Genome Editing as a Treatment for the Most Prevalent Causative Genes of Autosomal Dominant Retinitis Pigmentosa

 , ,
, , 
Abstract
:
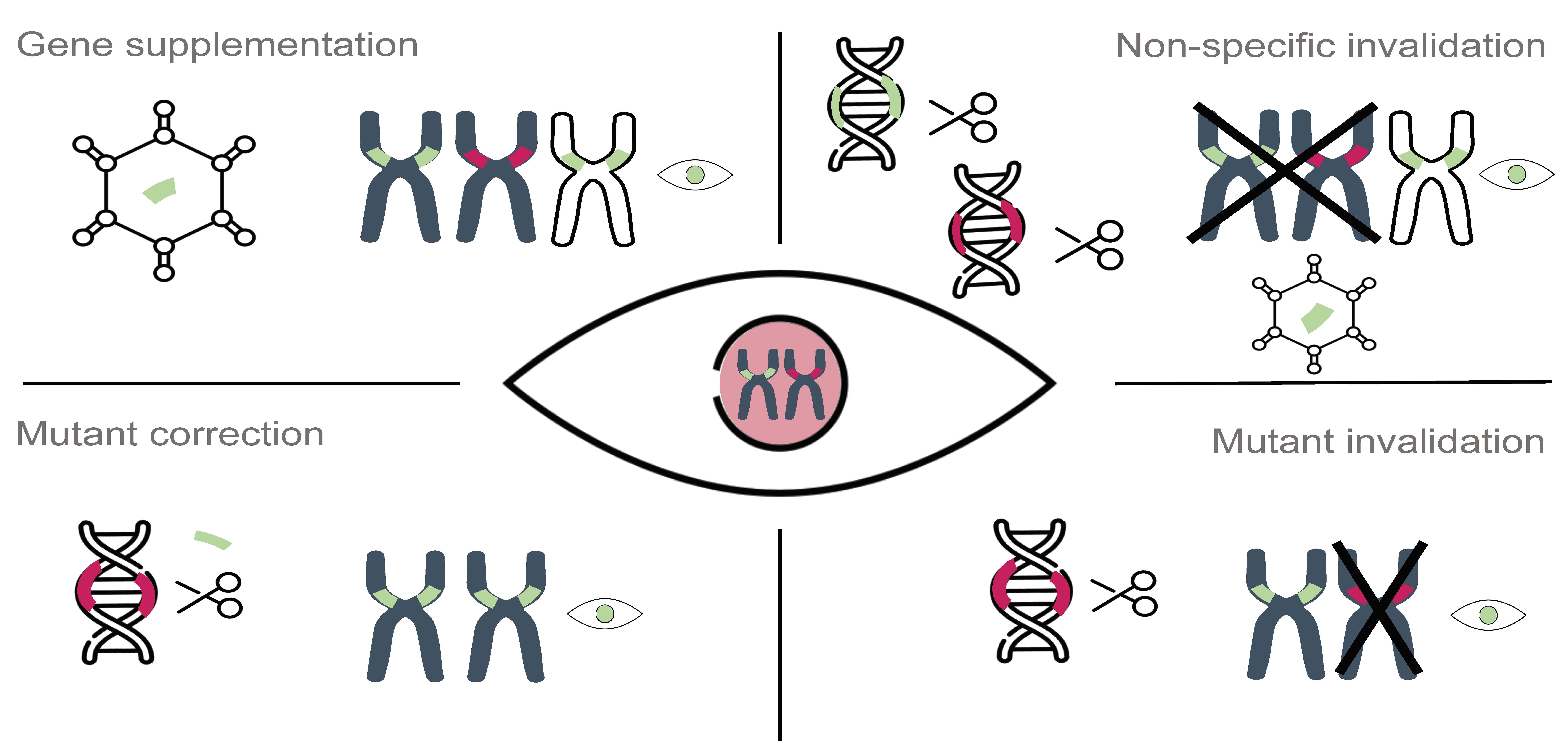
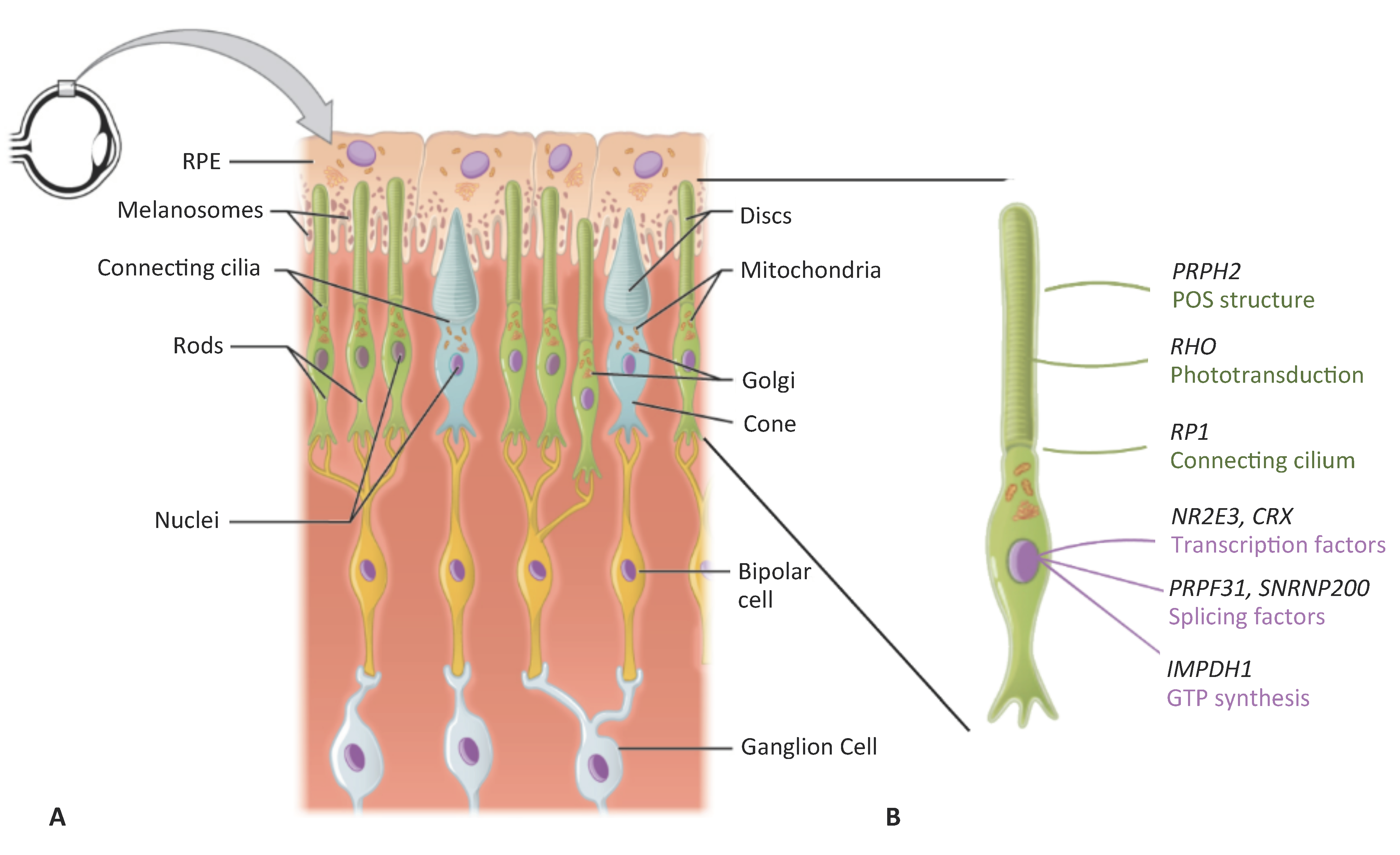
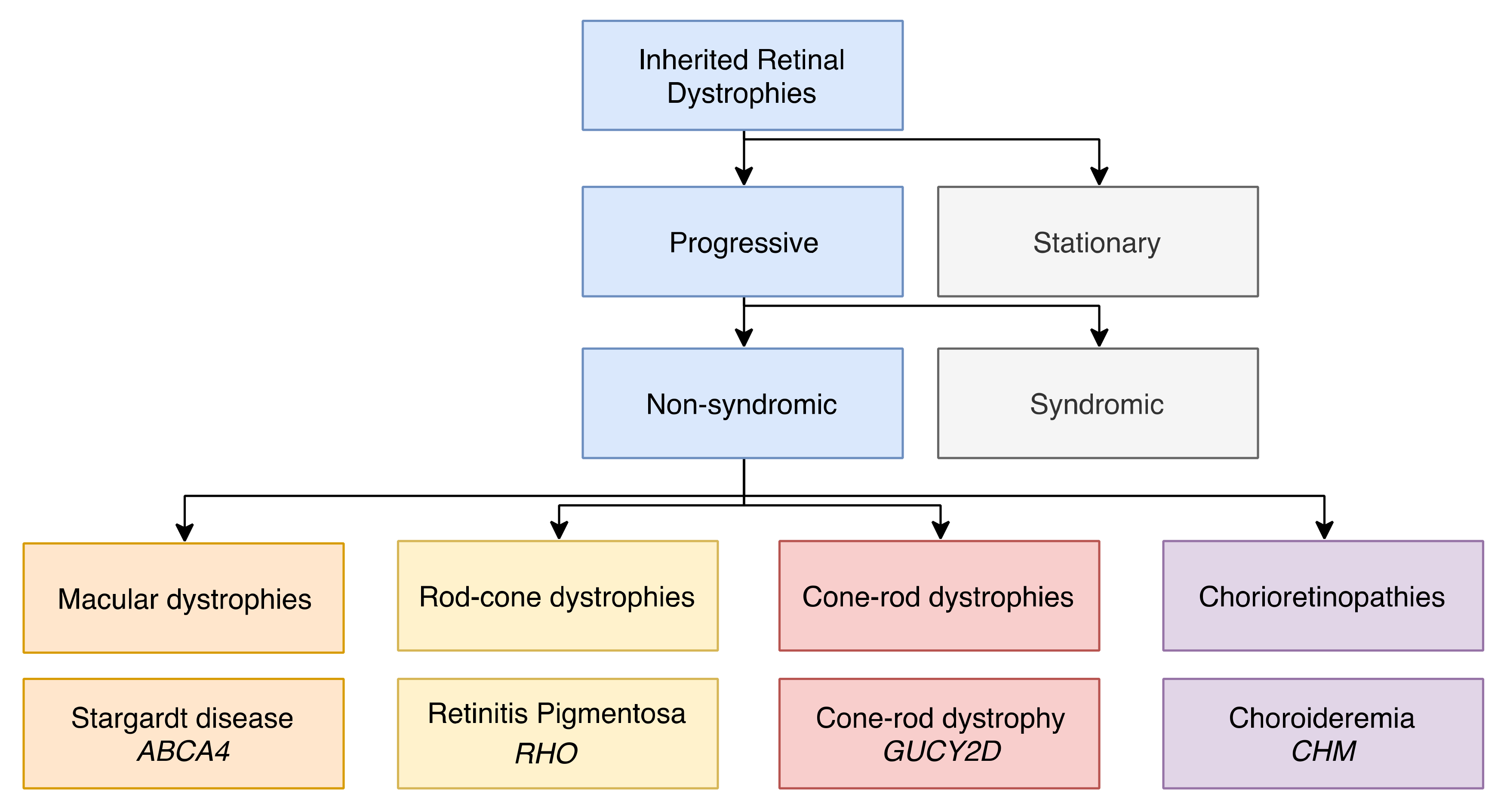
1. Inherited Retinal Dystrophies
2. Mutation Type and Compatible Therapeutic Approaches
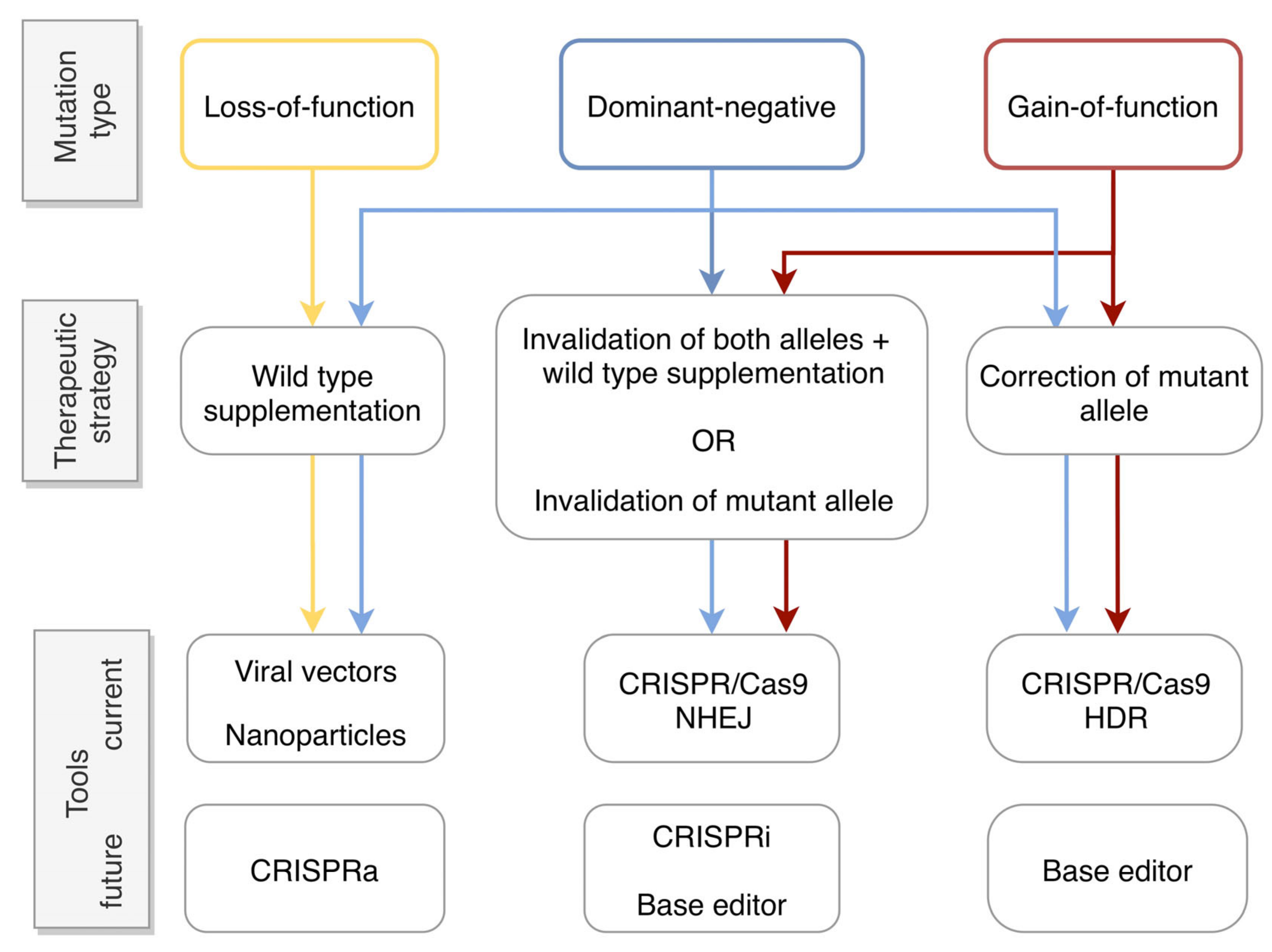
2.1. Types of Dominant Mutations
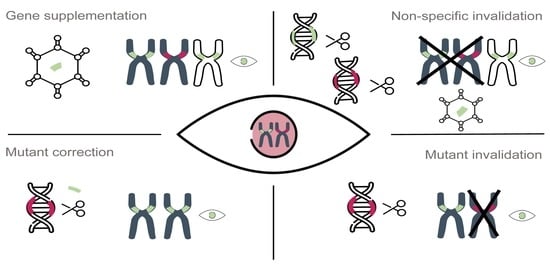
2.2. Gene Therapy Approaches
3. Genome Editing via the CRISPR/Cas System
4. Genome Editing for Autosomal Dominant Retinitis Pigmentosa
4.1. RHO
4.2. PRPF31
4.3. RP1
4.4. PRPH2
4.5. IMPDH1
4.6. NR2E3
4.7. SNRNP200
4.8. CRX
5. Discussion/Insights
Funding
Acknowledgments
Conflicts of Interest
References
- Sullivan, L.S.; Daiger, S.P. Inherited retinal degeneration: Exceptional genetic and clinical heterogeneity. Mol. Med. Today 1996, 380–386. [Google Scholar] [CrossRef]
- Sohocki, M.M.; Daiger, S.P.; Bowne, S.J.; Rodriquez, J.A.; Northrup, H.; Heckenlively, J.R.; Birch, D.G.; Mintz-Hittner, H.; Ruiz, R.S.; Lewis, R.A.; et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum. Mutat. 2001, 17, 42–51. [Google Scholar] [CrossRef]
- Cepko, C.; Vandenberghe, L.H. Retinal Gene Therapy Coming of Age. Hum. Gene Ther. 2013, 244, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Berger, W.; Kloeckener-Gruissem, B.; Neidhardt, J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010, 29, 335–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef]
- Gill, J.S.; Georgiou, M.; Kalitzeos, A.; Moore, A.T.; Michaelides, M. Progressive cone and cone-rod dystrophies: Clinical features, molecular genetics and prospects for therapy. Br. J. Ophthalmol. 2019, 103, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; De Roach, J.N.; McLaren, T.L.; Lamey, T.M. A Mini-Review: Leber Congenital Amaurosis: Identification of Disease-Causing Variants and Personalised Therapies. Adv. Exp. Med. Biol. 2018, 1074, 265–271. [Google Scholar] [PubMed]
- Farrar, G.J.; Carrigan, M.; Dockery, A.; Millington-Ward, S.; Palfi, A.; Chadderton, N.; Humphries, M.; Kiang, A.S.; Kenna, P.F.; Humphries, P. Toward an elucidation of the molecular genetics of inherited retinal degenerations. Hum. Mol. Genet. 2017, 26, R2–R11. [Google Scholar] [CrossRef] [PubMed]
- Scholl, H.P.N.; Strauss, R.W.; Singh, M.S.; Dalkara, D.; Roska, B.; Picaud, S.; Sahel, J.-A. Emerging therapies for inherited retinal degeneration. Sci. Transl. Med. 2016, 8, 368rv6. [Google Scholar] [CrossRef]
- Colella, P.; Cotugno, G.; Auricchio, A. Ocular gene therapy: Current progress and future prospects. Trends Mol. Med. 2009, 15, 23–31. [Google Scholar] [CrossRef]
- Cunha-Vaz, J.; Bernardes, R.; Lobo, C. Blood-Retinal Barrier. Eur. J. Ophthalmol. 2011, 21, 3–9. [Google Scholar] [CrossRef]
- Auricchio, A.; Smith, A.J.; Ali, R.R. The Future Looks Brighter After 25 Years of Retinal Gene Therapy. Hum. Gene Ther. 2017, 28, 982–987. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.H.; Wensel, T.G. The Nature of Dominant Mutations of Rhodopsin and Implications for Gene Therapy. Mol. Neurobiol. 2003, 28, 149–158. [Google Scholar] [CrossRef]
- Deutschbauer, A.M.; Jaramillo, D.F.; Proctor, M.; Kumm, J.; Hillenmeyer, M.E.; Davis, R.W.; Nislow, C.; Giaever, G. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics 2005, 169, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Mendes, H.F.; Van Der Spuy, J.; Chapple, J.P.; Cheetham, M.E. Mechanisms of cell death in rhodopsin retinitis pigmentosa: Implications for therapy. Trends Mol. Med. 2005, 11, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Veitia, R.A. Exploring the Molecular Etiology of Dominant-Negative Mutations. Plant Cell 2007, 19, 3843. [Google Scholar] [CrossRef] [PubMed]
- Audo, I.; Mohand-Saïd, S.; Dhaenens, C.-M.; Germain, A.; Orhan, E.; Antonio, A.; Hamel, C.; Sahel, J.-A.; Bhattacharya, S.S.; Zeitz, C. RP1 and autosomal dominant rod-cone dystrophy: Novel mutations, a review of published variants, and genotype-phenotype correlation. Hum. Mutat. 2012, 33, 73–80. [Google Scholar] [CrossRef]
- Chen, L.J.; Lai, T.Y.Y.; Tam, P.O.S.; Chiang, S.W.Y.; Zhang, X.; Lam, S.; Lai, R.Y.K.; Lam, D.S.C.; Pang, C.P. Compound heterozygosity of two novel truncation mutations in RP1 causing autosomal recessive retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2236–2242. [Google Scholar] [CrossRef]
- Pierce, E.A.; Saveliev, A.; Collin, R.W.J.; van den Born, I.; Liu, Q.E. Mutations in RP1 cause dominant retinitis pigmentosa via a dominant-negative mechanism (Abstract/Program 2147/W). In Proceedings of the 60th Annual Meeting of The American Society of Human Genetics, Washington, DC, USA, 3 November 2010. [Google Scholar]
- Mao, H.; James, T.; Schwein, A.; Shabashvili, A.E.; Hauswirth, W.W.; Gorbatyuk, M.S.; Lewin, A.S. AAV Delivery of Wild-Type Rhodopsin Preserves Retinal Function in a Mouse Model of Autosomal Dominant Retinitis Pigmentosa. Hum. Gene Ther. 2010, 22, 567–575. [Google Scholar] [CrossRef]
- Price, B.A.; Sandoval, I.M.; Chan, F.; Nichols, R.; Roman-Sanchez, R.; Wensel, T.G.; Wilson, J.H. Rhodopsin Gene Expression Determines Rod Outer Segment Size and Rod Cell Resistance to a Dominant-Negative Neurodegeneration Mutant. PLoS ONE 2012, 7, e49889. [Google Scholar] [CrossRef]
- Mendes, H.F.; Cheetham, M.E. Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 2008, 17, 3043–3054. [Google Scholar] [CrossRef] [Green Version]
- Farrar, G.J.; Millington-Ward, S.; Chadderton, N.; Humphries, P.; Kenna, P.F. Gene-based therapies for dominantly inherited retinopathies. Gene Ther. 2012, 19, 137–144. [Google Scholar] [CrossRef]
- Rossmiller, B.; Mao, H.; Lewin, A.S. Gene therapy in animal models of autosomal dominant retinitis pigmentosa. Mol. Vis. 2012, 18, 2479–2496. [Google Scholar]
- Mussolino, C.; Sanges, D.; Marrocco, E.; Bonetti, C.; Di Vicino, U.; Marigo, V.; Auricchio, A.; Meroni, G.; Surace, E.M. Zinc-finger-based transcriptional repression of rhodopsin in a model of dominant retinitis pigmentosa. EMBO Mol. Med. 2011, 3, 118–128. [Google Scholar] [CrossRef]
- Botta, S.; Marrocco, E.; de Prisco, N.; Curion, F.; Renda, M.; Sofia, M.; Lupo, M.; Carissimo, A.; Bacci, M.L.; Gesualdo, C.; et al. Rhodopsin targeted transcriptional silencing by DNA-binding. Elife 2016, 5, e12242. [Google Scholar] [CrossRef]
- Gorbatyuk, M.S.; Pang, J.J.; Thomas, J.; Hauswirth, W.W.; Lewin, A.S. Knockdown of wild-type mouse rhodopsin using an AAV vectored ribozyme as part of an RNA replacement approach. Mol. Vis. 2005, 11, 648–656. [Google Scholar] [PubMed]
- Cideciyan, A.V.; Sudharsan, R.; Dufour, V.L.; Massengill, M.T.; Iwabe, S.; Swider, M.; Lisi, B.; Sumaroka, A.; Marinho, L.F.; Appelbaum, T.; et al. Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc. Natl. Acad. Sci. USA 2018, 115, E8547–E8556. [Google Scholar] [CrossRef] [PubMed]
- Bakondi, B.; Lv, W.; Lu, B.; Jones, M.K.; Tsai, Y.; Kim, K.J.; Levy, R.; Akhtar, A.A.; Breunig, J.J.; Svendsen, C.N.; et al. In Vivo CRISPR/Cas9 Gene Editing Corrects Retinal Dystrophy in the S334ter-3 Rat Model of Autosomal Dominant Retinitis Pigmentosa. Mol. Ther. 2016, 24, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Latella, M.C.; Di Salvo, M.T.; Cocchiarella, F.; Benati, D.; Grisendi, G.; Comitato, A.; Marigo, V.; Recchia, A. In vivo Editing of the Human Mutant Rhodopsin Gene by Electroporation of Plasmid-based CRISPR/Cas9 in the Mouse Retina. Mol. Ther. Nucleic Acids 2016, 5, e389. [Google Scholar] [CrossRef]
- Burnight, E.R.; Gupta, M.; Wiley, L.A.; Anfinson, K.R.; Tran, A.; Triboulet, R.; Hoffmann, J.M.; Klaahsen, D.L.; Andorf, J.L.; Jiao, C.; et al. Using CRISPR-Cas9 to Generate Gene-Corrected Autologous iPSCs for the Treatment of Inherited Retinal Degeneration. Mol. Ther. 2017, 25, 1999–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drenser, K.A.; Timmers, A.M.; Hauswirth, W.W.; Lewin, A.S. Ribozyme-targeted destruction of RNA associated with autosomal-dominant retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 1998, 39, 681–689. [Google Scholar]
- Murray, S.F.; Jazayeri, A.; Matthes, M.T.; Yasumura, D.; Yang, H.; Peralta, R.; Watt, A.; Freier, S.; Hung, G.; Adamson, P.S.; et al. Allele-Specific Inhibition of Rhodopsin With an Antisense Oligonucleotide Slows Photoreceptor Cell Degeneration. Investig. Opthalmology Vis. Sci. 2015, 56, 6362. [Google Scholar] [CrossRef]
- Greenwald, D.L.; Cashman, S.M.; Kumar-Singh, R. Engineered zinc finger nuclease-mediated homologous recombination of the human rhodopsin gene. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6374–6380. [Google Scholar] [CrossRef]
- Foltz, L.P.; Howden, S.E.; Thomson, J.A.; Clegg, D.O. Functional Assessment of Patient-Derived Retinal Pigment Epithelial Cells Edited by CRISPR/Cas9. Int. J. Mol. Sci. 2018, 19, 4127. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef] [Green Version]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.; Embden, J.; Gaastra, W.; Schouls, L. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Klenk, H.; Clayton, R.A.; Tomb, J.; Dodson, R.J.; Gwinn, M.; Hickey, E.K.; Fleischmann, R.D.; Quackenbush, J.; Lee, N.H.; Dougherty, B.A.; et al. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon. Nature 1998, 390, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.; Diez-Villasenor, C.; Soria, E.; Juez, G. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol. Microbiol. 2000, 36, 244–246. [Google Scholar] [CrossRef]
- Mojica, F.J.M.; Ferrer, C.; Juez, G.; Rodriguez-Valera, F. Long streches of short tandem repeats are present in the largest replicons of the archeaea Haloferax mediterranei and Haloferax volcanii and could be involved in replican partitioning. Mol. Microbiol. 1995, 17, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.E.; Clayton, R.A.; Gill, S.R.; Gwinn, M.L.; Dodson, R.J.; Haft, D.H.; Hickey, E.K.; Peterson, J.D.; Nelson, W.C.; Ketchum, K.A.; et al. Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature 1999, 399, 323–329. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Gómez-González, B.; Aguilera, A. DNA double-strand break repair: How to fix a broken relationship. Cell. Mol. Life Sci. 2009, 66, 1039–1056. [Google Scholar] [CrossRef]
- Yanik, M.; Müller, B.; Song, F.; Gall, J.; Wagner, F.; Wende, W.; Lorenz, B.; Stieger, K. In vivo genome editing as a potential treatment strategy for inherited retinal dystrophies. Prog. Retin. Eye Res. 2017, 56, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 756–761. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A. Multiplex Genome Engineering Using CRISPR/VCas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Heyer, W.-D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes Wolf-Dietrich. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef]
- Sanjurjo-Soriano, C.; Kalatzis, V. Guiding Lights in Genome Editing for Inherited Retinal Disorders: Implications for Gene and Cell Therapy. Neural Plast. 2018, 2018, 5056279. [Google Scholar] [CrossRef]
- Sullivan, L.S.; Bowne, S.J.; Reeves, M.J.; Blain, D.; Goetz, K.; Difor, V.N.; Vitez, S.; Wang, X.; Tumminia, S.J.; Daiger, S.P. Prevalence of mutations in eyeGENE probands with a diagnosis of autosomal dominant retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6255–6261. [Google Scholar] [CrossRef]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin. Eye Res. 2018, 62, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Daiger, S.P.; Bowne, S.J.; Sullivan, L.S. Genes and Mutations Causing Autosomal Dominant Retinitis Pigmentosa. Cold Spring Harb. Perspect. Med. 2014, 5, a017129. [Google Scholar] [CrossRef]
- Martin-Merida, I.; Aguilera-Garcia, D.; Fernandez-San Jose, P.; Blanco-Kelly, F.; Zurita, O.; Almoguera, B.; Garcia-Sandoval, B.; Avila-Fernandez, A.; Arteche, A.; Minguez, P.; et al. Toward the mutational landscape of autosomal dominant retinitis pigmentosa: A comprehensive analysis of 258 Spanish families. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2345–2354. [Google Scholar] [CrossRef]
- Hargrave, P.A.; McDowell, J.H. Rhodopsin and phototransduction: A model system for G protein-linked receptors. FASEB J. 1992, 6, 2323–2331. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, S.E.; Xu, D.; McGrew, D.; Hamaguchi, N.; Lim, H.C.; Bowne, S.J.; Daiger, S.P.; Hedstrom, L. IMP dehydrogenase type 1 associates with polyribosomes translating rhodopsin mRNA. J. Biol. Chem. 2008, 283, 36354–36360. [Google Scholar] [CrossRef]
- Audo, I.; Manes, G.; Mohand-Saïd, S.; Friedrich, A.; Lancelot, M.-E.; Antonio, A.; Moskova-Doumanova, V.; Poch, O.; Zanlonghi, X.; Hamel, C.P.; et al. Spectrum of rhodopsin mutations in French autosomal dominant rod-cone dystrophy patients. Invest. Ophthalmol. Vis. Sci. 2010, 51, 3687–3700. [Google Scholar] [CrossRef] [PubMed]
- Illing, M.E.; Rajan, R.S.; Bence, N.F.; Kopito, R.R. A Rhodopsin Mutant Linked to Autosomal Dominant Retinitis Pigmentosa Is Prone to Aggregate and Interacts with the Ubiquitin Proteasome System. J. Biol. Chem. 2002, 277, 34150–34160. [Google Scholar] [CrossRef] [Green Version]
- Rajan, R.S.; Kopito, R.R. Suppression of Wild-type Rhodopsin Maturation by Mutants Linked to Autosomal Dominant Retinitis Pigmentosa. J. Biol. Chem. 2005, 280, 1284–1291. [Google Scholar] [CrossRef] [Green Version]
- Saliba, R.S.; Munro, P.M.G.; Luthert, P.J.; Cheetham, M.E. The cellular fate of mutant rhodopsin: Quality control, degradation and aggresome formation. J. Cell Sci. 2002, 115, 2907–2918. [Google Scholar] [PubMed]
- Lewin, A.S.; Rossmiller, B.; Mao, H. Gene augmentation for adRP mutations in RHO. Cold Spring Harb. Perspect. Med. 2014, 4, a017400. [Google Scholar] [CrossRef]
- Mao, H.; Gorbatyuk, M.S.; Rossmiller, B.; Hauswirth, W.W.; Lewin, A.S. Long-Term Rescue of Retinal Structure and Function by Rhodopsin RNA Replacement with a Single Adeno-Associated Viral Vector in P23H RHO Transgenic Mice. Hum. Gene Ther. 2012, 23, 356–366. [Google Scholar] [CrossRef]
- Millington-Ward, S.; Chadderton, N.; O’Reilly, M.; Palfi, A.; Goldmann, T.; Kilty, C.; Humphries, M.; Wolfrum, U.; Bennett, J.; Humphries, P.; et al. Suppression and replacement gene therapy for autosomal dominant disease in a murine model of dominant retinitis pigmentosa. Mol. Ther. 2011, 19, 642–649. [Google Scholar] [CrossRef]
- Mitra, R.N.; Zheng, M.; Weiss, E.R.; Han, Z. Genomic form of rhodopsin DNA nanoparticles rescued autosomal dominant Retinitis pigmentosa in the P23H knock-in mouse model. Biomaterials 2018, 157, 26–39. [Google Scholar] [CrossRef]
- Tsai, Y.-T.; Wu, W.-H.; Lee, T.-T.; Wu, W.-P.; Xu, C.L.; Park, K.S.; Cui, X.; Justus, S.; Lin, C.-S.; Jauregui, R.; et al. Clustered Regularly Interspaced Short Palindromic Repeats-Based Genome Surgery for the Treatment of Autosomal Dominant Retinitis Pigmentosa. Ophthalmology 2018, 125, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, S.G.; Luoni, M.; Castoldi, V.; Massimino, L.; Cabassi, T.; Angeloni, D.; Demontis, G.C.; Leocani, L.; Andreazzoli, M.; Broccoli, V. Cas9/sgRNA selective targeting of the P23H Rhodopsin mutant allele for treating retinitis pigmentosa by intravitreal AAV9.PHP.B-based delivery. Hum. Mol. Genet. 2018, 27, 761–779. [Google Scholar] [CrossRef]
- Li, P.; Kleinstiver, B.P.; Leon, M.Y.; Prew, M.S.; Navarro-Gomez, D.; Greenwald, S.H.; Pierce, E.A.; Joung, J.K.; Liu, Q. Allele-Specific CRISPR-Cas9 Genome Editing of the Single-Base P23H Mutation for Rhodopsin-Associated Dominant Retinitis Pigmentosa. Cris. J. 2018, 1, 55–64. [Google Scholar] [CrossRef]
- Dickinson, M.E.; Flenniken, A.M.; Ji, X.; Teboul, L.; Wong, M.D.; White, J.K.; Meehan, T.F.; Weninger, W.J.; Westerberg, H.; Adissu, H.; et al. High-throughput discovery of novel developmental phenotypes. Nature 2016, 537, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Deery, E.C.; Vithana, E.N.; Newbold, R.J.; Gallon, V.A.; Bhattacharya, S.S.; Warren, M.J.; Hunt, D.M.; Wilkie, S.E. Disease mechanism for retinitis pigmentosa (RP11) caused by mutations in the splicing factor gene PRPF31. Hum. Mol. Genet. 2002, 11, 3209–3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, D.; Cieply, B.; Carstens, R.; Ramamurthy, V.; Stoilov, P. The Musashi 1 Controls the Splicing of Photoreceptor-Specific Exons in the Vertebrate Retina. PLOS Genet. 2016, 12, e1006256. [Google Scholar] [CrossRef]
- Rio Frio, T.; Wade, N.M.; Ransijn, A.; Berson, E.L.; Beckmann, J.S.; Rivolta, C. Premature termination codons in PRPF31 cause retinitis pigmentosa via haploinsufficiency due to nonsense-mediated mRNA decay. J. Clin. Investig. 2008, 118, 1519–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Yin, X.; Wu, L.; Chen, N.; Zhang, H.; Li, G.; Ma, Z. Targeted exome capture and sequencing identifies novel PRPF31 mutations in autosomal dominant retinitis pigmentosa in Chinese families. BMJ Open 2013, 3, e004030. [Google Scholar] [CrossRef]
- Venturini, G.; Rose, A.M.; Shah, A.Z.; Bhattacharya, S.S.; Rivolta, C. CNOT3 is a modifier of PRPF31 mutations in retinitis pigmentosa with incomplete penetrance. PLoS Genet. 2012, 8, e1003040. [Google Scholar] [CrossRef]
- Rose, A.M.; Shah, A.Z.; Venturini, G.; Krishna, A.; Chakravarti, A.; Rivolta, C.; Bhattacharya, S.S. Transcriptional regulation of PRPF31 gene expression by MSR1 repeat elements causes incomplete penetrance in retinitis pigmentosa. Sci. Rep. 2016, 6, 19450. [Google Scholar] [CrossRef]
- Frio, T.R.; Civic, N.; Ransijn, A.; Beckmann, J.S.; Rivolta, C. Two trans-acting eQTLs modulate the penetrance of PRPF31 mutations. Hum. Mol. Genet. 2008, 17, 3154–3165. [Google Scholar] [CrossRef] [Green Version]
- Hafler, B.P.; Comander, J.; Weigel DiFranco, C.; Place, E.M.; Pierce, E.A. Course of Ocular Function in PRPF31 Retinitis Pigmentosa. Semin. Ophthalmol. 2016, 31, 49–52. [Google Scholar] [CrossRef]
- Buskin, A.; Zhu, L.; Chichagova, V.; Basu, B.; Mozaffari-Jovin, S.; Dolan, D.; Droop, A.; Collin, J.; Bronstein, R.; Mehrotra, S.; et al. Disrupted alternative splicing for genes implicated in splicing and ciliogenesis causes PRPF31 retinitis pigmentosa. Nat. Commun. 2018, 9, 4234. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhou, J.; Daiger, S.P.; Farber, D.B.; Heckenlively, J.R.; Smith, J.E.; Sullivan, L.S.; Zuo, J.; Milam, A.H.; Pierce, E.A. Identification and subcellular localization of the RP1 protein in human and mouse photoreceptors. Invest. Ophthalmol. Vis. Sci. 2002, 43, 22–32. [Google Scholar]
- Liu, Q.; Collin, R.W.J.; Cremers, F.P.M.; den Hollander, A.I.; van den Born, L.I.; Pierce, E.A. Expression of wild-type Rp1 protein in Rp1 knock-in mice rescues the retinal degeneration phenotype. PLoS ONE 2012, 7, e43251. [Google Scholar] [CrossRef] [PubMed]
- Stuck, M.W.; Conley, S.M.; Naash, M.I. PRPH2/RDS and ROM-1: Historical context, current views and future considerations. Prog. Retin. Eye Res. 2016, 52, 47–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manes, G.; Guillaumie, T.; Vos, W.L.; Devos, A.; Audo, I.; Zeitz, C.; Marquette, V.; Zanlonghi, X.; Defoort-Dhellemmes, S.; Puech, B.; et al. High Prevalence of PRPH2 in Autosomal Dominant Retinitis Pigmentosa in France and Characterization of Biochemical and Clinical Features. Am. J. Ophthalmol. 2015, 159, 302–314. [Google Scholar] [CrossRef]
- Conley, S.M.; Naash, M.I. Gene therapy for PRPH2-Associated ocular disease: Challenges and prospects. Cold Spring Harb. Perspect. Med. 2014, 4, a017376. [Google Scholar] [CrossRef]
- Boon, C.J.F.; den Hollander, A.I.; Hoyng, C.B.; Cremers, F.P.M.; Klevering, B.J.; Keunen, J.E.E. The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog. Retin. Eye Res. 2008, 27, 213–235. [Google Scholar] [CrossRef] [PubMed]
- Kajiwara, K.; Berson, E.; Dryja, T. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science 1994, 264, 1604–1608. [Google Scholar] [CrossRef]
- Ding, X.-Q.; Naash, M.I.; Quimbao, A.; Fliesler, S.J.; Stricker, H.M. The Cys 214 →Ser mutation in peripherin/rds causes a loss-of-function phenotype in transgenic mice. Biochem. J. 2005, 388, 605–613. [Google Scholar]
- Nour, M.; Fliesler, S.J.; Naash, M.I. Genetic Supplementation of RDS Alleviates a Loss-of-function Phenotype in C214S Model of Retinitis Pigmentosa. Adv. Exp. Med. Biol. 2008, 129–138. [Google Scholar]
- Kedzierski, W.; Nusinowitz, S.; Birch, D.; Clarke, G.; McInnes, R.R.; Bok, D.; Travis, G.H. Deficiency of rds/peripherin causes photoreceptor death in mouse models of digenic and dominant retinitis pigmentosa. Proc. Natl. Acad. Sci. 2001, 98, 7718–7723. [Google Scholar] [CrossRef] [Green Version]
- Ali, R.R.; Sarra, G.M.; Stephens, C.; De Alwis, M.; Bainbridge, J.W.B.; Munro, P.M.; Fauser, S.; Reichell, M.B.; Kinnon, C.; Hunt, D.M.; et al. Restoration of photoreceptor ultrastructure and function in retinal degeneration slow mice by gene therapy. Nat. Genet. 2000, 25, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Conley, S.M.; Nash, Z.; Fliesler, S.J.; Cooper, M.J.; Naash, M.I. Gene delivery to mitotic and postmitotic photoreceptors via compacted DNA nanoparticles results in improved phenotype in a mouse model of retinitis pigmentosa. FASEB J. 2010, 24, 1178–1191. [Google Scholar] [CrossRef]
- Collart, F.R.; Huberman, E. Cloning and sequence analysis of the human and Chinese hamster inosine-5′-monophosphate dehydrogenase cDNAs. J. Biol. Chem. 1988, 263, 15769–15772. [Google Scholar] [PubMed]
- Spellicy, C.J.; Daiger, S.P.; Sullivan, L.S.; Zhu, J.; Liu, Q.; Pierce, E.A.; Bowne, S.J. Characterization of retinal inosine monophosphate dehydrogenase 1 in several mammalian species. Mol. Vis. 2007, 13, 1866–1872. [Google Scholar]
- Bowne, S.J.; Liu, Q.; Sullivan, L.S.; Zhu, J.; Spellicy, C.J.; Rickman, C.B.; Pierce, E.A.; Daiger, S.P. Why do mutations in the ubiquitously expressed housekeeping gene IMPDH1 cause retina-specific photoreceptor degeneration? Invest. Ophthalmol. Vis. Sci. 2006, 47, 3754–3765. [Google Scholar] [CrossRef] [PubMed]
- Bowne, S.J.; Sullivan, L.S.; Mortimer, S.E.; Hedstrom, L.; Zhu, J.; Spellicy, C.J.; Gire, A.I.; Hughbanks-Wheaton, D.; Birch, D.G.; Lewis, R.A.; et al. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 2006, 47, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Kozhevnikova, E.N.; van der Knaap, J.A.; Pindyurin, A.V.; Ozgur, Z.; van Ijcken, W.F.J.; Moshkin, Y.M.; Verrijzer, C.P. Metabolic Enzyme IMPDH Is Also a Transcription Factor Regulated by Cellular State. Mol. Cell 2012, 47, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Mortimer, S.E.; Hedstrom, L. Autosomal dominant retinitis pigmentosa mutations in inosine 5′-monophosphate dehydrogenase type I disrupt nucleic acid binding. Biochem. J. 2005, 390, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Buey, R.M.; Ledesma-Amaro, R.; Velázquez-Campoy, A.; Balsera, M.; Chagoyen, M.; de Pereda, J.M.; Revuelta, J.L. Guanine nucleotide binding to the Bateman domain mediates the allosteric inhibition of eukaryotic IMP dehydrogenases. Nat. Commun. 2015, 6, 8923. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Takezawa, S.; Hara, K.; Yu, R.T.; Umesono, Y.; Agata, K.; Taniwaki, M.; Yasuda, K.; Umesono, K. Identification of a photoreceptor cell-specific nuclear receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 4814–4819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milam, A.H.; Rose, L.; Cideciyan, A.V.; Barakat, M.R.; Tang, W.-X.; Gupta, N.; Aleman, T.S.; Wright, A.F.; Stone, E.M.; Sheffield, V.C.; et al. The nuclear receptor NR2E3 plays a role in human retinal photoreceptor differentiation and degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 473–478. [Google Scholar] [CrossRef]
- Cheng, H.; Khanna, H.; Oh, E.C.T.; Hicks, D.; Mitton, K.P.; Swaroop, A. Photoreceptor-specific nuclear receptor NR2E3 functions as a transcriptional activator in rod photoreceptors. Hum. Mol. Genet. 2004, 13, 1563–1575. [Google Scholar] [CrossRef]
- Peng, G.H.; Ahmad, O.; Ahmad, F.; Liu, J.; Chen, S. The photoreceptor-specific nuclear receptor Nr2e3 interacts with Crx and exerts opposing effects on the transcription of rod versus cone genes. Hum. Mol. Genet. 2005, 14, 747–764. [Google Scholar] [CrossRef] [Green Version]
- Haider, N.B.; Jacobson, S.G.; Cideciyan, A.V.; Swiderski, R.; Streb, L.M.; Searby, C.; Beck, G.; Hockey, R.; Hanna, D.B.; Gorman, S.; et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat. Genet. 2000, 24, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, F.; Leroy, B.P.; Beysen, D.; Hellemans, J.; De Bosscher, K.; Haegeman, G.; Robberecht, K.; Wuyts, W.; Coucke, P.J.; De Baere, E. REPORT Recurrent Mutation in the First Zinc Finger of the Orphan Nuclear Receptor NR2E3 Causes Autosomal Dominant Retinitis Pigmentosa. Am. J. Hum. Genet. Am. J. Hum. Genet 2007, 8181, 147–157. [Google Scholar] [CrossRef]
- Blanco-Kelly, F.; García Hoyos, M.; Lopez Martinez, M.A.; Lopez-Molina, M.I.; Riveiro-Alvarez, R.; Fernandez-San Jose, P.; Avila-Fernandez, A.; Corton, M.; Millan, J.M.; García Sandoval, B.; et al. Dominant Retinitis Pigmentosa, p.Gly56Arg Mutation in NR2E3: Phenotype in a Large Cohort of 24 Cases. PLoS ONE 2016, 11, e0149473. [Google Scholar] [CrossRef] [PubMed]
- Gire, A.I.; Sullivan, L.S.; Bowne, S.J.; Birch, D.G.; Hughbanks-Wheaton, D.; Heckenlively, J.R.; Daiger, S.P. The Gly56Arg mutation in NR2E3 accounts for 1-2% of autosomal dominant retinitis pigmentosa. Mol. Vis. 2007, 13, 1970–1975. [Google Scholar] [PubMed]
- Escher, P.; Gouras, P.; Roduit, R.; Tiab, L.; Bolay, S.; Delarive, T.; Chen, S.; Tsai, C.C.; Hayashi, M.; Zernant, J.; et al. Mutations in NR2E3 can cause dominant or recessive retinal degenerations in the same family. Hum. Mutat. 2009, 30, 342–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roduit, R.; Escher, P.; Schorderet, D.F. Mutations in the DNA-binding domain of NR2E3 affect in vivo dimerization and interaction with CRX. PLoS ONE 2009, 4, e7379. [Google Scholar] [CrossRef] [PubMed]
- Lem, J.; Krasnoperova, N.V.; Calvert, P.D.; Kosaras, B.; Cameron, D.A.; Nicolò, M.; Makino, C.L.; Sidman, R.L. Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc. Natl. Acad. Sci. USA 1999, 96, 736–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cvačková, Z.; Matějů, D.; Staněk, D. Retinitis Pigmentosa Mutations of SNRNP200 Enhance Cryptic Splice-Site Recognition. Hum. Mutat. 2014, 35, 308–317. [Google Scholar] [CrossRef]
- Ezquerra-Inchausti, M.; Barandika, O.; Anasagasti, A.; Irigoyen, C.; López de Munain, A.; Ruiz-Ederra, J. High prevalence of mutations affecting the splicing process in a Spanish cohort with autosomal dominant retinitis pigmentosa. Sci. Rep. 2017, 7, 39652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cauwenbergh, C.; Coppieters, F.; Roels, D.; De Jaegere, S.; Flipts, H.; De Zaeytijd, J.; Walraedt, S.; Claes, C.; Fransen, E.; Van Camp, G.; et al. Mutations in splicing factor genes are a major cause of autosomal dominant retinitis pigmentosa in belgian families. PLoS ONE 2017, 12, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pena, V.; Jovin, S.M.; Fabrizio, P.; Orlowski, J.; Bujnicki, J.M.; Lührmann, R.; Wahl, M.C. Common Design Principles in the Spliceosomal RNA Helicase Brr2 and in the Hel308 DNA Helicase. Mol. Cell 2009, 35, 454–466. [Google Scholar] [CrossRef] [Green Version]
- Hahn, D.; Kudla, G.; Tollervey, D.; Beggs, J.D. Brr2p-mediated conformational rearrangements in the spliceosome during activation and substrate repositioning. Genes Dev. 2012, 26, 2408. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, T.; Maeder, C.; Bud, L.-O.; Shanks, J.; Nix, J.; Guthrie, C.; Pleiss, J.A.; Zhao, R. Structural evidence for consecutive Hel308-like modules in the spliceosomal ATPase Brr2. Nat. Struct. Mol. Biol. 2009, 16, 731–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Bellur, D.L.; Lu, S.; Zhao, F.; Grassi, M.A.; Bowne, S.J.; Sullivan, L.S.; Daiger, S.P.; Chen, L.J.; Pang, C.P.; et al. Autosomal-Dominant Retinitis Pigmentosa Caused by a Mutation in SNRNP200, a Gene Required for Unwinding of U4/U6 snRNAs. Am. J. Hum. Genet. 2009, 85, 617–627. [Google Scholar] [CrossRef] [Green Version]
- Santos, K.F.; Jovin, S.M.; Weber, G.; Pena, V.; Lührmann, R.; Wahl, M.C. Structural basis for functional cooperation between tandem helicase cassettes in Brr2-mediated remodeling of the spliceosome. Proc. Natl. Acad. Sci. USA 2012, 109, 17418. [Google Scholar] [CrossRef]
- Bujakowska, K.M.; Fernandez-Godino, R.; Place, E.; Consugar, M.; Navarro-Gomez, D.; White, J.; Bedoukian, E.C.; Zhu, X.; Xie, H.M.; Gai, X.; et al. Copy-number variation is an important contributor to the genetic causality of inherited retinal degenerations. Genet. Med. 2017, 19, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, T.; Morrow, E.M.; Li, T.; Davis, F.C.; Cepko, C.L. Retinopathy and attenuated circadian entrainment in Crx-deficient mice. Nat. Genet. 1999, 23, 466–470. [Google Scholar] [CrossRef]
- Silva, E.; Yang, J.M.; Li, Y.; Dharmaraj, S.; Sundin, O.H.; Maumenee, I.H. A CRX null mutation is associated with both Leber congenital amaurosis and a normal ocular phenotype. Invest. Ophthalmol. Vis. Sci. 2000, 41, 2076–2079. [Google Scholar]
- Tran, N.M.; Chen, S. Mechanisms of blindness: Animal models provide insight into distinct CRX-associated retinopathies. Dev. Dyn. 2014, 243, 1153–1166. [Google Scholar] [CrossRef]
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A.; Manuscript, A.; Houk, A.R.; Jilkine, A.; et al. Repurposing CRISPR as an RNA-Guided Platform for Sequence- Specific Control of Gene Expression. Curr. Opin. Cell Biol. 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, A.; Tuttle, M.; Pruitt, B.W.; Ewen-Campen, B.; Chari, R.; Ter-Ovanesyan, D.; Haque, S.J.; Cecchi, R.J.; Kowal, E.J.K.; Buchthal, J.; et al. Comparison of Cas9 activators in multiple species. Nat. Methods 2016. [Google Scholar] [CrossRef]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing CRISPR–Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016, 17, 5–15. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of T to G C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef]
- Nelson, C.E.; Wu, Y.; Gemberling, M.P.; Oliver, M.L.; Waller, M.A.; Bohning, J.D.; Robinson-Hamm, J.N.; Bulaklak, K.; Castellanos Rivera, R.M.; Collier, J.H.; et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019, 25, 427–432. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Kulcsár, P.I.; Tálas, A.; Huszár, K.; Ligeti, Z.; Tóth, E.; Weinhardt, N.; Fodor, E.; Welker, E. Crossing enhanced and high fidelity SpCas9 nucleases to optimize specificity and cleavage. Genome Biol. 2017, 18, 190. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Fagerlund, R.D.; Staals, R.H.J.; Fineran, P.C. The Cpf1 CRISPR-Cas protein expands genome-editing tools. Genome Biol. 2015, 16, 1–3. [Google Scholar] [CrossRef]
- Friedland, A.E.; Baral, R.; Singhal, P.; Loveluck, K.; Shen, S.; Sanchez, M.; Marco, E.; Gotta, G.M.; Maeder, M.L.; Kennedy, E.M.; et al. Characterization of Staphylococcus aureus Cas9: A smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol. 2015, 16, 257. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65 -mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
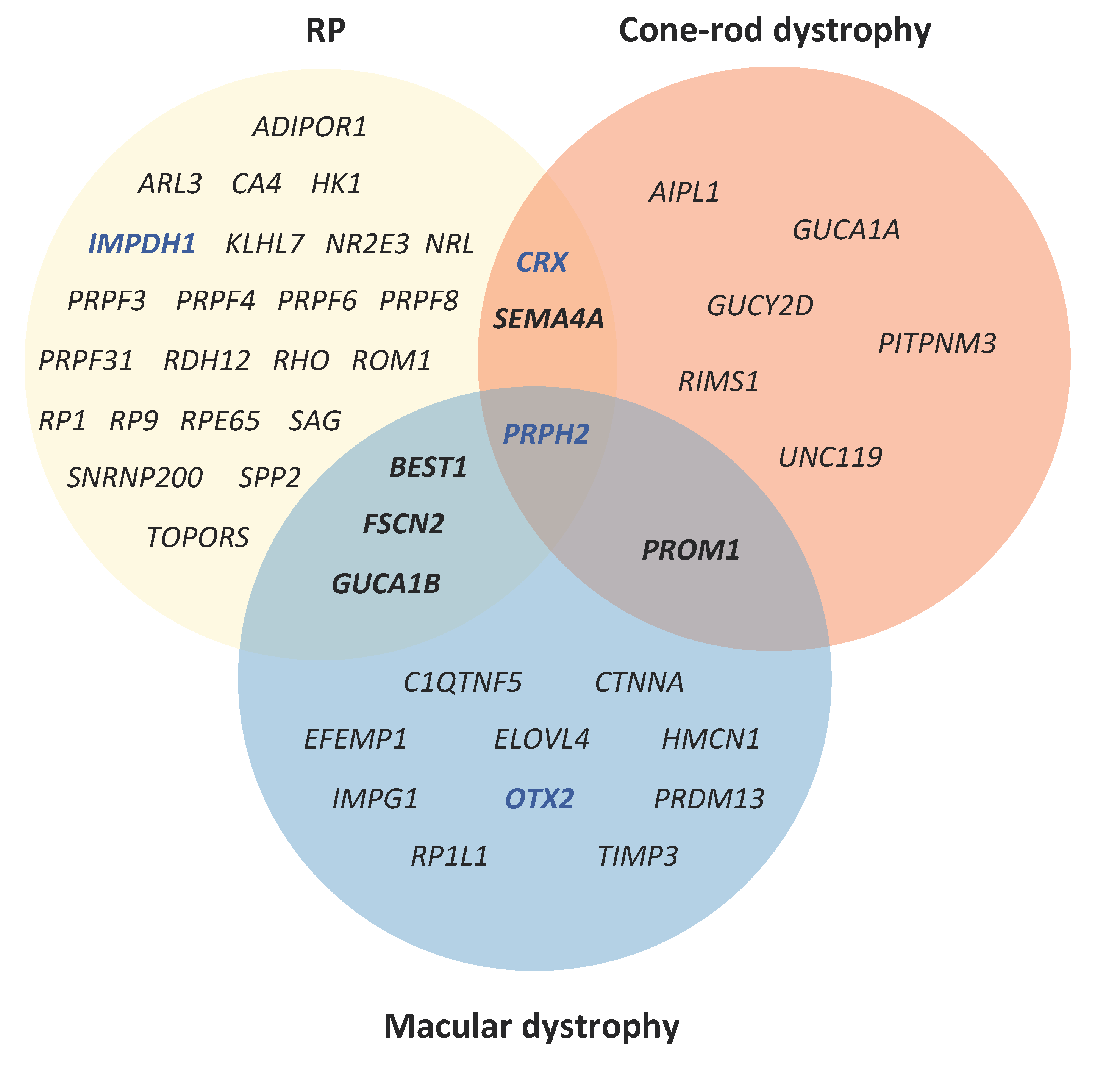
| Autosomal Dominant IRDs | ||||||||||
| Chorioretinal atrophy | PRDM13 | RGR | TEAD1 | |||||||
| Cone(-rod) dystrophy | AIPL1 | CRX | GUCA1A | GUCY2D | PITPNM3 | PROM1 | PRPH2 | RIMS1 | SEMA4A | UNC119 |
| LCA | CRX | IMPDH1 | OTX2 | |||||||
| Macular dystrophy | BEST1 | C1QTNF5 | CTNNA1 | EFEMP1 | ELOVL4 | FSCN2 | GUCA1B | HMCN1 | IMPG1 | OTX2 |
| PRDM13 | PROM1 | PRPH2 | RP1L1 | TIMP3 | ||||||
| RP | ADIPOR1 | ARL3 | BEST1 | CA4 | CRX | FSCN2 | GUCA1B | HK1 | IMPDH1 | KLHL7 |
| NR2E3 | NRL | PRPF3 | PRPF4 | PRPF6 | PRPF8 | PRPF31 | PRPH2 | RDH12 | RHO | |
| ROM1 | RP1 | RP9 | RPE65 | SAG | SEMA4A | SNRNP200 | SPP2 | TOPORS | ||
| Autosomal Recessive IRDs | ||||||||||
| Cone(-rod) dystrophy | ABCA4 | ADAM9 | ATF6 | C21orf2 | C8orf37 | CACNA2D4 | CDHR1 | CERKL | CNGA3 | CNGB3 |
| CNNM4 | GNAT2 | IFT81 | KCNV2 | PDE6C | PDE6H | POC1B | RAB28 | RAX2 | RDH5 | |
| RPGRIP1 | TTLL5 | |||||||||
| LCA | AIPL1 | CABP4 | CCT2 | CEP290 | CLUAP1 | CRB1 | CRX | DTHD1 | GDF6 | GUCY2D |
| IFT140 | IQCB1 | KCNJ13 | LCA5 | LRAT | NMNAT1 | PRPH2 | RD3 | RDH12 | RPE65 | |
| RPGRIP1 | SPATA7 | TULP1 | ||||||||
| Macular dystrophy | ABCA4 | CFH | DRAM2 | IMPG1 | MFSD8 | |||||
| RP | ABCA4 | AGBL5 | ARHGEF18 | ARL6 | ARL2BP | BBS1 | BBS2 | BEST1 | C2orf71 | C8orf37 |
| CERKL | CLRN1 | CNGA1 | CNGB1 | CRB1 | CYP4V2 | DHDDS | DHX38 | EMC1 | EYS | |
| FAM161A | GPR125 | HGSNAT | IDH3B | IFT140 | IFT172 | IMPG2 | KIAA1549 | KIZ | LRAT | |
| MAK | MERTK | MVK | NEK2 | NEUROD1 | NR2E3 | NRL | PDE6A | PDE6B | PDE6G | |
| POMGNT1 | PRCD | PROM1 | RBP3 | REEP6 | RGR | RHO | RLBP1 | RP1 | RP1L1 | |
| RPE65 | SAG | SAMD11 | SLC7A14 | SPATA7 | TRNT1 | TTC8 | TULP1 | USH2A | ZNF408 | |
| ZNF513 | ||||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diakatou, M.; Manes, G.; Bocquet, B.; Meunier, I.; Kalatzis, V. Genome Editing as a Treatment for the Most Prevalent Causative Genes of Autosomal Dominant Retinitis Pigmentosa. Int. J. Mol. Sci. 2019, 20, 2542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102542
Diakatou M, Manes G, Bocquet B, Meunier I, Kalatzis V. Genome Editing as a Treatment for the Most Prevalent Causative Genes of Autosomal Dominant Retinitis Pigmentosa. International Journal of Molecular Sciences. 2019; 20(10):2542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102542
Chicago/Turabian StyleDiakatou, Michalitsa, Gaël Manes, Beatrice Bocquet, Isabelle Meunier, and Vasiliki Kalatzis. 2019. "Genome Editing as a Treatment for the Most Prevalent Causative Genes of Autosomal Dominant Retinitis Pigmentosa" International Journal of Molecular Sciences 20, no. 10: 2542. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102542