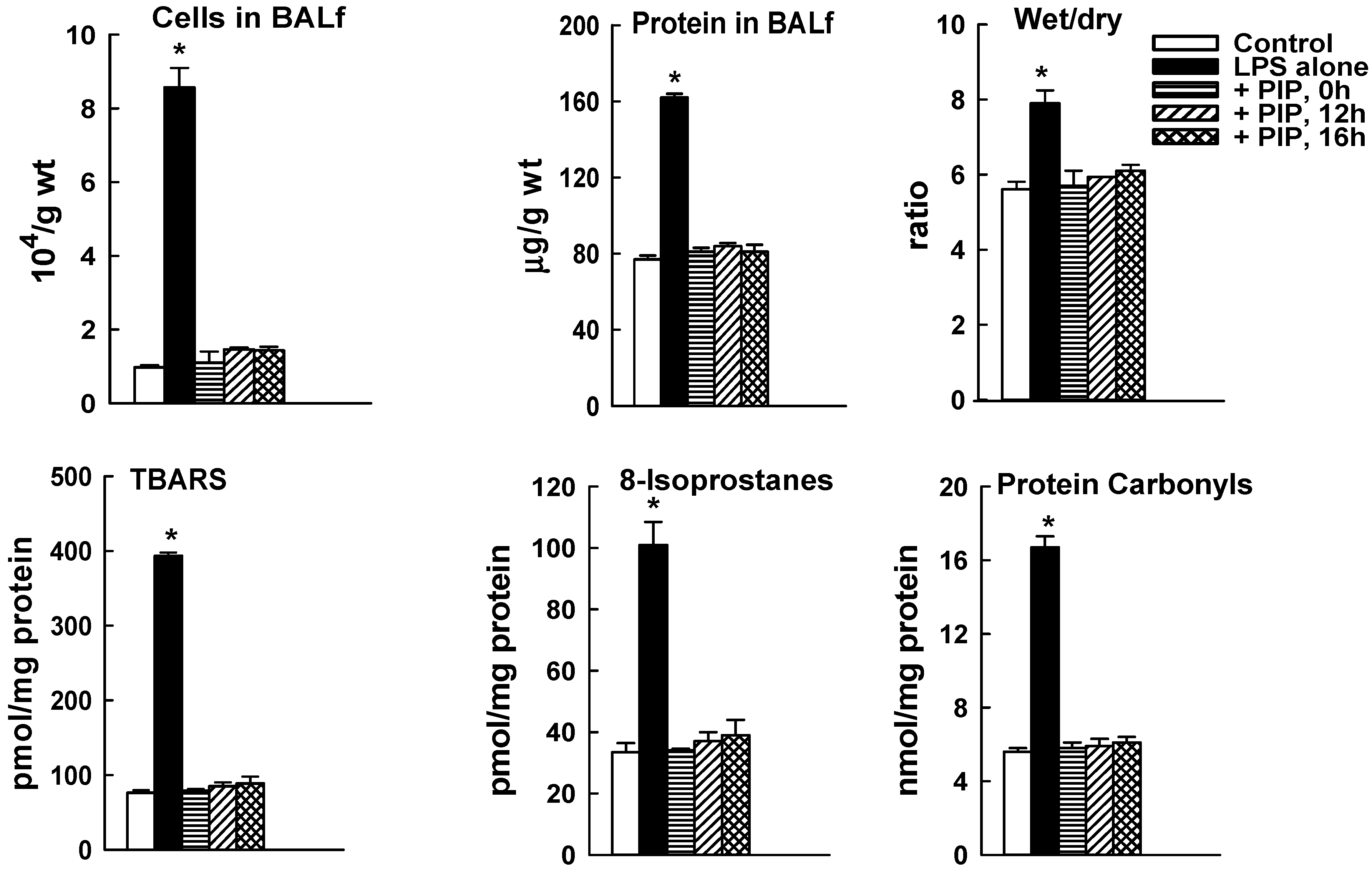
2.3. Effect of PIP-2 on LPS-Mediated Lung Injury
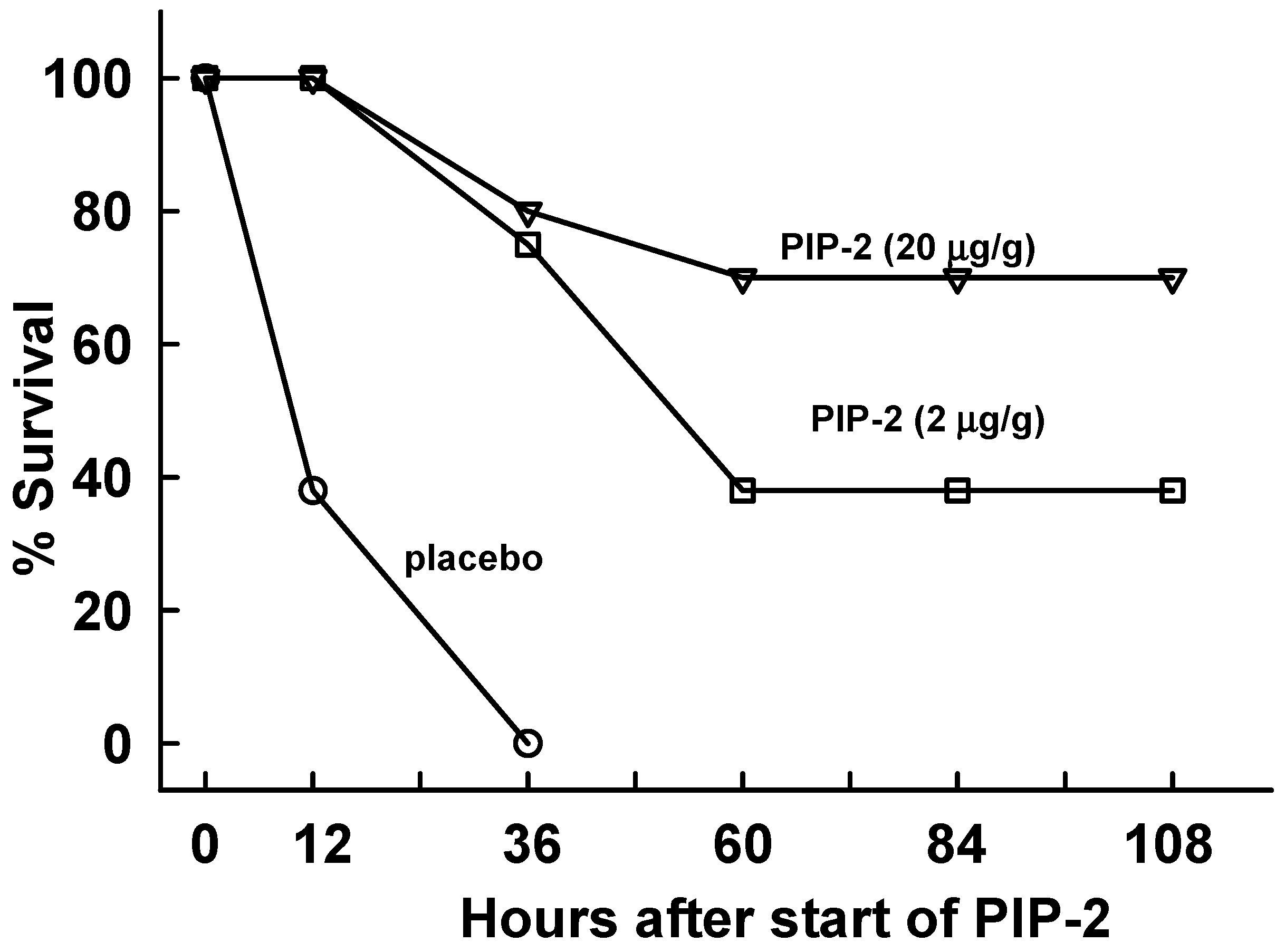
2.4. PIP-2 Treatment Prevents Mouse Mortality with High Dose LPS
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Reagents
4.3. Administration of LPS and PIP-2
4.4. Evaluation of Lung Injury
4.5. Measurement of Lung ROS Production and aiPLA2 Activity
4.6. Statistical Analysis
5. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AFU | arbitrary fluorescence units |
| aiPLA2 | PLA2 activity of Prdx6 |
| ALI | acute lung injury |
| AngII | angiotensin II |
| AM | alveolar macrophage |
| BALf | bronchoalveolar lavage fluid |
| DFFDA | difluorofuorescein diacetate |
| DPPC | dipalmitoyl phosphatidylcholine |
| DUOX | dual oxidase |
| EC | endothelial cell |
| HRP | horseradish peroxidase |
| IT | intratracheal |
| IV | intravenous |
| LPA | lysophosphatidic acid |
| LPAR | LPA receptor |
| LPC | lysoPC |
| lysoPLD | lysophospholipase D |
| LPS | lipopolysaccharide |
| Marcks | myristoylated alanine-rich C kinase substrate protein |
| NOX | NADPH oxidase |
| PC | phosphatidylcholine |
| PG | phosphatidyl glycerol |
| PLA2 | phospholipase A2 |
| PIP | Prdx6-PLA2 inhibitory peptide |
| PMA | phorbol myristic acid |
| PMN | polymorphonuclear leokocyte; pPrdx6, phosphorylated Prdx6 |
| Prdx | peroxiredoxin |
| ROS | reactive O2 species |
| SP-A | surfactant protein A |
| TBARS | thiobarbituric reactive substances |
| WT | wild type |
References
- Bhattacharya, J.; Matthay, M.A. Regulation and repair of the alveolar-capillary barrier in acute lung injury. Annu. Rev. Physiol. 2013, 75, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.L.; Downey, G.P. Neutrophil activation and acute lung injury. Curr. Opin. Crit. Care 2001, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.F.; Ward, P.A. Role of oxidants in lung injury during sepsis. Antioxid. Redox Signal. 2007, 9, 1991–2002. [Google Scholar] [CrossRef]
- Aldridge, A.J. Role of the neutrophil in septic shock and the adult respiratory distress syndrome. Eur. J. Surg. 2002, 168, 204–214. [Google Scholar] [CrossRef]
- Kilpatrick, L.E.; Sun, S.; Li, H.; Vary, T.C.; Korchak, H.M. Regulation of TNF-induced oxygen radical production in human neutrophils. J. Leukoc Biol. 2010, 87, 153–164. [Google Scholar] [CrossRef]
- Chabot, F.; Mitchell, J.A.; Gutteridge, J.M.; Evans, T.W. Reactive oxygen species in acute lung injury. Eur. Respir. J. 1998, 11, 745–757. [Google Scholar]
- Gandhirajan, R.K.; Meng, S.; Chandramoorthy, H.C.; Mallilankaraman, K.; Mancarella, S.; Gao, H.; Razmpour, R.; Yang, X.F.; Houser, S.R.; Chen, J.; et al. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J. Clin. Investig. 2013, 123, 887–902. [Google Scholar] [CrossRef] [Green Version]
- Lassegue, B.; San Martin, A.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef]
- Sato, K.; Kadiiska, M.B.; Ghio, A.J.; Corbett, J.; Fann, Y.C.; Holland, S.M.; Thurman, R.G.; Mason, R.P. In vivo lipid-derived free radical formation by NADPH oxidase in acute lung injury induced by lipopolysaccharide: A model for ARDS. FASEB J. 2002, 16, 1713–1720. [Google Scholar] [CrossRef]
- Lambeth, J.; Cheng, G.; Arnold, R.S.; Edens, W.A. Novel homologs of gp91phox. Trends Biochem. Sci. 2000, 25, 459–461. [Google Scholar] [CrossRef]
- Fisher, A.B.; Zhang, G. NADPH and NADPH oxidase. In Encyclopedia of Respiratory Medicine; Laurent, G.J., Shapiro, S.D., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; pp. 77–83. [Google Scholar]
- Nauseef, W.M. Biological roles for the NOX family NADPH oxidases. J. Biol. Chem. 2008, 283, 16961–16965. [Google Scholar] [CrossRef]
- Babior, B.M.; Lambeth, J.D.; Nauseef, W. The neutrophil NADPH oxidase. Arch. Biochem. Biophys. 2002, 397, 342–344. [Google Scholar] [CrossRef]
- Lambeth, J.D. NOX enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy. Free Radic. Biol. Med. 2007, 43, 332–347. [Google Scholar] [CrossRef]
- Wang, W.; Suzuki, Y.; Tanigaki, T.; Rank, D.R.; Raffin, T.A. Effect of the NADPH oxidase inhibitor apocynin on septic lung injury in guinea pigs. Am. J. Respir. Criti. Care Med. 1994, 150, 1449–1452. [Google Scholar] [CrossRef]
- Dodd-o, J.M.; Welsh, L.E.; Salazar, J.D.; Walinsky, P.L.; Peck, E.A.; Shake, J.G.; Caparrelli, D.J.; Ziegelstein, R.C.; Zweier, J.L.; Baumgartner, W.A.; et al. Effect of NADPH oxidase inhibition on cardiopulmonary bypass-induced lung injury. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H927–H936. [Google Scholar] [CrossRef] [Green Version]
- Xiang, L.; Lu, S.; Mittwede, P.N.; Clemmer, J.S.; Hester, R.L. Inhibition of NADPH oxidase prevents acute lung injury in obese rats following severe trauma. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H684–H689. [Google Scholar] [CrossRef] [Green Version]
- Davidson, B.A.; Vethanayagam, R.R.; Grimm, M.J.; Mullan, B.A.; Raghavendran, K.; Blackwell, T.S.; Freeman, M.L.; Ayyasamy, V.; Singh, K.K.; Sporn, M.B.; et al. NADPH oxidase and NRF2 regulate gastric aspiration-induced inflammation and acute lung injury. J. Immunol. 2013, 190, 1714–1724. [Google Scholar] [CrossRef]
- Dodd-O, J.M.; Pearse, D.B. Effect of the NADPH oxidase inhibitor apocynin on ischemia-reperfusion lung injury. Am. J. Physiol. 2000, 279, H303–H312. [Google Scholar] [CrossRef]
- Luo, G.; Zhu, G.; Yuan, D.; Yao, W.; Chi, X.; Hei, Z. Propofol alleviates acute lung injury following orthotopic autologous liver transplantation in rats via inhibition of the NADPH oxidase pathway. Mol. Med. Rep. 2015, 11, 2348–2354. [Google Scholar] [CrossRef]
- Gao, X.P.; Standiford, T.J.; Rahman, A.; Newstead, M.; Holland, S.M.; Dinauer, M.C.; Liu, Q.H.; Malik, A.B. Role of NADPH oxidase in the mechanism of lung neutrophil sequestration and microvessel injury induced by gram-negative sepsis: Studies in p47phox-/- and gp91phox-/- mice. J. Immunol. 2002, 168, 3974–3982. [Google Scholar] [CrossRef]
- Kilpatrick, L.E.; Standage, S.W.; Li, H.; Raj, N.R.; Korchak, H.M.; Wolfson, M.R.; Deutschman, C.S. Protection against sepsis-induced lung injury by selective inhibition of protein kinase c-delta (delta-PKC). J. Leukoc. Biol. 2011, 89, 3–10. [Google Scholar] [CrossRef]
- Lee, I.; Dodia, C.; Chatterjee, S.; Zagorski, J.; Mesaros, C.; Blair, I.A.; Feinstein, S.I.; Jain, M.; Fisher, A.B. A novel nontoxic inhibitor of the activation of NADPH oxidase reduces reactive oxygen species production in mouse lung. J. Pharmacol. Exp. Ther. 2013, 345, 284–296. [Google Scholar] [CrossRef]
- Vazquez-Medina, J.P.; Dodia, C.; Weng, L.; Mesaros, C.; Blair, I.; Feinstein, S.I.; Chatterjee, C.; Fisher, A. The phospholipase A2 activity of peroxiredoxin 6 modulates NADPH oxidase 2 activation via lysophosphatidic acid receptor signaling in the pulmonary endothelium and alveolar macrophages. FASEB J. 2016, 30, 2885–2898. [Google Scholar] [CrossRef]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Sulciner, D.J.; Gutkind, J.S.; Irani, K.; Goldschmidt-Clermont, P.J.; Finkel, T. Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. Biochem. J. 1996, 318 Pt 2, 379–382. [Google Scholar] [CrossRef]
- Chatterjee, S.; Feinstein, S.I.; Dodia, C.; Sorokina, E.; Lien, Y.C.; Nguyen, S.; Debolt, K.; Speicher, D.; Fisher, A.B. Peroxiredoxin 6 phosphorylation and subsequent phospholipase A2 activity are required for agonist-mediated activation of NADPH oxidase in mouse pulmonary microvascular endothelium and alveolar macrophages. J. Biol. Chem. 2011, 286, 11696–11706. [Google Scholar] [CrossRef]
- Lee, I.; Dodia, C.; Chatterjee, S.; Feinstein, S.I.; Fisher, A.B. Protection against LPS-induced acute lung injury by a mechanism based inhibitor of NADPH oxidase (type 2). Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, 635–644. [Google Scholar] [CrossRef]
- Kwon, J.; Wang, A.; Burke, D.J.; Boudreau, H.E.; Lekstrom, K.J.; Korzeniowska, A.; Sugamata, R.; Kim, Y.S.; Yi, L.; Ersoy, I.; et al. Peroxiredoxin 6 (Prdx6) supports NADPH oxidase1 (Nox1)-based superoxide generation and cell migration. Free Radic. Biol. Med. 2016, 96, 99–115. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.Z.; Manevich, Y.; Baldwin, J.L.; Dodia, C.; Yu, K.; Feinstein, S.I.; Fisher, A.B. Interaction of surfactant protein a with peroxiredoxin 6 regulates phospholipase A2 activity. J. Biol. Chem. 2006, 281, 7515–7525. [Google Scholar] [CrossRef]
- Fisher, A.B.; Dodia, C.; Chander, A. Inhibition of lung calcium-independent phospholipase A2 by surfactant protein A. Am. J. Physiol. Lung Cell Mol. Physiol. 1994, 267, L335–L341. [Google Scholar] [CrossRef]
- Krishnaiah, S.Y.; Dodia, C.; Sorokina, E.M.; Li, H.; Feinstein, S.I.; Fisher, A.B. Binding sites for interaction of peroxiredoxin 6 with surfactant protein A. Biochim. Biophys. Acta 2016, 1864, 419–425. [Google Scholar] [CrossRef]
- Fisher, A.B.; Dodia, C.; Feinstein, S.I. Identification of small peptides that inhibit NADPH oxidase (NOX2) activation. Antioxidants 2018, 7, 181. [Google Scholar] [CrossRef]
- Matute-Bello, G.; Frevert, C.W.; Martin, T.R. Animal models of acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L379–L399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Medina, J.P.; Tao, J.Q.; Patel, P.; Bannitz-Fernandes, R.; Dodia, C.; Sorokina, E.M.; Feinstein, S.I.; Chatterjee, S.; Fisher, A.B. Genetic inactivation of the phospholipase A2 activity of peroxiredoxin 6 in mice protects against LPS-induced acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L656–L668. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; He, Q.; Wang, F.; Redington, A.N. Acute and chronic remote ischemic conditioning attenuate septic cardiomyopathy, improve cardiac output, protect systemic organs, and improve mortality in a lipopolysaccharide-induced sepsis model. Basic Res. Cardiol. 2019, 114, 15. [Google Scholar] [CrossRef]
- Lewis, A.J.; Seymour, C.W.; Rosengart, M.R. Current murine models of sepsis. Surg. Infect. (Larchmt.) 2016, 17, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Hood, E.D.; Greineder, C.F.; Dodia, C.; Han, J.; Mesaros, C.; Shuvaev, V.V.; Blair, I.A.; Fisher, A.B.; Muzykantov, V.R. Antioxidant protection by PECAM-targeted delivery of a novel NADPH-oxidase inhibitor to the endothelium in vitro and in vivo. J. Control. Release 2012, 163, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. Antiox. Redox Signal. 2011, 15, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Pendyala, S.; Natarajan, V.; Garcia, J.G.; Jacobson, J.R. Endothelial cell barrier protection by simvastatin: GTPase regulation and NADPH oxidase inhibition. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L575–L583. [Google Scholar] [CrossRef] [Green Version]
- Altintas, N.D.; Atilla, P.; Iskit, A.B.; Topeli, A. Long-term simvastatin attenuates lung injury and oxidative stress in murine acute lung injury models induced by oleic acid and endotoxin. Respir. Care 2011, 56, 1156–1163. [Google Scholar] [CrossRef]
- Grommes, J.; Vijayan, S.; Drechsler, M.; Hartwig, H.; Morgelin, M.; Dembinski, R.; Jacobs, M.; Koeppel, T.A.; Binnebosel, M.; Weber, C.; et al. Simvastatin reduces endotoxin-induced acute lung injury by decreasing neutrophil recruitment and radical formation. PLoS ONE 2012, 7, e38917. [Google Scholar] [CrossRef]
- Abdelmageed, M.E.; El-Awady, M.S.; Suddek, G.M. Apocynin ameliorates endotoxin-induced acute lung injury in rats. Int. Immunopharmacol. 2016, 30, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Suh, G.J.; Kwon, W.Y.; Kim, K.S.; Park, M.J.; Kim, T.; Ko, J.I. Apocynin suppressed the nuclear factor-KappaB pathway and attenuated lung injury in a rat hemorrhagic shock model. J. Trauma Acute Care Surg. 2017, 82, 566–574. [Google Scholar] [CrossRef]
- Yin, Q.; Fang, S.; Park, J.; Crews, A.L.; Parikh, I.; Adler, K.B. An inhaled inhibitor of myristoylated alanine-rich C kinase substrate reverses LPS-induced acute lung injury in mice. Am. J. Respir. Cell Mol. Biol. 2016, 55, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Kettle, A.J. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid. Redox Signal. 2013, 18, 642–660. [Google Scholar] [CrossRef]
- Van Acker, H.; Coenye, T. The role of reactive oxygen species in antibiotic-mediated killing of bacteria. Trends Microbiol. 2017, 25, 456–466. [Google Scholar] [CrossRef]
- Fan, E.; Brodie, D.; Slutsky, A.S. Acute respiratory distress syndrome: Advances in diagnosis and treatment. JAMA 2018, 319, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Benipal, B.; Feinstein, S.I.; Chatterjee, S.; Dodia, C.; Fisher, A.B. Inhibition of the phospholipase A2 activity of peroxiredoxin 6 prevents lung damage wih exposure to hyperoxia. Redox Biol. 2015, 4, 321–327. [Google Scholar] [CrossRef]
- Rich, J.T.; Neely, J.G.; Paniello, R.C.; Voelker, C.C.; Nussenbaum, B.; Wang, E.W. A practical guide to understanding Kaplan-Meier curves. Otolaryngol. Head Neck Surg. 2010, 143, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Debski, D.; Smulik, R.; Zielonka, J.; Michalowski, B.; Jakubowska, M.; Debowska, K.; Adamus, J.; Marcinek, A.; Kalyanaraman, B.; Sikora, A. Mechanism of oxidative conversion of amplex(r) red to resorufin: Pulse radiolysis and enzymatic studies. Free Radic. Biol. Med. 2016, 95, 323–332. [Google Scholar] [CrossRef]
- Noel, J.; Wang, H.; Hong, N.; Tao, J.Q.; Yu, K.; Sorokina, E.M.; Debolt, K.; Heayn, M.; Rizzo, V.; Delisser, H.; et al. PECAM-1 and caveolae form the mechanosensing complex necessary for NOX2 activation and angiogenic signaling with stopped flow in pulmonary endothelium. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L805–L818. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BALf Cells ×104/g Body wt. | BALf Protein µg/g wt. | Wet/Dry Ratio | TBARS pmol/mg prot | 8-Isoprostanes pmol/mg prot | Protein Carbonyls nmol/mg prot | |
|---|---|---|---|---|---|---|
| Control (no LPS) | 0.95 ± 0.04 | 75 ± 1.3 | 5.59 ± 0.03 | 76 ± 6 | 34 ± 3 | 5.6 ± 0.2 |
| LPS + PIP-2 | 0.96 ± 0.40 | 78 ± 2.2 | 5.34 ± 0.03 | 77 ± 1 | 34 ± 3 | 5.6 ± 0.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fisher, A.B.; Dodia, C.; Chatterjee, S.; Feinstein, S.I. A Peptide Inhibitor of NADPH Oxidase (NOX2) Activation Markedly Decreases Mouse Lung Injury and Mortality Following Administration of Lipopolysaccharide (LPS). Int. J. Mol. Sci. 2019, 20, 2395. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102395
Fisher AB, Dodia C, Chatterjee S, Feinstein SI. A Peptide Inhibitor of NADPH Oxidase (NOX2) Activation Markedly Decreases Mouse Lung Injury and Mortality Following Administration of Lipopolysaccharide (LPS). International Journal of Molecular Sciences. 2019; 20(10):2395. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102395
Chicago/Turabian StyleFisher, Aron B., Chandra Dodia, Shampa Chatterjee, and Sheldon I. Feinstein. 2019. "A Peptide Inhibitor of NADPH Oxidase (NOX2) Activation Markedly Decreases Mouse Lung Injury and Mortality Following Administration of Lipopolysaccharide (LPS)" International Journal of Molecular Sciences 20, no. 10: 2395. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102395