Selection of Biophysical Methods for Characterisation of Membrane Proteins

 ,
,  , and
, and 
Abstract
:1. Introduction
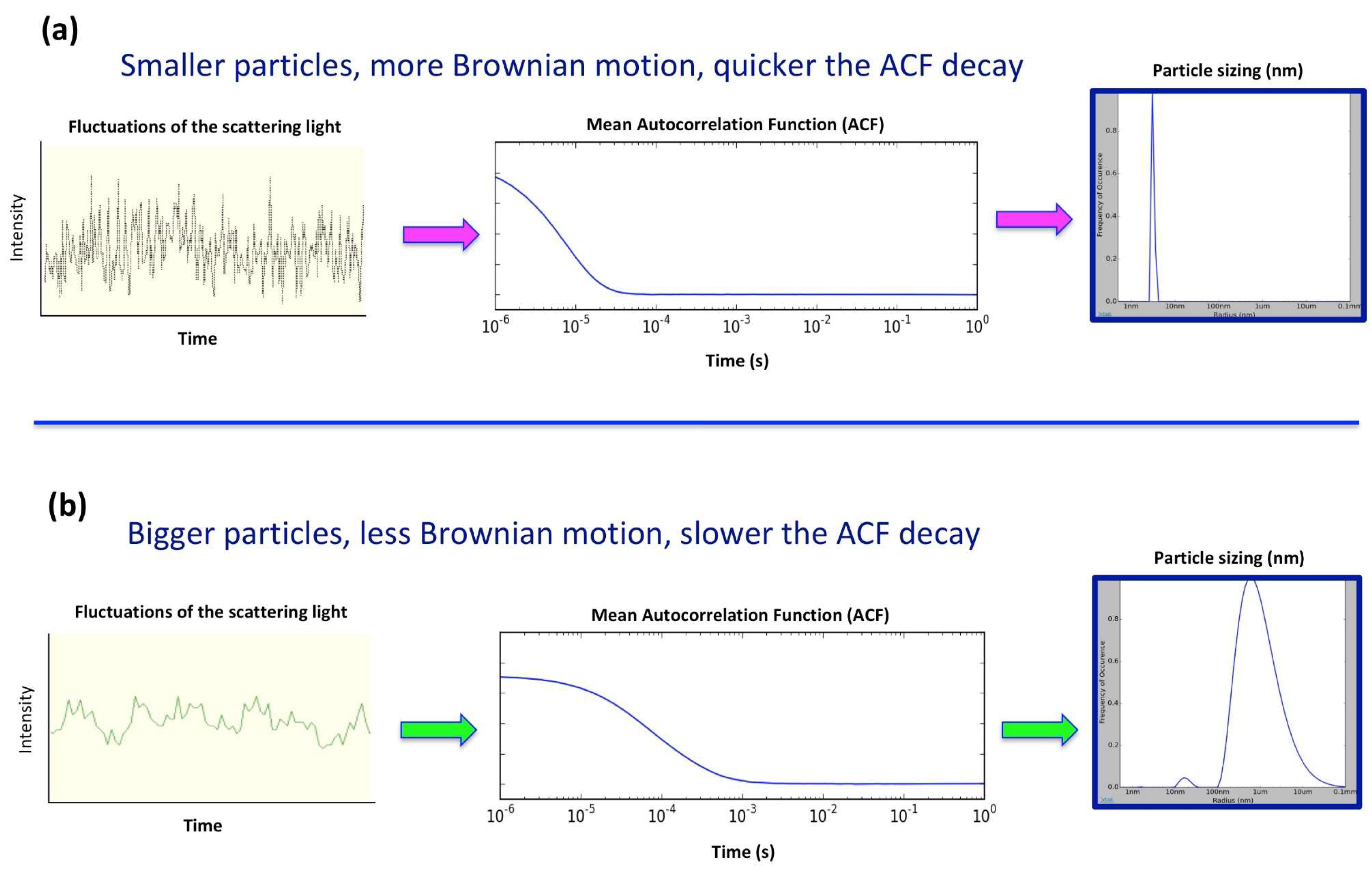
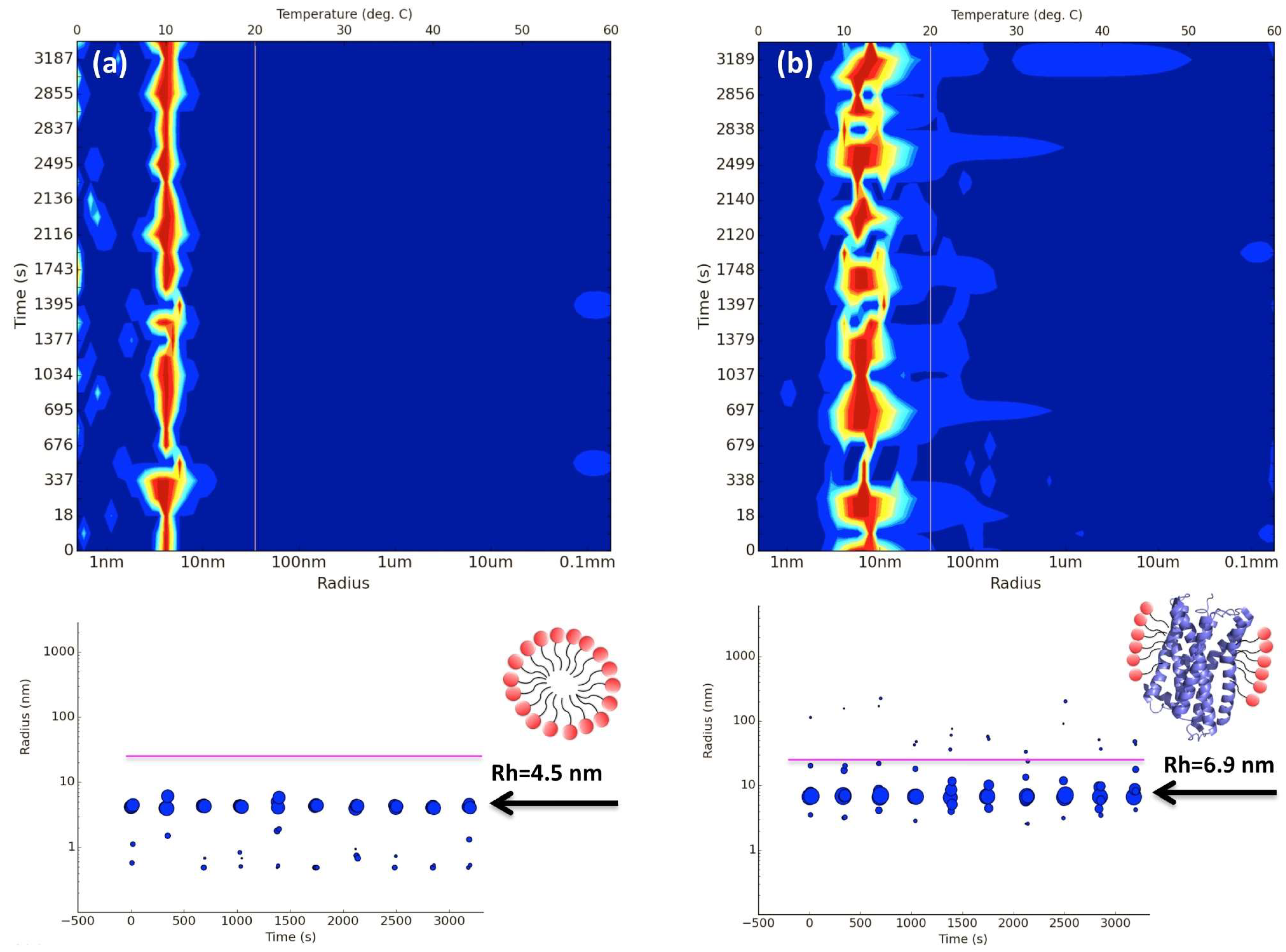
2. In Situ Dynamic Light Scattering (DLS)
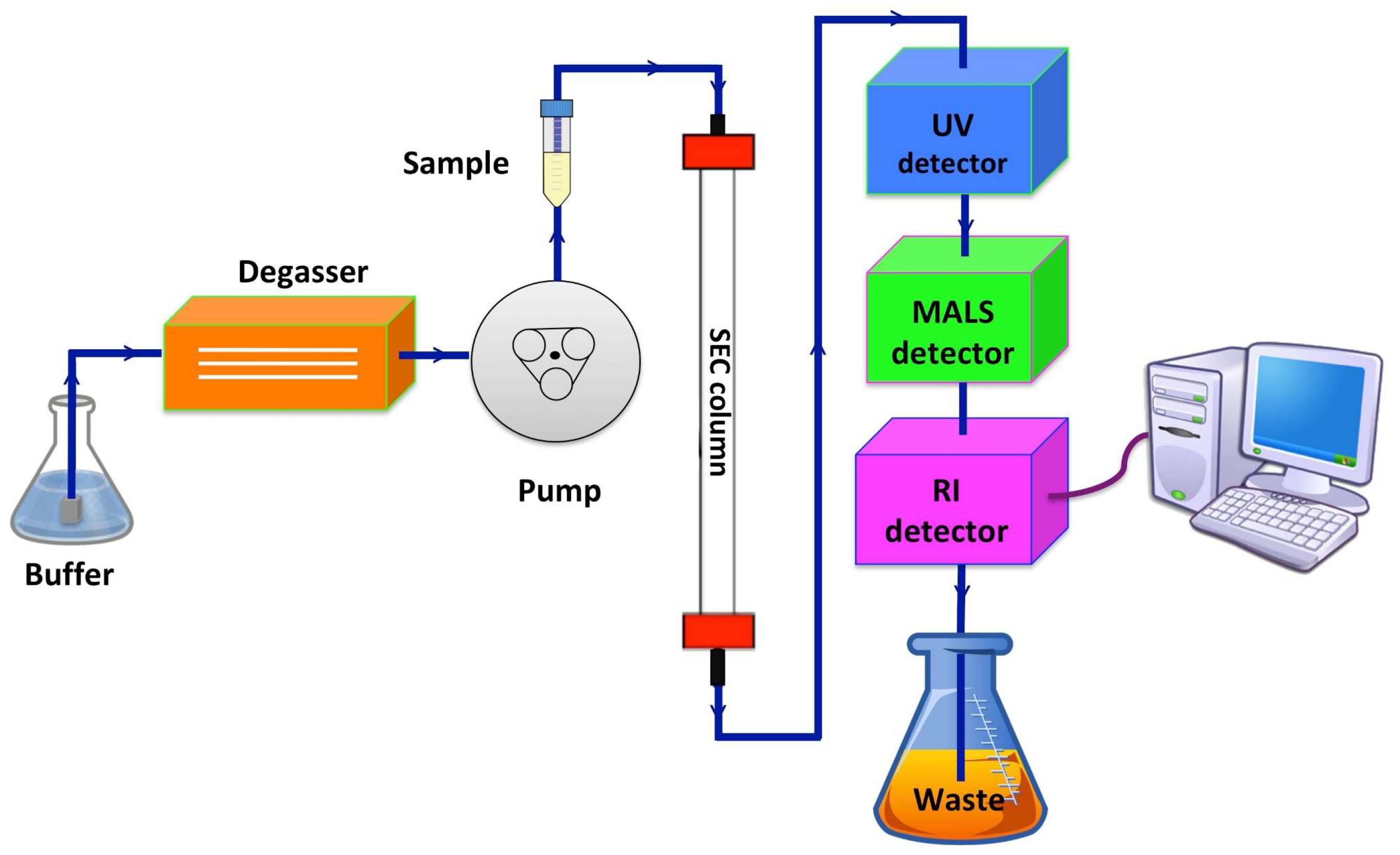
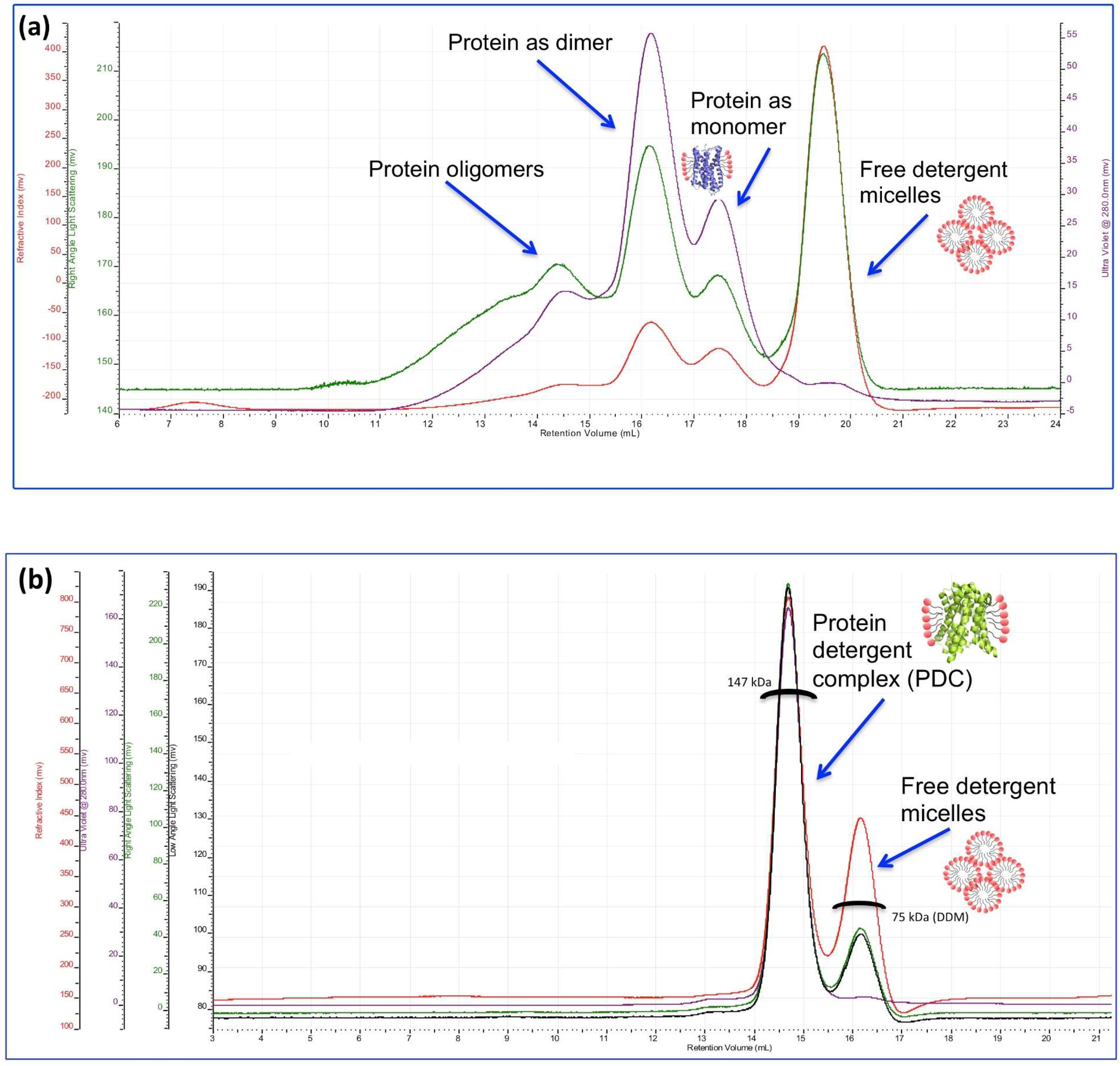
3. Characterisation of Membrane Proteins by Size-Exclusion Chromatography Multi-Angle Light Scattering (SEC-MALS)
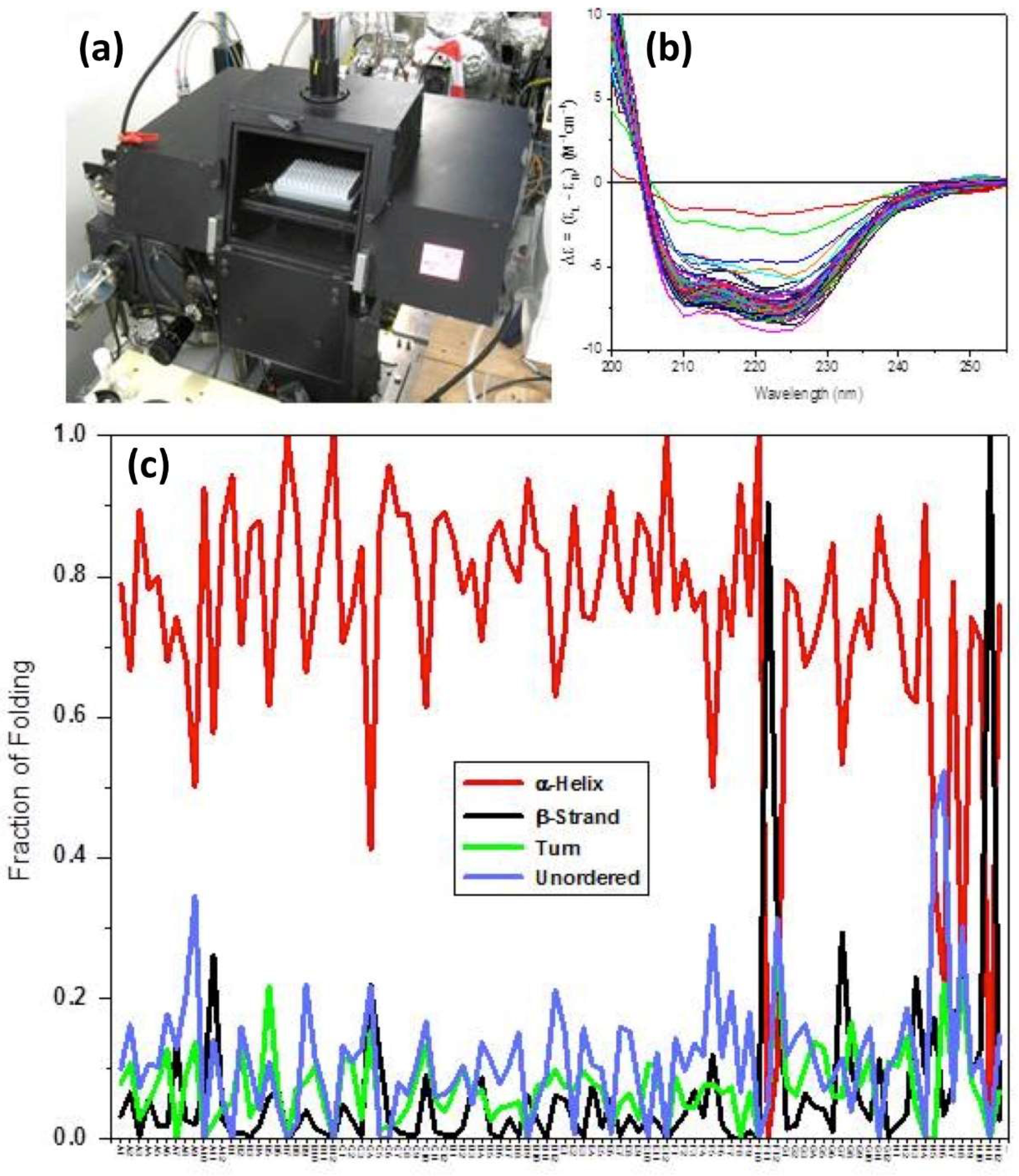
4. Circular Dichroism (CD)/Synchrotron Radiation Circular Dichroism (SRCD) of Membrane Proteins
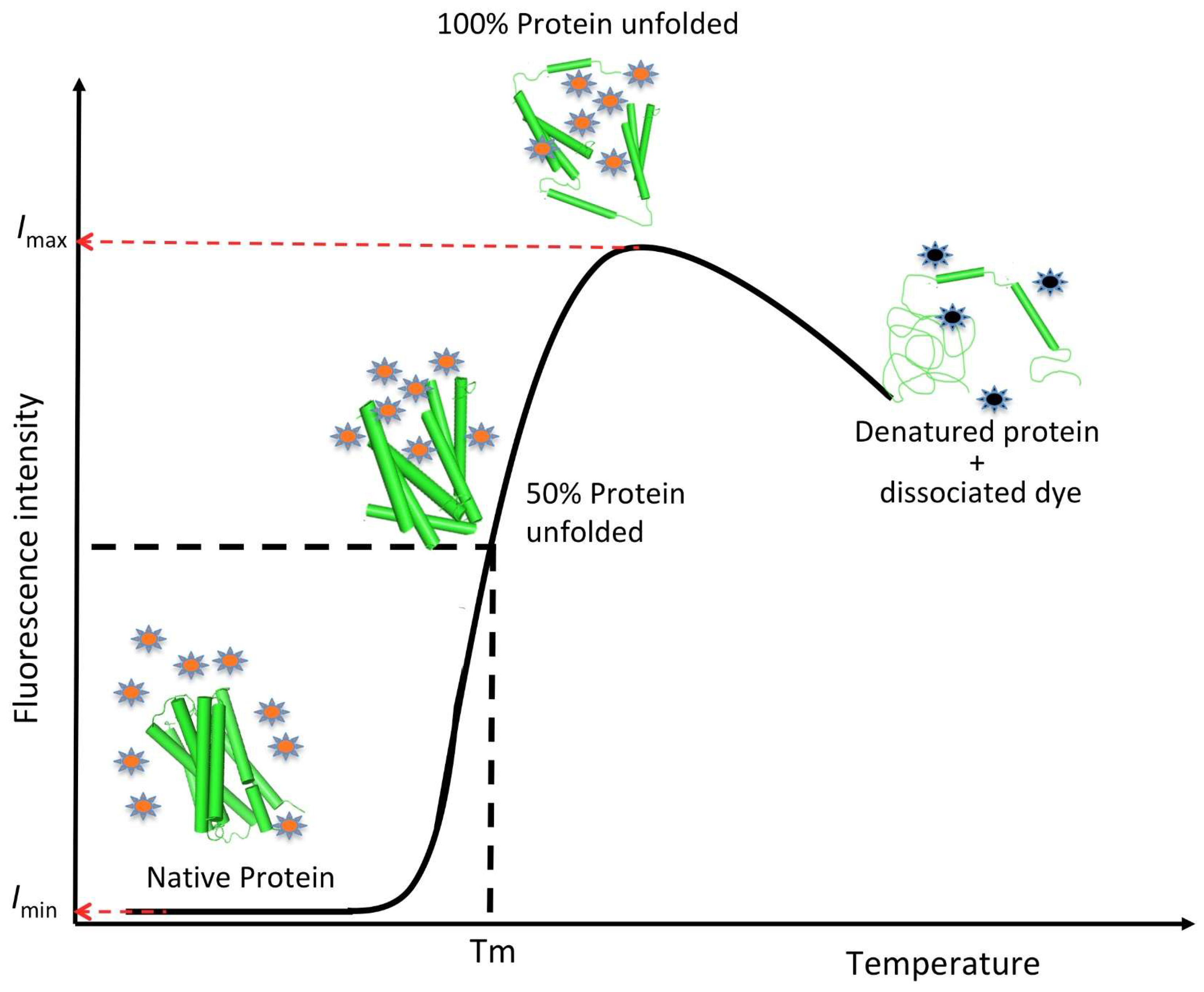
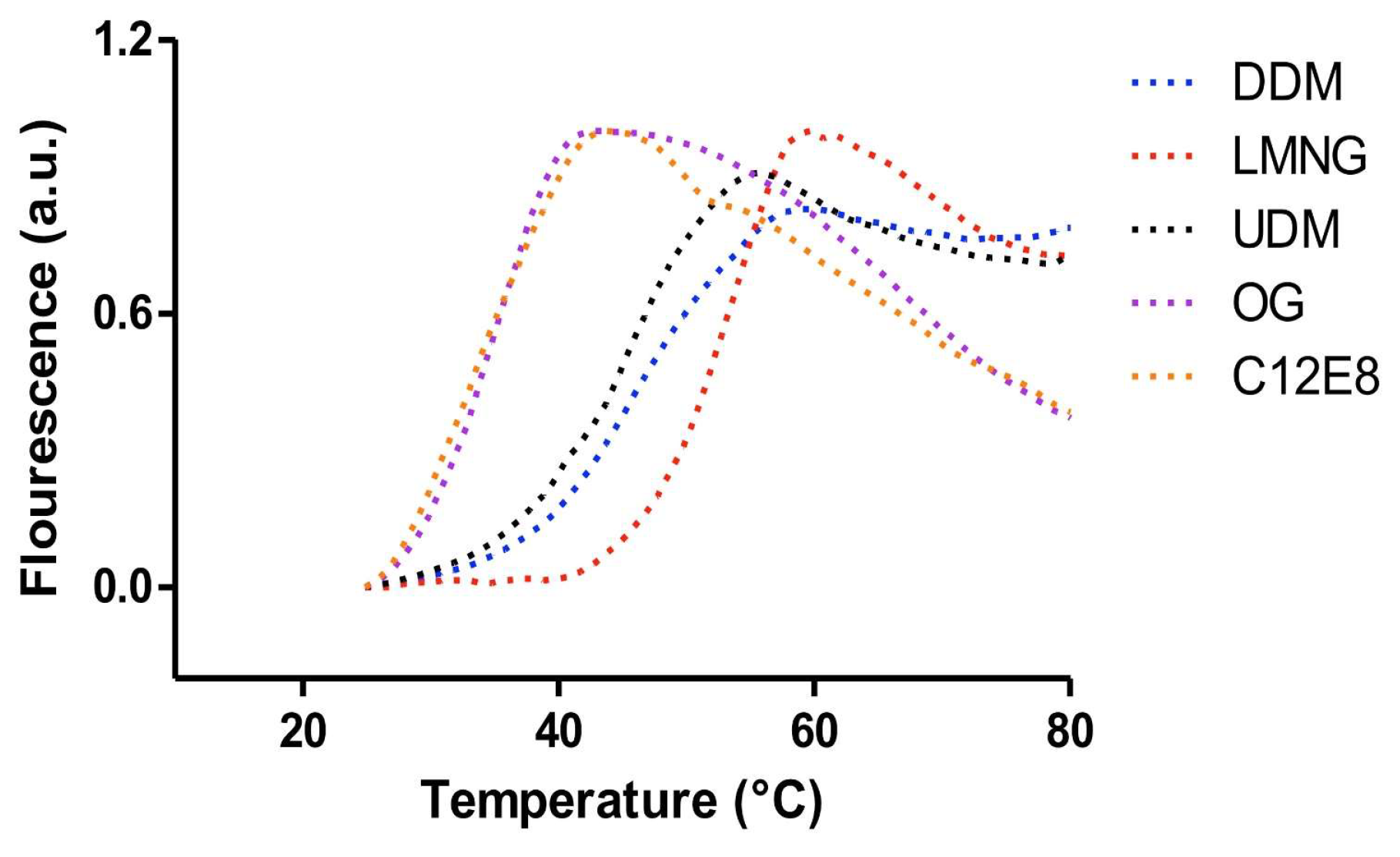
5. Fluorescence Dye-Based Differential Scanning Fluorimetry (DSF) Assay
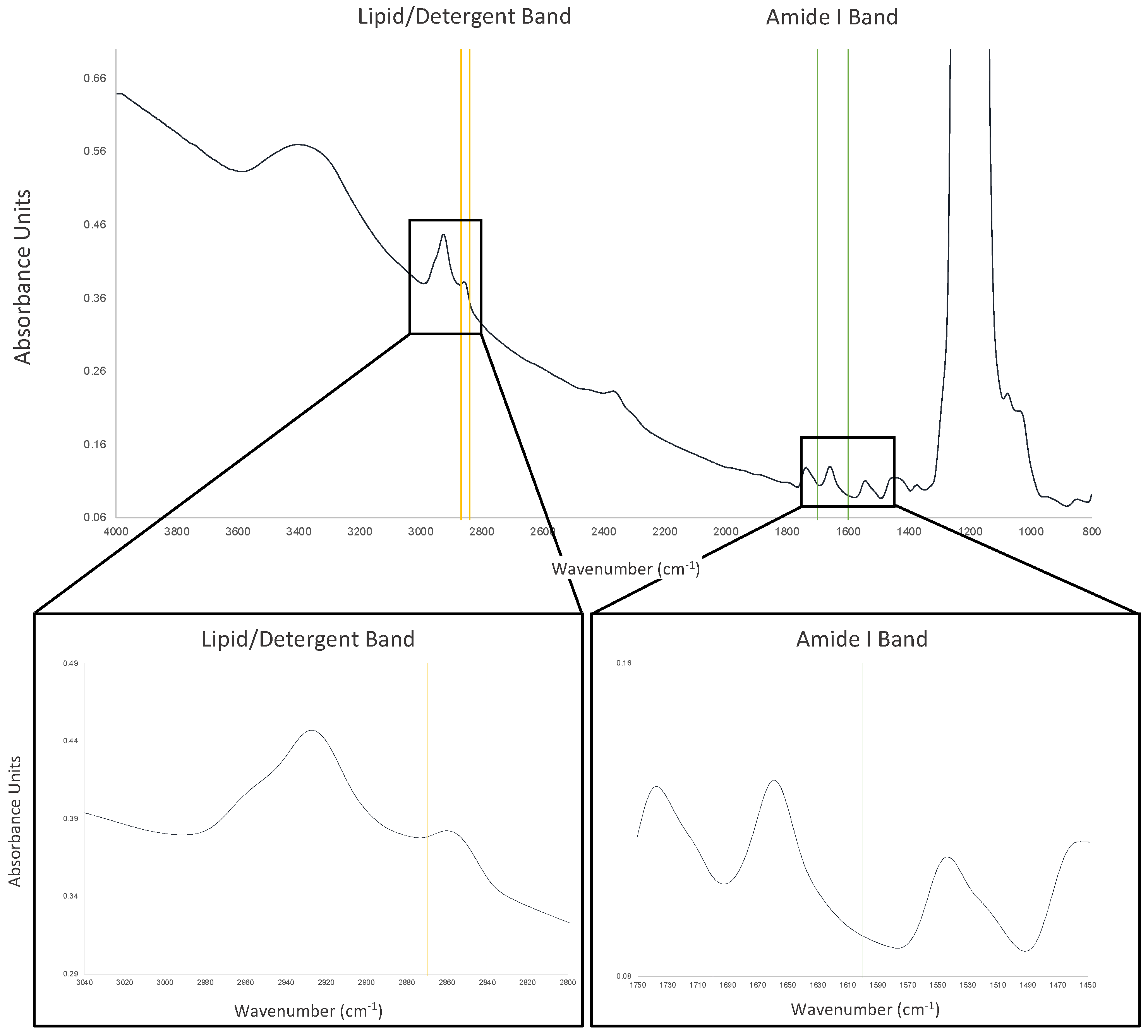
6. Mid-Infrared Spectroscopy (MIR)
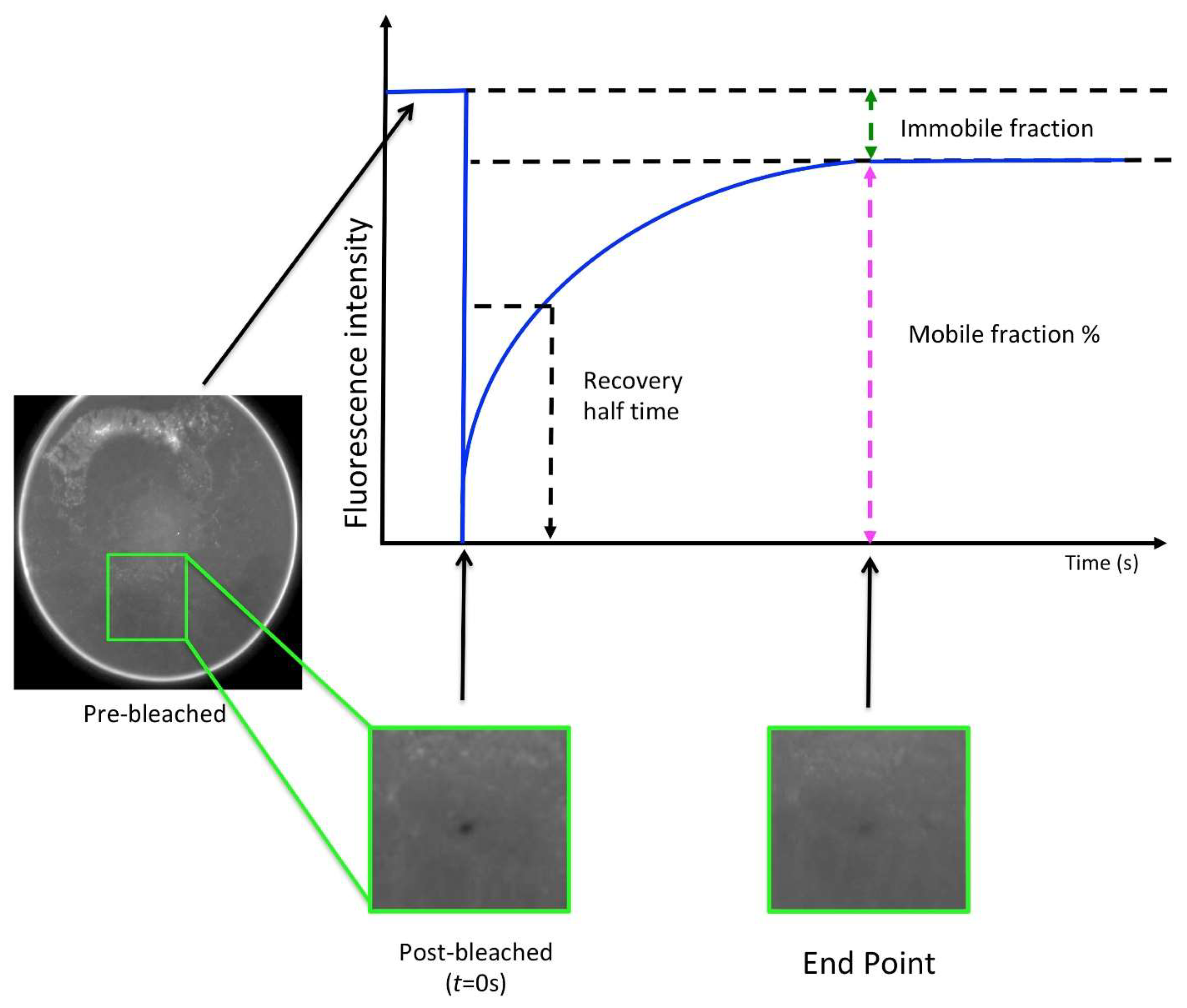
7. Lipidic Cubic Phase Fluorescence Recovery After Photobleaching (LCP-FRAP)
8. Summary and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Cy3 | 5, 5′-disulfato-1′-ethyl-3, 3, 3, 3- tetramethylindocarbocyanine |
| C8E5 | Pentaethylene glycol monooctyl ether |
| C12E8 | Dodecyl octaethylene glycol ether |
| CHS | Cholesteryl hemisuccinate |
| CMC | Critical micelle concentration |
| CPM | N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide |
| Cryo-EM | Cryo-electron microscopy |
| Cymal-6 | 6-Cyclohexyl-1-Hexyl-β-d-Maltoside |
| DDM | n-Dodecyl β-d-maltoside |
| DLS | Dynamic light scattering |
| DM | n-Decyl-β-d-Maltoside |
| - (dRFU)/dT | Negative first derivative of the relative fluorescence units with respect to temperature |
| DSF | Differential scanning fluorimetry |
| GPCR | G-protein-coupled receptors |
| HTCD | High-throughput circular dichroism |
| IR | Infrared radiation |
| LALS | Low angle light scattering |
| LCP-FRAP | Lipidic cubic phase fuorescence recovery after photobleaching |
| LDAO | Lauryldimethylamine-N-oxide |
| LMNG | Lauryl maltose neopentyl glycol |
| LS | Light scattering |
| MIR | Mid-infrared spectroscopy |
| NBEs | New biological entities |
| NMEs | New molecular entities |
| NMR | Nuclear magnetic resonance |
| OG | n-Octyl-β-d-Glucoside |
| PDB | Protein Data Bank |
| PDC | Protein detergent complexes |
| Phe | Phenylalanine |
| PTFE | Polytetrafluoroethylene |
| RALS | Right angle light scattering |
| RI | Refracting index |
| RT-PCR | Real time polymerase chain reaction |
| SAXS | Small angle X-ray scattering |
| SDS-PAGE | Sodium dodecyl sulphate polyacrylamide gel electrophoresis |
| SEC | Size-exclusion chromatography |
| SEC-MALS | Size-exclusion chromatography multi-angle light scattering |
| SMALPS | poly(styrene-co-maleic acid) lipid particles |
| SMILPS | poly(styrene-co-maleimide) lipid particles |
| Trp | Tryptophan |
| Tyr | Tyrosine |
| UDM | n-Undecyl-β-d-Maltopyranoside |
| SRCD | Synchrotron radiation circular dichroism |
| UV | Ultraviolet |
References
- Almén, M.S.; Nordström, K.J.; Fredriksson, R.; Schiöth, H.B. Mapping the human membrane proteome: A majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009, 7, 50. [Google Scholar] [CrossRef]
- Wallin, E.; Heijne, G.V. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998, 7, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Cordwell, S.J.; Thingholm, T.E. Technologies for plasma membrane proteomics. Proteomics 2010, 10, 611–627. [Google Scholar] [CrossRef]
- Moran, M.M. TRP channels as potential drug targets. Annu. Rev. Pharmacol. 2018, 58, 309–330. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR drug targets. Cell 2018, 172, 41–54. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Bull, S.C.; Doig, A.J. Properties of protein drug target classes. PLoS ONE 2015, 10, e0117955. [Google Scholar] [CrossRef] [PubMed]
- Overington, J.; Al-Lazikani, B.; Hopkins, A. Opinion–How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef]
- Walker, A.L.; Imam, S.Z.; Roberts, R.A. Drug discovery and development: Biomarkers of neurotoxicity and neurodegeneration. Exp. Biol. Med. 2018, 243, 1037–1045. [Google Scholar] [CrossRef] [Green Version]
- Shih, H.P.; Zhang, X.; Aronov, A.M. Drug discovery effectiveness from the standpoint of therapeutic mechanisms and indications. Nat. Rev. Drug Discov. 2018, 17, 19–33. [Google Scholar] [CrossRef]
- Reis, R.; Moraes, I. Structural biology and structure–function relationships of membrane proteins. Biochem. Soc. Trans. 2018, 47, 47–61. [Google Scholar] [CrossRef]
- Renaud, J.P.; Chung, C.W.; Danielson, U.H.; Egner, U.; Hennig, M.; Hubbard, R.E.; Nar, H. Biophysics in drug discovery: Impact, challenges and opportunities. Nat. Rev. Drug Discov. 2016, 15, 679. [Google Scholar] [CrossRef] [PubMed]
- The Next Generation in Membrane Protein Structure Determination; Advances in Experimental Medicine and Biology; Springer International; Moraes, I. (Ed.) Springer: Berlin/Heidelberg, Germany, 2016; Volume 922. [Google Scholar]
- Sørensen, T.M.; Hjorth-Jensen, S.J.; Oksanen, E.; Andersen, J.L.; Olesen, C.; Møller, J.V.; Nissen, P. Membrane-protein crystals for neutron diffraction. Acta Crystallogr. D Struct. Biol. 2018, 74, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Wolf, M. Cryo-electron microscopy for structural analysis of dynamic biological macromolecules. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 324–334. [Google Scholar] [CrossRef]
- Shoemaker, S.C.; Ando, N. X-rays in the cryo-electron microscopy era: Structural biology’s dynamic future. Biochemistry 2018, 57, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Thonghin, N.; Kargas, V.; Clews, J.; Ford, R.C. Cryo-electron microscopy of membrane proteins. Methods 2018, 147, 176–186. [Google Scholar] [CrossRef] [Green Version]
- Cressey, D.; Callaway, E. Cryo-electron microscopy wins chemistry Nobel. Nature 2017, 550, 167. [Google Scholar] [CrossRef] [PubMed]
- Danev, R.; Tegunov, D.; Baumeister, W. Using the Volta phase plate with defocus for cryo-EM single particle analysis. Elife 2017, 6, e23006. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.C.; McMullan, G.; Scheres, S.H. How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 2015, 40, 49–57. [Google Scholar] [CrossRef]
- De Zorzi, R.; Mi, W.; Liao, M.; Walz, T. Single-particle electron microscopy in the study of membrane protein structure. J. Electron Microsc. 2015, 65, 81–96. [Google Scholar] [CrossRef] [Green Version]
- Ishchenko, A.; Gati, C.; Cherezov, V. Structural biology of G protein-coupled receptors: New opportunities from XFELs and cryoEM. Curr. Opin. Struct. Biol. 2018, 51, 44–52. [Google Scholar] [CrossRef]
- Johansson, L.C.; Stauch, B.; Ishchenko, A.; Cherezov, V. A bright future for serial femtosecond crystallography with XFELs. Trends Biochem. Sci. 2017, 42, 749–762. [Google Scholar] [CrossRef]
- Spence, J.C.H. XFELs for structure and dynamics in biology. IUCrJ 2017, 4, 322–339. [Google Scholar] [CrossRef]
- Jaeger, K.; Dworkowski, F.; .Nogly, P.; Milne, C.; Wang, M.; Standfuss, J. Serial millisecond crystallography of membrane proteins. In The Next Generation in Membrane Protein Structure Determination; Moraes, I., Ed.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 137–149. [Google Scholar]
- Nango, E.; Royant, A.; Kubo, M.; Nakane, T.; Wickstrand, C.; Kimura, T.; Tanaka, T.; Tono, K.; Song, C.; Tanaka, R.; et al. A three-dimensional movie of structural changes in bacteriorhodopsin. Science 2016, 354, 1552–1557. [Google Scholar] [CrossRef]
- Zhu, L.; Weierstall, U.; Cherezov, V.; Liu, W. Serial femtosecond crystallography of membrane proteins. In The Next Generation in Membrane Protein Structure Determination; Moraes, I., Ed.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 151–160. [Google Scholar]
- Chapman, H.N.; Fromme, P.; Barty, A.; White, T.A.; Kirian, R.A.; Aquila, A.; Hunter, M.S.; Schulz, J.; DePonte, D.P.; Weierstall, U.; et al. Femtosecond X-ray protein nanocrystallography. Nature 2011, 470, 73–77. [Google Scholar] [CrossRef]
- Schlichting, I. Serial femtosecond crystallography: The first five years. IUCrJ 2015, 2, 246–255. [Google Scholar] [CrossRef]
- Neutze, R. Opportunities and challenges for time-resolved studies of protein structural dynamics at X-ray free-electron lasers. Philos. Trans. Royal Soc. B 2014, 369, 20130318. [Google Scholar] [CrossRef]
- Dilworth, M.V.; Piel, M.S.; Bettaney, K.E.; Ma, P.; Luo, J.; Sharples, D.; Poyner, D.R.; Gross, S.R.; Moncoq, K.; Henderson, P.J.; et al. Microbial expression systems for membrane proteins. Methods 2018, 147, 3–39. [Google Scholar] [CrossRef]
- McKenzie, E.A.; Abbott, W.M. Expression of recombinant proteins in insect and mammalian cells. Methods 2018, 147, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.K.; Tullman-Ercek, D. Engineering expression and function of membrane proteins. Methods 2018, 147, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Hardy, D.; Mandon, E.D.; Rothnie, A.; Jawhari, A. The yin and yang of solubilization and stabilization for wild-type and full-length membrane protein. Methods 2018, 147, 118–125. [Google Scholar] [CrossRef]
- Moraes, I.; Evans, G.; Sanchez-Weatherby, J.; Newstead, S.; Stewart, P.D.S. Membrane protein structure determination—the next generation. Biochim. Biophys. Acta Biomembr. 2014, 1838, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Pecora, R. Dynamic Light Scattering: Applications of Photon Correlation Spectroscopy; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Lorber, B.; Fischer, F.; Bailly, M.; Roy, H.; Kern, D. Protein analysis by dynamic light scattering: Methods and techniques for students. Biochem. Mol. Biol. Educ. 2012, 40, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Jachimska, B.; Wasilewska, M.; Adamczyk, Z. Characterization of globular protein solutions by dynamic light scattering, electrophoretic mobility, and viscosity measurements. Langmuir 2008, 24, 6866–6872. [Google Scholar] [CrossRef]
- International Standard ISO22412 Particle Size Analysis – Dynamic Light Scattering, International Organisation for Standardisation (ISO), 2008. Available online: https://www.iso.org/standard/40942.html (accessed on 5 May 2019).
- Murphy, R.M. Static and dynamic light scattering of biological macromolecules: What can we learn? Curr. Opin. Biotecnhol. 1997, 8, 25–30. [Google Scholar] [CrossRef]
- Provencher, S.W.; Stepanek, P. Global analysis of dynamic light scattering autocorrelation functions. Part. Syst. Charact. 1996, 13, 291–294. [Google Scholar] [CrossRef]
- International Standard ISO13321. Methods for Determination of Particle Size Distribution Part 8: Photon Correlation Spectroscopy. International Organisation for Standardisation (ISO). 1996. Available online: https://www.iso.org/obp/ui/#iso:std:iso:13321:ed-1:v1:en (accessed on 5 May 2019).
- Brown, W. Dynamic Light Scattering: The Method and Some Applications; Clarendon Press: Oxford, UK, 1993; Volume 313. [Google Scholar]
- Stetefeld, J.; McKenna, S.A.; Patel, T.R. Dynamic light scattering: A practical guide and applications in biomedical sciences. Biophys. Rev. 2016, 8, 409–427. [Google Scholar] [CrossRef]
- Oberthuer, D.; Melero-García, E.; Dierks, K.; Meyer, A.; Betzel, C.; Garcia-Caballero, A.; Gavira, J.A. Monitoring and scoring counter-diffusion protein crystallization experiments in capillaries by in situ dynamic light scattering. PLoS ONE. 2012, 7, e33545. [Google Scholar] [CrossRef]
- Dierks, K.; Meyer, A.; Einspahr, H.; Betzel, C. Dynamic light scattering in protein crystallization droplets: Adaptations for analysis and optimization of crystallization processes. Cryst. Growth Des. 2008, 8, 1628–1634. [Google Scholar] [CrossRef]
- Wilson, W.W. Light scattering as a diagnostic for protein crystal growth—A practical approach. J. Struct. Biol. 2003, 142, 56–65. [Google Scholar] [CrossRef]
- Ferré-D’Amaré, A.R.; Burley, S.K. Use of dynamic light scattering to assess crystallizability of macromolecules and macromolecular assemblies. Structure 1994, 2, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Kadima, W.; McPherson, A.; Dunn, M.F.; Jurnak, F.A. Characterization of precrystallization aggregation of canavalin by dynamic light scattering. Biophys. J. 1990, 57, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Birch, J.; Axford, D.; Foadi, J.; Meyer, A.; Eckardt, A.; Thielmann, Y.; Moraes, I. The fine art of integral membrane protein crystallisation. Methods 2018, 147, 150–162. [Google Scholar] [CrossRef]
- Meyer, A.; Dierks, K.; Hussein, R.; Brillet, K.; Brognaro, H.; Betzel, C. Systematic analysis of protein–detergent complexes applying dynamic light scattering to optimize solutions for crystallization trials. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 2015, 71, 75–81. [Google Scholar] [CrossRef]
- Aivaliotis, M.; Samolis, P.; Neofotistou, E.; Remigy, H.; Rizos, A.K.; Tsiotis, G. Molecular size determination of a membrane protein in surfactants by light scattering. Biochim. Biophys. Acta Biomembr. 2003, 1615, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Anandan, A.; Vrielink, A. Detergents in membrane protein purification and crystallisation. In The Next Generation in Membrane Protein Structure Determination; Moraes, I., Ed.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 13–28. [Google Scholar]
- Privé, G.G. Detergents for the stabilization and crystallization of membrane proteins. Methods 2007, 41, 388–397. [Google Scholar] [CrossRef]
- Otzen, D.E. Protein unfolding in detergents: Effect of micelle structure, ionic strength, pH, and temperature. Biophys. J. 2002, 83, 2219–2230. [Google Scholar] [CrossRef]
- Breyton, C.; Tribet, C.; Olive, J.; Dubacq, J.P.; Popot, J.L. Dimer to monomer conversion of the cytochrome b6 f complex. Causes and consequences. J. Biol. Chem. 1997, 272, 21892–21900. [Google Scholar] [CrossRef]
- Subburaj, Y.; Cosentino, K.; Axmann, M.; Pedrueza-Villalmanzo, E.; Hermann, E.; Bleicken, S.; Spatz, J.; García-Saéz, A.J. Bax Monomers Form Dimer Units in the Membrane That Further Self-Assemble into Multiple Oligomeric Species. Nat. Commun. 2015, 6, 8042. [Google Scholar] [CrossRef]
- Arkhipov, A.; Shan, Y.; Das, R.; Endres, N.F.; Eastwood, M.P.; Wemmer, D.E.; Kuriyan, J.; Shaw, D.E. Architecture and membrane interactions of the EGF receptor. Cell 2013, 152, 557–569. [Google Scholar] [CrossRef]
- Rath, A.; Glibowicka, M.; Nadeau, V.G.; Chen, G.; Deber, C.M. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 1760–1765. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Matsui, H.; Takagi, T. Membrane protein molecular weight determined by low-angle laser light-scattering photometry coupled with high-performance gel chromatography. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1989; Volume 172, pp. 514–528. [Google Scholar]
- Moraes, I.; Archer, M. Methods for the successful crystallization of membrane proteins. In Structural Proteomics: High-Throughput Methods, Methods in Molecular Biology; Owens, R.J., Ed.; Humana Press: New York, NY, USA, 2015; Volume 1261, pp. 211–230. [Google Scholar]
- Miercke, L.J.; Robbins, R.A.; Stroud, R.M. Tetra detector analysis of membrane proteins. Curr. Protoc. Protein Sci. 2014, 77, 29.10.1–29.10.30. [Google Scholar] [CrossRef]
- Sahin, E.; Roberts, C.J. Size-exclusion chromatography with multi-angle light scattering for elucidating protein aggregation mechanisms. In Therapeutic Proteins; Voynov, V., Caravella, J.A., Eds.; Humana Press: Totowa, NJ, USA, 2012; Volume 899, pp. 403–423. [Google Scholar]
- Slotboom, D.J.; Duurkens, R.H.; Olieman, K.; Erkens, G.B. Static light scattering to characterize membrane proteins in detergent solution. Methods 2008, 46, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Arakawa, T.; Philo, J.S. Size-exclusion chromatography with on-line light-scattering, absorbance, and refractive index detectors for studying proteins and their interactions. Anal. Biochem. 1996, 240, 155–166. [Google Scholar] [CrossRef]
- Wyatt, P. Light scattering and the absolute characterization of macromolecules. Anal. Chim. Acta 1993, 272, 1–40. [Google Scholar] [CrossRef]
- Dorwart, M.R.; Wray, R.; Brautigam, C.A.; Jiang, Y.; Blount, P. S. aureus MscL is a pentamer in vivo but of variable stoichiometries in vitro: Implications for detergent-solubilized membrane proteins. PLoS Biol. 2010, 8, e1000555. [Google Scholar] [CrossRef]
- Gill, S.C.; Von Hippel, P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989, 182, 319–326. [Google Scholar] [CrossRef]
- Hall, S.C.L.; Tognoloni, C.; Charlton, J.; Bragginton, É.C.; Rothnie, A.J.; Sridhar, P.; Wheatley, M.; Knowles, T.J.; Arnold, T.; Edler, K.J.; et al. An acid-compatible co-polymer for the solubilization of membranes and proteins into lipid bilayer-containing nanoparticles. Nanoscale 2018, 10, 10609–10619. [Google Scholar] [CrossRef] [Green Version]
- Hall, S.C.L.; Tognoloni, C.; Price, G.J.; Klumperman, B.; Edler, K.J.; Dafforn, T.R.; Arnold, T. Influence of Poly (styrene-co-maleic acid) Copolymer Structure on the Properties and Self-Assembly of SMALP Nanodiscs. Biomacromolecules 2017, 19, 761–772. [Google Scholar] [CrossRef]
- Parmar, M.; Rawson, S.; Scarff, C.A.; Goldman, A.; Dafforn, T.R.; Muench, S.P.; Postis, V.L. Using a SMALP platform to determine a sub-nm single particle cryo-EM membrane protein structure. Biochim. Biophys. Acta Biomembr. 2018, 1860, 378–383. [Google Scholar] [CrossRef]
- Berova, N.; Nakanishi, K.; Woody, R.W.; Woody, R. Circular Dichroism: Principles and Applications, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2000. [Google Scholar]
- Hussain, R.; Benning, K.; Javorfi, T.; Longo, E.; Rudd, T.R.; Pulford, B.; Siligardi, G. CDApps: Integrated software for experimental planning and data processing at beamline B23 Diamond Light Source. J. Synchrotron Radiat. 2015, 22, 465–468. [Google Scholar] [CrossRef]
- Sreerama, N.; Woody, R.W. On the analysis of membrane protein circular dichroism spectra. Protein Sci. 2004, 13, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Siligardi, G.; Hussain, R. CD spectroscopy: An essential tool for quality control of protein folding. In Structural Proteomics: High-Throughput Methods, Methods in Molecular Biology; Owens, R.J., Ed.; Humana Press: New York, NY, USA, 2015; Volume 1261, pp. 255–276. [Google Scholar]
- Siligardi, G.; Hussain, R. Biomolecules interactions and competitions by non-immobilised ligand interaction assay by circular dichroism. Enantiomer 1998, 3, 77–87. [Google Scholar]
- Siligardi, G.; Hussain, R. Applications of circular dichroism. In Encyclopedia of Spectroscopy and Spectrometry, 2nd ed.; Lindon, J., Tranter, G., Koppenaal, D., Eds.; Elsevier: Amsterdam, The Netherlands, 2010; Volume 1, pp. 9–14. [Google Scholar]
- Siligardi, G.; Panaretou, B.; Meyer, P.; Singh, S.; Woolfson, D.N.; Piper, P.W.; Pearl, L.H.; Prodromou, C. Regulation of Hsp90 ATPase activity by the co-chaperone Cdc37p/p50cdc37. J. Biol. Chem. 2002, 277, 20151–20159. [Google Scholar] [CrossRef]
- Miles, A.J.; Wallace, B.A. Circular dichroism spectroscopy of membrane proteins. Chem. Soc. Rev. 2016, 45, 4859–4872. [Google Scholar] [CrossRef] [Green Version]
- Phillips-Jones, M.K.; Patching, S.G.; Edara, S.; Nakayama, J.; Hussain, R.; Siligardi, G. Interactions of the intact FsrC membrane histidine kinase with the tricyclic peptide siamycin I revealed through synchrotron radiation circular dichroism. Phys. Chem. Chem. Phys. 2013, 15, 444–447. [Google Scholar] [CrossRef]
- Patching, S.G.; Edara, S.; Patching, S.G.; Edara, S.; Ma, P.; Nakayama, J.; Hussain, R.; Siligardi, G.; Phillips-Jones, M.K. Interactions of the intact FsrC membrane histidine kinase with its pheromone ligand GBAP revealed through synchrotron radiation circular dichroism. Biochim. Biophys. Acta Biomembr. 2012, 1818, 1595–1602. [Google Scholar] [CrossRef] [Green Version]
- Hussain, R.; Jávorfi, T.; Siligardi, G. Circular dichroism beamline B23 at the Diamond Light Source. J. Synchrotron Radiat. 2012, 19, 32–135. [Google Scholar] [CrossRef]
- Hussain, R.; Jávorfi, T.; Siligardi, G. Spectroscopic analysis: Synchrotron radiation circular dichroism. Compr. Chirality 2012, 8, 438–448. [Google Scholar]
- Jávorfi, T.; Hussain, R.; Myatt, D.; Siligardi, G. Measuring circular dichroism in a capillary cell using the B23 synchrotron radiation CD beamline at Diamond Light Source. Chirality 2010, 22, E149–E153. [Google Scholar] [CrossRef]
- Hussain, R.; Siligardi, G. Characterisation of conformational and ligand binding properties of membrane proteins using synchrotron radiation circular dichroism. In The Next Generation in Membrane Protein Structure Determination; Moraes, I., Ed.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 43–59. [Google Scholar]
- Abramson, J.; Smirnova, I.; Kasho, V.; Verner, G.; Kaback, H.R.; Iwata, S. Structure and mechanism of the lactose permease of Escherichia coli. Science 2003, 301, 610–615. [Google Scholar] [CrossRef]
- Mao, A.; Vaidehi, N.; Grisshammer, R.; Tate, C.G.; Robertson, N.; Bhattacharya, S.; Lee, S. How Do Short Chain Nonionic Detergents Destabilize G-Protein-Coupled Receptors? J. Am. Chem. Soc. 2016, 138, 15425–15433. [Google Scholar] [CrossRef] [Green Version]
- Bill, R.M.; Henderson, P.J.; Iwata, S.; Kunji, E.R.; Michel, H.; Neutze, R.; Newstead, S.; Poolman, B.; Tate, C.G.; Vogel, H. Overcoming barriers to membrane protein structure determination. Nat. Biotechnol. 2011, 29, 335–340. [Google Scholar] [CrossRef] [Green Version]
- Kulig, W.; Jurkiewicz, P.; Olżyńska, A.; Tynkkynen, J.; Javanainen, M.; Manna, M.; Rog, T.; Hof, M.; Vattulainen, I.; Jungwirth, P. Experimental determination and computational interpretation of biophysical properties of lipid bilayers enriched by cholesteryl hemisuccinate. Biochim. Biophys. Acta Biomembr. 2015, 1848, 422–432. [Google Scholar] [CrossRef]
- Hanson, M.A.; Cherezov, V.; Griffith, M.T.; Roth, C.B.; Jaakola, V.P.; Chien, E.Y.; Velasquez, J.; Kuhn, P.; Stevens, R.C. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure 2008, 16, 897–905. [Google Scholar] [CrossRef]
- Oates, J.; Faust, B. The role of cholesterol on the activity and stability of neurotensin receptor 1. Biochim. Biophys. Acta Biomembr. 2012, 1818, 2228–2233. [Google Scholar] [CrossRef] [Green Version]
- Rosenbusch, J.P. Stability of Membrane Proteins: Relevance for the Selection of Appropriate Methods for High-Resolution Structure Determinations. J. Struct. Biol. 2001, 136, 144–157. [Google Scholar] [CrossRef]
- Popov, P.; Peng, Y.; Shen, L.; Stevens, R.C.; Cherezov, V.; Liu, Z.J.; Katritch, V. Computational design of thermostabilizing point mutations for G protein-coupled receptors. eLife 2018, 7, e34729. [Google Scholar] [CrossRef]
- Magnani, F.; Serrano-Vega, M.J.; Shibata, Y.; Abdul-Hussein, S.; Lebon, G.; Miller-Gallacher, J.; Singhal, A.; Strege, A.; Thomas, J.A.; Tate, C.G. A mutagenesis and screening strategy to generate optimally thermostabilized membrane proteins for structural studies. Nat. Protoc. 2016, 11, 1554–15571. [Google Scholar] [CrossRef]
- Vaidehi, N.; Grisshammer, R.; Tate, C.G. How can mutations thermostabilize G-Protein-Coupled receptors? Trends Pharmacol. Sci. 2016, 37, 37–46. [Google Scholar] [CrossRef]
- Tate, C.G.; Schertler, G.F. Engineering G protein-coupled receptors to facilitate their structure determination. Curr. Opin. Struct. Biol. 2009, 19, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.C.; Aulabaugh, A.; Jin, G.; Cowling, R.; Bard, J.; Malamas, M.; Ellestad, G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 2004, 332, 153–159. [Google Scholar] [CrossRef]
- Pantoliano, M.W.; Petrella, E.C.; Kwasnoski, J.D.; Lobanov, V.S.; Myslik, J.; Graf, E.; Carver, T.; Asel, E.; Springer: Berlin/Heidelberg, B.A.; Lane, P.; et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen. 2001, 6, 429–440. [Google Scholar] [CrossRef]
- Zucker, F.H.; Stewart, C.; dela Rosa, J.; Kim, J.; Zhang, L.; Xiao, L.; Ross, J.; Napuli, A.J.; Mueller, N.; Castaneda, L.J.; et al. Prediction of protein crystallization outcome using a hybrid method. J. Struct. Biol. 2010, 171, 64–73. [Google Scholar] [CrossRef] [Green Version]
- Vedadi, M.; Arrowsmith, C.H.; Allali-Hassani, A.; Senisterra, G.; Wasney, G.A. Biophysical characterization of recombinant proteins: A key to higher structural genomics success. J. Struct. Biol. 2010, 172, 107–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef]
- Ericsson, U.B.; Hallberg, B.M.; Detitta, G.T.; Dekker, N.; Nordlund, P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 2006, 357, 289–298. [Google Scholar] [CrossRef]
- Vedadi, M.; Niesen, F.H.; Allali-Hassani, A.; Fedorov, O.Y.; Finerty, P.J.; Wasney, G.A.; Yeung, R.; Arrowsmith, C.; Ball, L.J.; Berglund, H.; et al. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. USA. 2006, 103, 15835–15840. [Google Scholar] [CrossRef] [Green Version]
- Kohlstaedt, M.; von der Hocht, I.; Hilbers, F.; Thielmann, Y.; Michel, H. Development of a Thermofluor assay for stability determination of membrane proteins using the Na+/H+ antiporter NhaA and cytochrome c oxidase. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 1112–1122. [Google Scholar] [CrossRef]
- Fan, J.; Heng, J.; Dai, S.; Shaw, N.; Zhou, B.; Huang, B.; He, Z.; Wang, Y.; Jiang, T.; Li, X.; et al. An efficient strategy for high throughput screening of recombinant integral membrane protein expression and stability. Protein Expr. Purif. 2011, 78, 6–13. [Google Scholar] [CrossRef]
- Liu, W.; Hanson, M.A.; Stevens, R.C.; Cherezov, V. LCP-Tm: An assay to measure and understand stability of membrane proteins in a membrane environment. Biophys. J. 2010, 98, 1539–1548. [Google Scholar] [CrossRef]
- Alexandrov, A.I.; Mileni, M.; Chien, E.Y.T.; Hanson, M.A.; Stevens, R.C. Microscale Fluorescent Thermal Stability Assay for Membrane Proteins. Structure 2008, 16, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Yeh, A.P.; McMillan, A.; Stowell, M.H. Rapid and simple protein-stability screens: Application to membrane proteins. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 451–457. [Google Scholar] [CrossRef]
- Vivoli, M.; Novak, H.R.; Littlechild, J.A.; Harmer, N.J. Determination of protein-ligand interactions using differential scanning fluorimetry. J. Vis. Exp. 2014, 91, e51809. [Google Scholar] [CrossRef]
- Bergsdorf, C.; Fiez-Vandal, C. An Alternative Thiol-Reactive Dye to Analyze Ligand Interactions with the Chemokine Receptor CXCR2 Using a New Thermal Shift Assay Format. J. Biomol Screen. 2016, 21, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Svilenov, H.; Markoja, U.; Winter, G. Isothermal chemical denaturation as a complementary tool to overcome limitations of thermal differential scanning fluorimetry in predicting physical stability of protein formulations. Eur. J. Pharm. Biopharm. 2018, 125, 106–113. [Google Scholar] [CrossRef]
- Wanner, R.; Breitsprecher, D.; Duhr, S.; Baaske, P.; Winter, G. Thermo-optical protein characterization for straightforward preformulation development. J. Pharm. Sci. 2017, 106, 2955–2958. [Google Scholar] [CrossRef]
- Alexander, C.G.; Wanner, R.; Johnson, C.M.; Breitsprecher, D.; Winter, G.; Duhr, S.; Baaske, P.; Ferguson, N. Novel microscale approaches for easy, rapid determination of protein stability in academic and commercial settings. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 2241–2250. [Google Scholar] [CrossRef] [Green Version]
- Boland, C.; Olatunji, S.; Bailey, J.; Howe, N.; Weichert, D.; Fetics, S.K.; Yu, X.; Merino-Gracia, J.; Delsaut, C.; Caffrey, M. Membrane (and Soluble) Protein Stability and Binding Measurements in the Lipid Cubic Phase Using Label-Free Differential Scanning Fluorimetry. Anal. Chem. 2018, 90, 12152–12160. [Google Scholar] [CrossRef]
- Goldring, J.P.D. Protein Quantification Methods to Determine Protein Concentration Prior to Electrophoresis in Protein Electrophoresis; Kurien, B.T., Scofield, R.H., Eds.; Humana Press: New York, NY, USA, 2012; pp. 29–35. [Google Scholar]
- Noble, J.E.; Bailey, M.J. Quantitation of protein. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2009; Volume 463, pp. 73–95. [Google Scholar]
- Swinehart, D.F. The beer-lambert law. J. Chem. Educ. 1962, 39, 333–335. [Google Scholar] [CrossRef]
- Mayerhöfer, T.G.; Popp, J. Beer’s Law–Why Absorbance Depends (Almost) Linearly on Concentration. Chem. Phys. Chem. 2019, 20, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, P.; Hansen, J.B.; Allen, M. Microvolume protein concentration determination using the NanoDrop 2000c spectrophotometer. J. Vis. Exp. 2009, 33, e1610. [Google Scholar] [CrossRef]
- Strug, I.; Utzat, C.; Cappione, A.; Gutierrez, S.; Amara, R.; Lento, J.; Capito, F.; Skudas, R.; Chernokalskaya, E.; Nadler, T. Development of a univariate membrane-based mid-infrared method for protein quantitation and total lipid content analysis of biological samples. J. Anal. Methods Chem. 2014, 657079. [Google Scholar] [CrossRef]
- Stuart, B.H. Infrared Spectroscopy of Biological Applications: An Overview. In Encyclopedia of Analytical Chemistry; John Wiley & Sons, Ltd.: Chichester, UK, 2012. [Google Scholar] [CrossRef]
- Singh, B.R. Basic Aspects of the Technique and Applications of Infrared Spectroscopy of Peptides and Proteins. In ACS Symposium Series; Washington, D.C., Ed.; American Chemical Society: Washington, WA, USA, 2000; Volume 750, pp. 2–37. [Google Scholar]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta Bioenerg. 2007, 1767, 1073–1101. [Google Scholar] [CrossRef] [Green Version]
- Fabian, H.; Mäntele, W. Infrared spectroscopy of proteins. In Handbook of Vibrational Spectroscopy; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar] [CrossRef]
- Dubois, J.; Shaw, R.A. IR Spectroscopy in Clinical and Diagnostic Applications. Anal. Chem. 2004, 76, 360A–367A. [Google Scholar] [CrossRef] [PubMed]
- Dacosta, C.J.B.; Baenziger, J.E. A rapid method for assessing lipid:protein and detergent:protein ratios in membrane-protein crystallization. Acta Crystallogr. D Biol. Crystallogr. 2003, 59, 77–83. [Google Scholar] [CrossRef]
- Pincet, F.; Adrien, V.; Yang, R.; Delacotte, J.; Rothman, J.E.; Urbach, W.; Tareste, D. FRAP to Characterize Molecular Diffusion and Interaction in Various Membrane Environments. PLoS ONE 2016, 11, e0158457. [Google Scholar] [CrossRef]
- Landau, E.M.; Rosenbusc, J.P. Lipidic cubic phases: A novel concept for the crystallization of membrane proteins. Proc. Natl. Acad. Sci. USA. 1996, 93, 14532–14535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dods, R.; Båth, P.; Arnlund, D.; Beyerlein, K.R.; Nelson, G.; Liang, M.; Harimoorthy, R.; Berntsen, P.; Malmerberg, E.; Johansson, L.; et al. From Macrocrystals to Microcrystals: A Strategy for Membrane Protein Serial Crystallography. Structure 2017, 25, 1461–1468. [Google Scholar] [CrossRef]
- Sugahara, M.; Nakane, T.; Masuda, T.; Suzuki, M.; Inoue, S.; Song, C.; Tanaka, R.; Nakatsu, T.; Mizohata, E.; Yumoto, F.; et al. Hydroxyethyl cellulose matrix applied to serial crystallography. Sci. Rep. 2017, 7, 703. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ishchenko, A.; Cherezov, V. Preparation of microcrystals in lipidic cubic phase for serial femtosecond crystallography. Nat. Protoc. 2014, 9, 2123–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weierstall, U.; James, D.; Wang, C.; White, T.A.; Wang, D.; Liu, W.; Spence, J.C.; Doak, R.B.; Nelson, G.; Fromme, P.; et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 2014, 5, 3309. [Google Scholar] [CrossRef]
- Nollert, P.; Qiu, H.; Caffrey, M.; Rosenbusch, J.P.; Landau, E. M Molecular mechanism for the crystallization of bacteriorhodopsin in lipidic cubic phases. FEBS Lett. 2001, 504, 179–186. [Google Scholar] [CrossRef]
- Chung, H.; Caffrey, M. The curvature elastic-energy function of the lipid-water cubic mesophase. Nature 1994, 368, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; An, S. Methods used to study the oligomeric structure of G-protein-coupled receptors. Bioscience Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenalti, G.; Abola, E.E.; Wang, C.; Wu, B.; Cherezov, V. Fluorescence recovery after photobleaching in Lipidic Cubic Phase (LCP-FRAP): A precrystallization assay for membrane proteins. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2015; Volume 557, pp. 417–437. [Google Scholar] [CrossRef]
- Cherezov, V.; Liu, J.; Griffith, M.; Hanson, M.A.; Stevens, R.C. LCP-FRAP Assay for Pre-Screening Membrane Proteins for in Meso Crystallization. Cryst. Growth. Des. 2008, 8, 4307–4315. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strengths | Limitations | Amount of Sample Required |
|---|---|---|
| In Situ Dynamic Light Scattering (DLS) | ||
|
| Minimum: 0.3 to 2.0 mg/mL. Sample volume of 0.5–2 μL per well |
| Size-Exclusion Chromatography Multi-Angle Light Scattering (SEC-MALS) | ||
|
| Minimum: 0.5 to 2.0 mg/mL. Requires a sample volume of ~160 μL |
| Circular Dichroism (CD)/Synchrotron Radiation Circular Dichroism (SRCD) | ||
|
| 1 mg/mL in a volume of ~25 μL when using a 0.1 mm cuvette |
| Fluorescence Dye-based Differential Scanning Fluorimetry Assay (DSF) | ||
|
| Minimum: 1 to 15 µg of protein in a volume of 50 µL reaction |
| Mid-Infrared Spectroscopy (MIR) | ||
|
| Protein range: 0.25 to 5.0 mg/mL Lipid/detergent range: 0.25 to 4.0 % Requires a sample volume of 2 µL |
| Lipidic Cubic Phase Fluorescence Recovery After Photobleaching (LCP-FRAP) | ||
|
| Minimum: 1 mg/mL of labelled protein reconstituted to LCP |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwan, T.O.C.; Reis, R.; Siligardi, G.; Hussain, R.; Cheruvara, H.; Moraes, I. Selection of Biophysical Methods for Characterisation of Membrane Proteins. Int. J. Mol. Sci. 2019, 20, 2605. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102605
Kwan TOC, Reis R, Siligardi G, Hussain R, Cheruvara H, Moraes I. Selection of Biophysical Methods for Characterisation of Membrane Proteins. International Journal of Molecular Sciences. 2019; 20(10):2605. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102605
Chicago/Turabian StyleKwan, Tristan O. C., Rosana Reis, Giuliano Siligardi, Rohanah Hussain, Harish Cheruvara, and Isabel Moraes. 2019. "Selection of Biophysical Methods for Characterisation of Membrane Proteins" International Journal of Molecular Sciences 20, no. 10: 2605. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102605