Lysophosphatidic Acid Signaling in Diabetic Nephropathy


1
College of Pharmacy, Gachon University, Incheon 21936, Korea
2
Lee Gil Ya Cancer and Diabetes Institute, Gachon University, Incheon 21999, Korea
3
Department of Food and Nutrition, Eulji University, Seongnam 13135, Korea
4
Gachon University Gil Medical Center, Gachon Medical and Convergence Institute, Incheon 21565, Korea
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(11), 2850; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112850
Submission received: 6 May 2019
/
Revised: 7 June 2019
/
Accepted: 8 June 2019
/
Published: 11 June 2019
(This article belongs to the Special Issue Kidney Inflammation, Injury and Regeneration)
Abstract
:Lysophosphatidic acid (LPA) is a bioactive phospholipid present in most tissues and body fluids. LPA acts through specific LPA receptors (LPAR1 to LPAR6) coupled with G protein. LPA binds to receptors and activates multiple cellular signaling pathways, subsequently exerting various biological functions, such as cell proliferation, migration, and apoptosis. LPA also induces cell damage through complex overlapping pathways, including the generation of reactive oxygen species, inflammatory cytokines, and fibrosis. Several reports indicate that the LPA–LPAR axis plays an important role in various diseases, including kidney disease, lung fibrosis, and cancer. Diabetic nephropathy (DN) is one of the most common diabetic complications and the main risk factor for chronic kidney diseases, which mostly progress to end-stage renal disease. There is also growing evidence indicating that the LPA–LPAR axis also plays an important role in inducing pathological alterations of cell structure and function in the kidneys. In this review, we will discuss key mediators or signaling pathways activated by LPA and summarize recent research findings associated with DN.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Diabetic nephropathy (DN) is a microvascular complication of diabetes and develops in approximately 20–40% patients with diabetes, including type 1 and type 2 diabetes patients [1,2]. It has become the main risk factor for the development of chronic kidney diseases, such that most DN patients progress to end-stage renal disease (ESRD) and require renal replacement in the end [2]. Thus, DN not only increases the health cost for individuals and the society at large but is also a risk factor for morbidity and mortality. Intensive glycemic control is a well-established strategy for the prevention of DN. Despite the efforts for the management of hyperglycemia and hypertension using current therapies, such as angiotensin converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) [3,4], the risk of DN progression has still not been reduced. Currently available drugs only delay the progress of the disease, rather than curing it. Therefore, a novel and effective therapeutic approach is urgently needed for patients with DN.
Phospholipids are generally known to be structural components of plasma membranes, but growing evidence indicates that membrane phospholipids also play crucial roles as signaling molecules and exert a wide range of physiological responses. Lysophosphatidic acid (LPA) is a small, naturally occurring glycerophospholipid, which is composed of a glycerol backbone with an ester-linked acyl chain and a phosphate group. It is also produced by the action of various lysophospholipases, including autotaxin (ATX), and phospholipases A1 or A2 (PLA1 and PLA2). LPA acts through specific LPA receptors (LPAR1 to LPAR6) coupled with G protein and is associated with a wide range of cell responses, such as proliferation and migration [5,6], and pathologies of a number of diseases, including fibrosis, cancer, neuronal disorders, and bone metabolism [5,7,8,9,10]. Hyperglycemia, advanced glycation end product (AGE), and pro-inflammatory cytokines are considered the three main initial mediators of DN development [11,12]. However, some recent studies also showed that the LPA–LPA receptor axis may play an important role in the pathogenesis of diabetic kidney disease.
In this review article, we review the key mediators and signaling pathways activated by LPA and summarize newly reported signaling pathways modulated by different LPA receptor antagonists in different DN models.
2. Biosynthesis and Degradation of LPA
LPA is the smallest bioactive lysophospholipid (MW 430–480 Da) derived from membrane phospholipids [13] and acts as an extracellular signaling molecule via its receptors, regulating various cellular processes, including cell proliferation, survival, migration, differentiation, remodeling, and cytokine/chemokine secretion [13]. LPA is known to be water soluble and is present in most tissues and biological fluids, such as plasma, saliva, tears, follicular fluid, and cerebrospinal fluid [5,14]. LPA is produced by several enzymes, either in the intracellular or extracellular compartment, as shown in Figure 1. In the intracellular compartment, LPA is naturally synthesized through the action of glycerol-3-phosphate acyltransferase (GPAT) during the process of triglyceride and phospholipid anabolism, which is found mainly in the mitochondria and endoplasmic reticulum of various cell types [15,16]. LPA is also synthesized by the action of intra/extra-cellular PLA1 or PLA2 from phosphatidic acid (PA). Ubiquitously expressed PLA1 and PLA2 in the body hydrolyze the bond between the fatty acid chain and the glycerol backbone at sn1 (saturated) or sn2 (unsaturated) positions of PA [17,18]. Membrane phospholipids are major sources for LPA production. Metabolically, they are first converted into lysophospholipids, such as lysophosphatidylethanolamine (LPE), lysophosphatidylcholine (LPC), lysophosphatidylserine (LPS), and then, subsequently cleaved by ATX, also known as ectonucleotidepyrophosphatases/ phosphodiesterases-2. This pathway is considered a determinant for the LPA level in plasma [19,20].
LPA is converted by several classes of enzymes, including lipid phosphate phosphatases (LPPs), LPA acyltransferase, and phospholipases [21,22,23], as shown in Figure 1. LPPs (LPP1, LPP2, and LPP3) exist extracellularly and intracellularly in the endoplasmic reticulum and Golgi, and dephosphorylate LPA and degrade it into monoacylglycerol (MAG). LPA can also be converted to PA by the action of the acylglycerophosphate acyltransferase (AGPAT) enzyme, also known as LPA acyltransferase [24]. The third alternative pathway for LPA degradation is mediated by the action of lysophospholipases, via formation of glycerol-3-phosphate [25].
3. LPA Receptors and Intracellular Signaling Pathways
LPA induces various cellular effects by binding to specific G protein-coupled LPA receptors (LPARs) and activates downstream intracellular signaling pathways, resulting in various physiological and pathophysiological responses [5]. Till now, six LPARs have been identified and classified into A rhodopsin-like G protein-coupled receptors [26]. They can be further grouped into two groups, according to their distinct protein homology, such as LPAR1 to LPAR3 belonging to the endothelial differentiation gene (Edg) family, and LPAR4 to LPAR6 belonging to the P2Y purinergic gene cluster [6,8]. These receptors have the ability to interact with at least one or more heterotrimeric G subunits, such as Gαi/o, Gαq/11, Gα12/13, and Gs [6]. LPAR1/Edg2 and LPAR2/Edg4 receptors couple with Gαi/o, Gαq/11, and Gα12/13. Once bound together, the complexes transduce extracellular signals into intracellular pathways through molecules, such as the Ras homologous (Rho) protein family of GTPases, phospholipase C (PLC), diacylglycerol (DAG), mitogen-activated protein kinase (MAPK), and phosphatidylinositol 3-kinase (PI3K)-protein kinase B (Akt). Activation of these receptors mostly ends up promoting cell proliferation, survival, and migration [27,28]. LPAR3/Edg7 couples with Gαi/o and Gαq/11, and participates in LPA-induced Ca2+ mobilization, PLC, adenylyl-cyclase inhibition, and MAPK activation [28]. LPAR4/GPR23/P2Y9 and LPAR5/GPR92 induce stress fiber formation and neurite retraction through Gα12/13 and downstream Rho/Rho-associated protein kinase (ROCK) pathway [29,30]. LPAR4 is known as the only LPA receptor that can increase intracellular cAMP accumulation by coupling to Gs [29]. LPAR5 interacts with Gαq/11 and increases intracellular Ca2+ levels [31]. LPA6/P2Y5 receptor binds to either Gαi/o or Gα12/13, and induces Rho-dependent alteration of cellular morphology [32,33]. In addition, other G protein-coupled receptors, including GPR35 [34] and P2Y10 [35], were also identified as LPARs, which induced Ca2+ responses by LPA stimulation. LPA can also bind to and activate non-GPCR targets, the receptor for advanced glycation end products (RAGE) [36], and the cation channel transient receptor potential vanilloid 1 (TRPV1) [37]. RAGE participates in LPA-induced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox), reactive oxygen species (ROS) induction, and activation of NFKB, serum response factor (SRF), PI3K, and Akt [38]. TRPV1 increases intracellular Ca2+ levels following LPA stimulation [37]. Another non-GPCR, peroxisome proliferator-activated receptor gamma (PPARγ), is the intracellular receptor for LPA, and is critically important for mediating the effects of LPA on vascular remodeling [39]. Among them, the expression of LPAR1-4 has been detected in renal tissue [40]. Although many LPA receptors and their signaling pathways have been identified, as shown in Figure 2, the functional role of each receptor is poorly understood. Further studies will be required.
4. Pathogenesis of DN
4.1. Definition of Chronic Kidney Disease
Kidney disease is a heterogeneous group of disorders affecting kidney structure and function, which can be further classified into two distinct syndromes, acute and chronic kidney disease (CKD) [6]. Acute kidney injury is characterized by rapid diminution of kidney function, resulting in excretion of creatinine [41]. It occurs over several hours or days but not more than three months, whereas chronic kidney injury is in cases of more than three months. Acute kidney injury often leads to the development of chronic kidney failure [41]. Conversely, chronic kidney disease increases the risk of incidence of acute kidney injury [42,43]. Whatever the initial injury, the final pathogenesis of CKD involves renal fibrosis, characterized by excessive extracellular matrix (ECM) accumulation in glomerular and tubular interstitial cells [44,45]. Renal fibrosis changes tissue architecture and function, which leads to kidney dysfunction and failure.
4.2. Glomerulosclerosis
Glomerulosclerosis is hardening of glomeruli in the kidney and is frequently referred to as focal segmental glomerulosclerosis or nodular glomerulosclerosis [46]. The initial pathogenic events start in glomerular endothelium, including the activation and dysfunction of endothelial cells, hemodynamic alterations, as well as loss of glomerular basement membrane (GBM) electrical charge. These alterations subsequently trigger an inflammatory response in the glomerular apparatus, which further affects mesangial cells and induces proliferation and dysfunction of mesangial cells [43,47]. Excessive ECM production is the underlying mechanism for mesangial expansion. Podocytes are the cells located inside the Bowman’s capsule of the kidney, which anatomically wrap around capillaries of the glomerulus [48]. The damage in podocytes decreases their number and increases foot process effacement and denudation of GBM. The accumulated damage increases cell death by apoptosis or necrosis, and replacement of glomerular cells by ECM, which, in turn, increases hardening of glomeruli and eventually leads to glomerulosclerosis [16,44].
4.3. Tubular Interstitial Fibrosis
Although the initial glomerular damage and glomerulosclerosis are the major factors for the development of CKDs, the fibrosis in the tubulo-interstitial compartment is also a major contributor [43,49]. Initial glomerular damage increases the leakage of abnormally filtered proteins, including albumin, complement, and cytokines. These proteins and immune effectors or molecules activate tubular cells and initiate an inflammatory response. Simultaneously, different types of inflammatory cells (macrophages, monocytes, lymphocytes, mast cells, and dendritic cells) are recruited from circulation by increasing the expression of chemokines (monocyte chemoattractant protein 1, CCL2/MCP1; regulated upon activation, normal T cell expressed, and secreted, CCL5/RANTES), and adhesion molecules (E and P selectins, intercellular adhesion molecule-1, ICAM-1; vascular cell adhesion molecule-1, VCAM-1) [50]. In particular, the recruited monocytes transdifferentiate into macrophages in the kidney interstitium. Myofibroblasts are responsible for producing fibrillar matrix in the renal interstitium [51]. Under normal conditions, a lower population of myofibroblasts is maintained. However, they are activated, and their number is increased through various ways (proliferation, epithelial mesenchymal transition (EMT), and recruitment of circulating fibrocytes derived from bone marrow cells) in a pathological environment. In addition, accumulation of ECM proteins within the kidney interstitium is the consequence of imbalance between synthesis and degradation of ECM. Thus, the expression of plasminogen activator inhibitor-1 (PAI-1, a specific inhibitor of urokinase-type and tissue-type plasminogen activator (uPA and tPA)) and tissue inhibitor of metalloproteinase 1 (tissue inhibitor matrix metalloproteinase-1, TIMP-1; a specific inhibitor of MMPs) are increased during fibrosis, resulting in the inhibition of ECM degradation [52,53,54]. Beyond the abovementioned pathological events, hemodynamic alterations also reduce post-glomerular blood flow to peritubular capillaries, which, in turn, leads to hypoxic conditions and damage to tubular epithelium [55].
5. Cellular Signaling Pathways Involved in Pathogenesis of DN
In the diabetic milieu, such as hyperglycemia and dyslipidemia, metabolic alteration increases the production of ROS mainly through the Polyol and/or Nox pathway [56,57]. In addition, an increased intracellular glucose level increases AGE production and ROS by activating the AGEs receptor (RAGE) axis [58]. These abnormal metabolic and physiological consequences directly induce both vascular endothelial cell dysfunction and hemodynamic alterations, including activation of the renin–angiotensin system. Subsequently, this impact triggers a number of cellular signaling cascades, including the protein kinase c (PKC), mitogen-activated protein kinases (MAPKs; p38 and c-Jun N-terminal kinases (JNK)), Janus kinase/signal transducer and activator of transcription protein (JAK/STAT), and transforming growth factor (TGF)-β/Smad, thereby inducing a cellular response via activation of key transcription factors, such as NF-κB [56,57]. In particular, several vasoactive factors (angiotensin II, thromboxane, and endothelin-1) enhance their fibrotic action in diabetic renal diseases via secondary induction of TGF-β expression [56,59].
The Smad signaling pathway has also been reported to be associated with the renal hypertrophy and accumulation of ECM molecules through TGF-β signaling in DN [56,60,61]. Accumulation of AGEs within the glomerular apparatus, mesangial matrix, and tubular cells also directly causes serious alteration of diabetic kidney structure by reacting with plasma proteins and extra vascular proteins. As a consequence, AGEs lead to the transcriptional upregulation of TGF-β1, IL-6, and NF-κB, possibly via activation of PKC and/or oxidative stress [59,62]. PKCs are important players for the onset and progression of DN via hyperglycemia-induced upregulation of vascular endothelial growth factor (VEGF) expression in the mesangial cells [63]. In response to such molecules or signals, chemokines, growth factors, and profibrotic factors are ultimately upregulated in renal cells, such as tubular epithelial cells, podocytes, and mesangial cells, which contribute to cellular injury, progressive fibrosis, and loss of glomerular filtration rate, thereby increasing proteinuria during the development and progression of DN [56,57].
Recently, new molecules or potential signaling pathways have been added to the established pathways, which has made the pathogenesis of DN more complex. However, new observations simultaneously increase the possibility of identifying new targets for the treatment of DN. Interleukin (IL)-33-mediated suppression of the tumorigenicity 2 receptor (ST2) axis is one such pathway, which is very attractive due to its association with inflammatory response. IL-33 was first identified as a member of the IL-1 family in 2005 [64]. IL-33 acts through ST2 [65], which plays an important role in the immunity against pathogens, type 2 inflammation, tissue homeostasis, and repair [66,67]. Soluble ST2 level elevates in the serum of CKD and is associated with the severity of the disease [68], possibly suggesting its role in the development of CKD. However, these studies are very limited.
Sodium–glucose cotransporter (SGLT)-2 may be a potential contributor to CKD. Under normal physiological conditions, urine does not contain glucose, owing to its effective reabsorption by two transporters, SGLT-1 and SGLT-2 [69]. The SGLT-2 transporter is located on the luminal side of the first segment of the proximal tubule in the kidney. It is a high-capacity, low-affinity transporter, but is responsible for the reabsorption of approximately 90% of all filtered glucose [70]. SGLT-2 inhibitor induces glycosuria and natriuresis by blocking reabsorption of glucose and sodium in the proximal tubule [71]. As a consequence, afferent arteriolar vasoconstriction is induced, thereby reducing intraglomerular pressure, decreasing hyperfiltration, and improving renal function [72].
Current evidence also suggests that autophagy is critical in kidney physiology and homeostasis. In the diabetic milieu, increased oxidative stress, inflammation, and mitochondrial dysfunction modulate the autophagy activation and inhibition as well as lead to cellular recycling dysfunction [73]. Reduction of autophagy induces loss of podocytes, damage in proximal tubular cells, and glomerulosclerosis [74,75]. High glucose treatment has activated autophagy in podocytes and protected the podocytes from hyperglycemia-related apoptosis [76]. Similarly, autophagy related 5 (Atg5)-knockout diabetic mice (deficiency of autophagy activation) showed more severe proteinuria and impaired renal function [77,78]. Autophagy also protects mesangial cells from apoptosis induced by TGF-β1 via transforming growth factor β-activated kinase (TAK)1 and PI3K–AKT-dependent pathways [79]. Several studies have consistently indicated that DN is associated with decreased autophagy and increased apoptosis [80,81,82].
6. Chronic Kidney Injury and LPA–LPAR Axis
CKD has been becoming a major public health problem globally. It involves a progressive loss of kidney function over a period of months or years, which frequently leads to end-stage renal failure [1]. The main risk factors of CKD include high blood pressure, diabetes, cardiac disease, and a family history of kidney failure with genetic problems [83]. The potential role of LPA in the pathogenesis of kidney disease was suggested for the first time following the discovery of a positive correlation between circulation LPA levels and renal dysfunction in patients with kidney disorder at the end of the 1990s [84]. Later on, Grove KJ et al. also reported that the LPA level was elevated in glomeruli of eNOS (–/–) db/db mice, a robust model of DN [85]. Furthermore, LPA induced renal tubulointerstitial fibrosis, a classical hallmark of CKD, in a mouse model of unilateral ureteral obstruction [86]. More recently, LPA was identified as a biomarker of CKD in metabolic screening assay, using plasma samples from CKD patients with diverse etiologies and two different CKD rodent models [87]. Consistent with results of other studies, the LPA and LPC forms (16:0, 18:0, 18:1, and 18:2) were also detected in urine of DN patients [88]. On the contrary, several studies showed similar or reduced LPA levels in the plasma, but an increased urinary LPA level in CKD patients and animal models, suggesting that the local expression levels of LPA and LPARs are more important for the development of CKD [88,89,90]. LPA regulates various biological responses by binding to G-protein–coupled receptors (LPAR1–LPAR6) [5].
Recently, we and another group reported that the LPAR1 and/or LPAR3 were upregulated in different DN mouse models [91,92,93]. Treatment with a dual LPAR1/3 antagonist (Ki16425 or BMS002) or AM095 (a novel antagonist of LPAR1) reduced renal injury in the db/db mice (leptin receptor-deficient mouse with human type 2 diabetic phenotype) and streptozotocin (STZ)-induced diabetic mice (pancreatic beta cell-destroyed mouse with human type 1 diabetic phenotype) [91,92,93]. The expression of ATX and LPA production increases and activates the LPAR1-mediated signaling pathway in mesangial cells’ exposure to high-glucose media and in the kidney cortex of diabetic db/db mice [91]. LPA–LPAR1 activation increases the phosphorylation of glycogen synthase kinase (GSK)3β at serine 9 residue (Ser9) and induces translocation of sterol regulatory element-binding protein (SREBP) 1 into the nucleus [91]. Subsequently, it induces TGF-β expression, which contributes to the development of glomerular injury in db/db mice. However, treatment of ki16425 reduces proteinuria, glomerular tuft area and volume, and mesangial matrix expansion by regulating the LPA–GSK3β–SREBP1 axis [91]. In line with our observation, LPAR1 deletion in unilateral ureteral obstruction-induced mice prevents renal fibrosis by suppressing the expression of connective tissue growth factor in proximal tubular epithelial cells [86]. Additionally, Diao et al. reported that LPAR3 deletion affects the spatiotemporal expression of collagen types I, III, IV, and VI in peri-implantation of the mouse uterus [94]. All of these studies suggest that the LPA–LPAR axis regulates renal fibrosis differently for different kidney cell types or different tissues, and that at least two receptors, LPAR1 and LPAR3, might be important contributors to renal fibrosis.
Similarly, Zhang et al. showed improvements in renal function, using a different dual LPAR1/3 antagonist, BMS002, in the endothelial nitric oxide synthase-deficient (eNOS (–/–)) db/db mouse [92]. The expressions of ATX, LPAR1, and LPAR3 proteins in these mice significantly increased in the glomerular podocytes and tubular epithelial cells in the renal cortex [92]. Blockading the LPAR1/3 activity ameliorated glomerular injury and reverses kidney dysfunction by reducing podocyte loss and increased the phosphorylation of Akt2 (known to be essential for maintaining podocyte viability and function) [95], which prevented reduction in the glomerular filtration rate and reduced proteinuria without affecting blood pressure [92]. However, the underlying molecular mechanism in their study was not fully addressed and further studies will be needed.
Furthermore, our studies showed that increased LPA levels in STZ-induced diabetic mice increases Toll-like receptor (TLR) 4 expression and directly activates the NF-κB, the master transcription factor responsible for inflammatory cytokine expression. Simultaneously, activated TLR4 also produces ROS through the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase system [93]. All these intracellular changes synergistically activate NF-κB and/or JNK by increasing phosphorylation of p65 and JNK [93]. Ultimately, it leads to renal fibrosis via upregulation of pro-inflammatory cytokines and fibrotic factors, including TGF-β1, TIMP-1, and fibronectin. In contrast, AM095 treatment attenuates DN by downregulation of these signaling pathways in vivo. In mesangial cells, LPA treatment activates these signaling pathways similar to that in mice; however, this degree of activation was reduced by AM095 treatment [93]. We also observed some beneficial effects of AM095 treatment on glycemic control. However, the underlying mechanism should be addressed in future studies.
Mesangial cell proliferation and accumulation in the pathogenesis of DN is a major risk factor contributing to glomerulosclerosis. LPA treatment increased the proliferation of mouse mesangial cells (SV40 MES13), concomitant with the increased expression levels of cyclin D1 and CDK4 and decreased expression of p27Kip1. The expression of Krüppel-like factor 5 (KLF5) was upregulated by activating MAPK and elevating the expression of early growth response 1 (Egr1) in the kidney cortex of db/db mice and LPA-treated SV40 MES13 cells. Moreover, LPA significantly increased the activity of Rac1 GTPase in SV40 MES13 cells, and the dominant–negative form of Rac1 blunted the phosphorylation of p38, the upregulation of Egr1, and the LPA-mediated induction of KLF5, indicating that the downstream pathway of Rac1 was involved in LPA-induced mesangial cell proliferation [96]. Taken together, these observations suggested that the Rac1/MAPK/KLF5 signaling pathway is one of the underlying mechanisms contributing to glomerular hyper proliferation during the progression of DN. Recently, Guo et al. showed that LPA activates β-catenin, a downstream mediator of Wnt signaling, in colon cancer cells and KLF5 plays a critical role in β-catenin activation [97]. Several studies directly indicated that the Wnt/β-catenin signaling pathway was implicated in renal fibrosis and apoptosis in CKD models [98,99]. Thus, the regulation of these pathways may provide a potential therapeutic target for the treatment of DN.
Some studies have also suggested that LPA may bind to the receptor for RAGE, a member of the immunoglobulin superfamily [36,100]. Under in vitro conditions, this interaction is essential for LPA-induced ovarian tumor implantation and metastasis, as well as for LPA-mediated proliferation and migration of vascular smooth muscle cells [36]. In addition, LPA failed to activate vascular Akt signaling in mice that were administered soluble RAGE or genetic deletion of RAGE [36]. These observations indicated that RAGE-mediated signal transduction may play an important role in diabetic microvascular complication, including DN. Zaslaysky et al. also revealed that three major LPARs (LPAR1-3) form homo- or hetero-dimers within the LPAR subgroup and hetero-dimers with sphingosine 1 phosphate receptor (S1PR), pH-sensing G protein-coupled receptor (GPR4), and ovarian cancer G-protein coupled receptor-1 (OGR1/GPR68) [101], thereby activating the downstream signaling pathways linked to inflammation and fibrosis. S1PR signaling activation promotes renal fibrosis in the diabetic condition [102,103,104]. In the terminal ileum of inflammatory bowel diseases, the expression of OGR1/GPR68 positively increases the expression of pro-fibrotic genes and collagen deposition, suggesting the potential involvement of these receptors, signaling for the pathogenesis of DN [105]. However, there is no direct evidence connecting DN pathogenesis with dimer formation between LPARs and these receptors. Another potential LPA receptor is the TRPV1 ion channel [37]. However, there have been no reports demonstrating its role in DN.
7. Conclusions and Future Research Directions
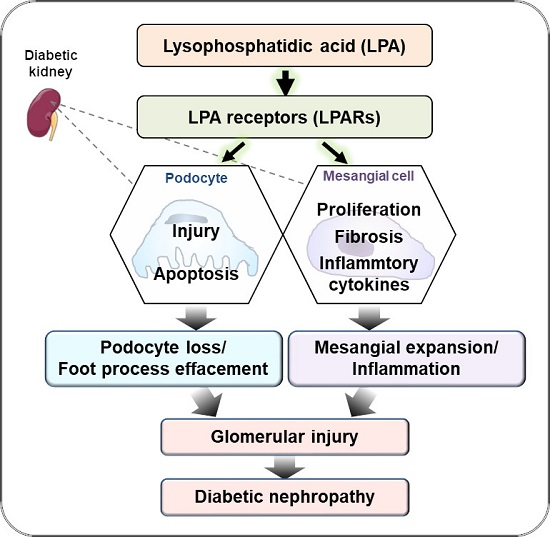
DN is the main risk factor for chronic kidney diseases, which mostly progress to the development of ESRD in the end. The best treatment regimen for DN is kidney replacement; however, it is highly restricted, due to various reasons, such as rare donors and rejection of the transplanted organ. Several medications are currently available for DN, but they are not sufficient for the recovery from kidney injury and restoration of kidney function in DN patients. Alternative drugs are thus required. Accumulated evidence indicates that the LPA–LPAR axis plays an important role in inducing pathological alterations of cell structure and function in the kidneys. Current studies showed that LPAR antagonism using pharmacological inhibitors significantly decreases the abnormality of kidney structure, such as GBM thickness, and increases of renal function, such as reducing proteinuria in different types of diabetic mouse models by regulating several signaling pathways, as shown in Figure 3.
The currently available data suggest that LPA signaling may regulate fibrosis, proliferation, and the inflammatory response in mesangial cells and podocytes or induce apoptosis via the following signaling pathways: 1) PI3K-AKT-GSK3β-TGF-β axis for fibrosis; 2) Rac1GTPase-MAPK-KLF5-CDK4/Cyclin D1 axis for proliferation; 3) TLR4-NADPH oxidase-ROS–NF-KB/MAPK or TLR4-NF-KB/MAPK axis for inflammatory response; 4) PI3K-AKT-GSK3α axis for apoptosis; 5) LPA-RAGE axis for glomerular injury; and 6) Wnt/β-catenin axis for fibrosis and apoptosis. For further details, please see above. LPAR: LPA receptor, RAGE: AGE receptor, Rac1: Ras-related C3 botulinum toxin substrate 1, NADPH: Nicotinamide adenine dinucleotide phosphate oxidase, TLR4: Toll-like receptor 4, NF-KB: Nuclear factor-KB, KLF5: Krüppel-like factor 5.
However, there are still many unanswered questions. Further studies should be performed in the following directions: Since the kidney consists of heterogeneous cells (such as podocytes, mesangial microvascular endothelial cells, and renal proximal tubule epithelial cells) and the LPAR expression level may be different in different cell types, further studies should first determine the LPAR expression level under different physiological and pathophysiological conditions. In addition, the studies should investigate the effects of LPAR antagonism on each type of kidney cell, using not only pharmacological inhibitors, but also genetically engineered in vivo models. Moreover, there are many different forms of LPA, which exhibit different affinities for each LPAR type. Each LPAR also exhibits various interaction affinities with one or more G protein subunits, thereby promoting different biological effects. These aspects should be further elucidated. Furthermore, the LPA–LPAR-mediated signaling pathway is also modulated by a cross-talk with other signaling pathways, such as epidermal growth factor (EGF) or vasoactive (angiotensin II) receptor-mediated signals [106], which will be defined in future studies. Since LPA regulates local blood flow, systemic blood pressure, and platelet function, and these factors directly play a critical role in renal function [40,107], future studies need to investigate the effect of LPA on the physiological and pathophysiological conditions in the renal vasculature and the tubular system. In addition, there are no studies demonstrating the relationship between the LPA–LPAR axis and the IL-33-ST2 axis and SGLT2 and autophagy. These new avenues should also be investigated in the future. Further understanding of the precise mechanisms underlying LPA action under physiological and pathophysiological conditions may facilitate the development of new therapeutic targets for DN.
Author Contributions
J.H.L. and H.-S.J. collected information and wrote the manuscript; J.H.L, D.K, Y.S.O, and H.-S.J. edited and revised the manuscript; J.H.L. and H.-S.J. approved the final version of the manuscript.
Funding
This work was supported by the Ministry of Education of Korea under a Basic Science Research Program Grant of the National Research Foundation of Korea (NRF-2016R1A2B2013347, NRF-2018R1C1B6000998, NRF-2017R1D1A1B03036210).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| DN | Diabetic nephropathy |
| ESRD | End-stage renal disease |
| ACE | Angiotensin converting enzyme |
| ARBs | Angiotensin II receptor blockers |
| LPA | Lysophosphatidic acid |
| ATX | Autotaxin |
| AGE | Advanced glycation end (product) |
| GPAT | Glycerol-3-phosphate acyltransferase |
| PLA1 and PLA2 | Phospholipases A1 or A2 |
| PA | Phosphatidic acid |
| LPE | Lysophosphatidylethanolamine |
| LPC | Lysophosphatidylcholine |
| LPS | Lysophosphatidylserine |
| LPPs | Lipid phosphate phosphatases |
| MAG | Monoacylglycerol |
| AGPAT | Acylglycerophosphate acyltransferase |
| PLC | Phospholipase C |
| DAG | Diacylglycerol |
| MAPK | Mitogen-activated protein kinase |
| PI3K | Phosphatidylinositol 3 kinase |
| PKB | Protein kinase B |
| ROCK | Rho-associated protein kinase |
| RAGE | Receptor for advanced glycation end products |
| Nox | NADPH oxidase |
| PPARγ | Peroxisome proliferator-activated receptor γ |
| CKD | Chronic kidney disease |
| ECM | Extracellular matrix |
| GBM | Glomerular basement membrane |
| EMT | Epithelial mesenchymal transition |
| CCL2/MCP1 | Monocyte chemoattractant protein 1 |
| CCL5/RANTES | Regulated upon activation, normal T cell expressed, and secreted |
| ICAM-1 | Intercellular adhesion molecule-1 |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| PAI-1 | Plasminogen activator inhibitor-1 |
| uPA and tPA | Urokinase-type and tissue-type plasminogen activator |
| TIMP-1 | Tissue inhibitor matrix metalloproteinase-1 |
| JNK | c-Jun N-terminal kinases |
| VEGF | Vascular endothelial growth factor |
| SGLT-2 | Sodium–glucose cotransporter-2 |
| GSK3β | Glycogen synthase kinase 3β |
| SREBP 1 | Sterol regulatory element-binding protein1 |
| TGF-β | Transforming growth factor-β |
| TLR 4 | Toll-like receptor 4 |
| NF-KB | Nuclear factor-KB |
| KLF5 | Krüppel-like factor 5 |
| Egr1 | Early growth response 1 |
| S1PR | Sphingosine 1 phosphate receptor |
| GPR4 | pH-sensing G protein-coupled receptor |
| OGR1/GPR68 | Ovarian cancer G-protein coupled receptor-1 |
References
- Gheith, O.; Farouk, N.; Nampoory, N.; Halim, M.A.; Al-Otaibi, T. Diabetic kidney disease: World wide difference of prevalence and risk factors. J. Nephropharmacol. 2016, 5, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic kidney disease: Challenges, progress, and possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Dounousi, E.; Duni, A.; Leivaditis, K.; Vaios, V.; Eleftheriadis, T.; Liakopoulos, V. Improvements in the management of diabetic nephropathy. Rev. Diabet. Stud. 2015, 12, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Bash, L.D.; Selvin, E.; Steffes, M.; Coresh, J.; Astor, B.C. Poor glycemic control in diabetes and the risk of incident chronic kidney disease even in the absence of albuminuria and retinopathy: Atherosclerosis risk in communities (ARIC) study. Arch. Intern. Med. 2008, 168, 2440–2447. [Google Scholar] [CrossRef] [PubMed]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [PubMed]
- Park, F.; Miller, D.D. Role of lysophosphatidic acid and its receptors in the kidney. Physiol. Genom. 2017, 49, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, S.; Hashimoto, T.; Kano, K.; Aoki, J. Lysophosphatidic acid as a lipid mediator with multiple biological actions. J. Biochem. 2015, 157, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.W.; Mutoh, T.; Lin, M.E.; Teo, S.T.; Park, K.E.; Mosley, A.N.; et al. LPA receptors: Subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 157–186. [Google Scholar] [CrossRef]
- Budd, D.C.; Qian, Y. Development of lysophosphatidic acid pathway modulators as therapies for fibrosis. Future Med. Chem. 2013, 5, 1935–1952. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Wei, X.; Lu, S.; He, P. Autotaxin-lpa receptor axis in the pathogenesis of lung diseases. Int. J. Clin. Exp. Med. 2015, 8, 17117–17122. [Google Scholar] [PubMed]
- Rhee, S.Y.; Kim, Y.S. The role of advanced glycation end products in diabetic vascular complications. Diabetes Metab. J. 2018, 42, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Advanced glycation end products in the pathogenesis of chronic kidney disease. Kidney Int. 2018, 93, 803–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdes-Rives, S.A.; Gonzalez-Arenas, A. Autotaxin-lysophosphatidic acid: From inflammation to cancer development. Mediat. Inflamm. 2017, 2017, 9173090. [Google Scholar] [CrossRef]
- Aoki, J. Mechanisms of lysophosphatidic acid production. Semin. Cell Dev. Biol. 2004, 15, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Madiraju, S.R. Glycerolipid metabolism and signaling in health and disease. Endocr. Rev. 2008, 29, 647–676. [Google Scholar] [CrossRef] [PubMed]
- Mirzoyan, K. The Role of lpa in Kidney Pathologies. Ph.D. Thesis, Université Toulouse 3 Paul Sabatier, Toulouse, France, 2018. [Google Scholar]
- Nakajima, K.; Sonoda, H.; Mizoguchi, T.; Aoki, J.; Arai, H.; Nagahama, M.; Tagaya, M.; Tani, K. A novel phospholipase a1 with sequence homology to a mammalian sec23p-interacting protein, p125. J. Biol. Chem. 2002, 277, 11329–11335. [Google Scholar] [CrossRef] [PubMed]
- Richmond, G.S.; Smith, T.K. The role and characterization of phospholipase a1 in mediating lysophosphatidylcholine synthesis in trypanosoma brucei. Biochem. J. 2007, 405, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Aoki, J.; Inoue, A.; Okudaira, S. Two pathways for lysophosphatidic acid production. Biochim. Biophys. Acta 2008, 1781, 513–518. [Google Scholar] [CrossRef]
- Nakanaga, K.; Hama, K.; Aoki, J. Autotaxin—An LPA producing enzyme with diverse functions. J. Biochem. 2010, 148, 13–24. [Google Scholar] [CrossRef]
- Brindley, D.N.; Pilquil, C. Lipid phosphate phosphatases and signaling. J. Lipid Res. 2009, 50 (Suppl. 1), S225–S230. [Google Scholar] [CrossRef] [Green Version]
- Saba, J.D. Lysophospholipids in development: Miles apart and edging in. J. Cell. Biochem. 2004, 92, 967–992. [Google Scholar] [CrossRef] [PubMed]
- Kok, B.P.; Venkatraman, G.; Capatos, D.; Brindley, D.N. Unlike two peas in a pod: Lipid phosphate phosphatases and phosphatidate phosphatases. Chem. Rev. 2012, 112, 5121–5146. [Google Scholar] [CrossRef] [PubMed]
- Aguado, B.; Campbell, R.D. Characterization of a human lysophosphatidic acid acyltransferase that is encoded by a gene located in the class iii region of the human major histocompatibility complex. J. Biol. Chem. 1998, 273, 4096–4105. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Dennis, E.A. Mammalian lysophospholipases. Biochim. Biophys. Acta 1999, 1439, 1–16. [Google Scholar] [CrossRef]
- Pasternack, S.M.; von Kugelgen, I.; Al Aboud, K.; Lee, Y.A.; Ruschendorf, F.; Voss, K.; Hillmer, A.M.; Molderings, G.J.; Franz, T.; Ramirez, A.; et al. G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat. Genet. 2008, 40, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, N.; Kimura, Y.; Chun, J. A single receptor encoded by VZG-1/LPA1/EDG-2 couples to g proteins and mediates multiple cellular responses to lysophosphatidic acid. Proc. Natl. Acad. Sci. USA 1998, 95, 6151–6156. [Google Scholar] [CrossRef] [PubMed]
- Ishii, I.; Contos, J.J.; Fukushima, N.; Chun, J. Functional comparisons of the lysophosphatidic acid receptors, lp(a1)/vzg-1/edg-2, lp(a2)/edg-4, and lp(a3)/edg-7 in neuronal cell lines using a retrovirus expression system. Mol. Pharmacol. 2000, 58, 895–902. [Google Scholar] [CrossRef]
- Lee, C.W.; Rivera, R.; Dubin, A.E.; Chun, J. LPA(4)/GPR23 is a lysophosphatidic acid (LPA) receptor utilizing g(s)-, g(q)/g(i)-mediated calcium signaling and G(12/13)-mediated rho activation. J. Biol. Chem. 2007, 282, 4310–4317. [Google Scholar] [CrossRef]
- Noguchi, K.; Ishii, S.; Shimizu, T. Identification of P2Y9/GPR23 as a novel g protein-coupled receptor for lysophosphatidic acid, structurally distant from the edg family. J. Biol. Chem. 2003, 278, 25600–25606. [Google Scholar] [CrossRef]
- Lee, C.W.; Rivera, R.; Gardell, S.; Dubin, A.E.; Chun, J. GPR92 as a new G12/13- and GQ-coupled lysophosphatidic acid receptor that increases camp, lpa5. J. Biol. Chem. 2006, 281, 23589–23597. [Google Scholar] [CrossRef]
- Yanagida, K.; Masago, K.; Nakanishi, H.; Kihara, Y.; Hamano, F.; Tajima, Y.; Taguchi, R.; Shimizu, T.; Ishii, S. Identification and characterization of a novel lysophosphatidic acid receptor, P2Y5/LPA6. J. Biol. Chem. 2009, 284, 17731–17741. [Google Scholar] [CrossRef] [PubMed]
- Riaz, A.; Huang, Y.; Johansson, S. G-protein-coupled lysophosphatidic acid receptors and their regulation of akt signaling. Int. J. Mol. Sci. 2016, 17, 215. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Ota, R.; Shima, M.; Yamashita, A.; Sugiura, T. GPR35 is a novel lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2010, 395, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Shiraishi, A.; Tabata, K.; Fujita, N. Identification of the orphan gpcr, p2y(10) receptor as the sphingosine-1-phosphate and lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2008, 371, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Rai, V.; Toure, F.; Chitayat, S.; Pei, R.; Song, F.; Li, Q.; Zhang, J.; Rosario, R.; Ramasamy, R.; Chazin, W.J.; et al. Lysophosphatidic acid targets vascular and oncogenic pathways via rage signaling. J. Exp. Med. 2012, 209, 2339–2350. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Posadas, A.; Picazo-Juarez, G.; Llorente, I.; Jara-Oseguera, A.; Morales-Lazaro, S.; Escalante-Alcalde, D.; Islas, L.D.; Rosenbaum, T. Lysophosphatidic acid directly activates TRPV1 through a c-terminal binding site. Nat. Chem. Biol. 2011, 8, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Bowman, M.A.; Schmidt, A.M. The next generation of rage modulators: Implications for soluble rage therapies in vascular inflammation. J. Mol. Med. 2013, 91, 1329–1331. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, T.M.; Pontsler, A.V.; Silva, A.R.; St Hilaire, A.; Xu, Y.; Hinshaw, J.C.; Zimmerman, G.A.; Hama, K.; Aoki, J.; Arai, H.; et al. Identification of an intracellular receptor for lysophosphatidic acid (LPA): Lpa is a transcellular ppargamma agonist. Proc. Natl. Acad. Sci. USA 2003, 100, 131–136. [Google Scholar] [CrossRef]
- Pradere, J.P.; Gonzalez, J.; Klein, J.; Valet, P.; Gres, S.; Salant, D.; Bascands, J.L.; Saulnier-Blache, J.S.; Schanstra, J.P. Lysophosphatidic acid and renal fibrosis. Biochim. Biophys. Acta 2008, 1781, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Makris, K.; Spanou, L. Acute kidney injury: Definition, pathophysiology and clinical phenotypes. Clin. Biochem. Rev. 2016, 37, 85–98. [Google Scholar]
- Levey, A.S.; Levin, A.; Kellum, J.A. Definition and classification of kidney diseases. Am. J. Kidney Dis. 2013, 61, 686–688. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, T.D.; Holt, S.G.; Smith, E.R. Progression of tubulointerstitial fibrosis and the chronic kidney disease phenotype—Role of risk factors and epigenetics. Front. Pharmacol. 2017, 8, 520. [Google Scholar] [CrossRef] [PubMed]
- Ke, B.; Fan, C.; Yang, L.; Fang, X. Matrix metalloproteinases-7 and kidney fibrosis. Front. Physiol. 2017, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Chen, C.; Meng, T.; Zhang, W.; Zhou, Q. Resveratrol attenuates renal injury and fibrosis by inhibiting transforming growth factor-β pathway on matrix metalloproteinase 7. Exp. Biol. Med. 2016, 241, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.Z.; Kopp, J.B. Focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2017, 12, 502–517. [Google Scholar] [CrossRef]
- Humphreys, B.D. Mechanisms of renal fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Reiser, J.; Altintas, M.M. Podocytes. F1000 Res. 2016, 5. [Google Scholar] [CrossRef]
- Iwano, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. Curr. Opin. Nephrol. Hypertens. 2004, 13, 279–284. [Google Scholar] [CrossRef]
- Lee, S.B.; Kalluri, R. Mechanistic connection between inflammation and fibrosis. Kidney Int. Suppl. 2010, S22–S26. [Google Scholar] [CrossRef]
- Grgic, I.; Duffield, J.S.; Humphreys, B.D. The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr. Nephrol. 2012, 27, 183–193. [Google Scholar] [CrossRef]
- Ye, Y.; Vattai, A.; Zhang, X.; Zhu, J.; Thaler, C.J.; Mahner, S.; Jeschke, U.; von Schonfeldt, V. Role of plasminogen activator inhibitor type 1 in pathologies of female reproductive diseases. Int. J. Mol. Sci. 2017, 18, 1651. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Sun, L.; Xiao, L.; Han, Y.; Fu, X.; Xiong, X.; Xu, X.; Liu, Y.; Yang, S.; Liu, F.; et al. Insights into the mechanisms involved in the expression and regulation of extracellular matrix proteins in diabetic nephropathy. Curr. Med. Chem. 2015, 22, 2858–2870. [Google Scholar] [CrossRef] [PubMed]
- Hodgkins, K.S.; Schnaper, H.W. Tubulointerstitial injury and the progression of chronic kidney disease. Pediatr. Nephrol. 2012, 27, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.K.; Winocour, P.; Farrington, K. Mechanisms of disease: The hypoxic tubular hypothesis of diabetic nephropathy. Nat. Clin. Pract. Nephrol. 2008, 4, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, N.; Barma, S.; Konwar, N.; Dewanjee, S.; Manna, P. Mechanistic insight of diabetic nephropathy and its pharmacotherapeutic targets: An update. Eur. J. Pharmacol. 2016, 791, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Matoba, K.; Utsunomiya, K. Signaling pathways in diabetic nephropathy. Histol. Histopathol. 2016, 31, 1059–1067. [Google Scholar] [PubMed]
- Kanwar, Y.S.; Wada, J.; Sun, L.; Xie, P.; Wallner, E.I.; Chen, S.; Chugh, S.; Danesh, F.R. Diabetic nephropathy: Mechanisms of renal disease progression. Exp. Biol. Med. 2008, 233, 4–11. [Google Scholar] [CrossRef]
- Nowotny, K.; Jung, T.; Hohn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef]
- Sureshbabu, A.; Muhsin, S.A.; Choi, M.E. TGF-β signaling in the kidney: Profibrotic and protective effects. Am. J. Physiol. Ren. Physiol. 2016, 310, F596–F606. [Google Scholar] [CrossRef]
- Chang, A.S.; Hathaway, C.K.; Smithies, O.; Kakoki, M. Transforming growth factor-β1 and diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2016, 310, F689–F696. [Google Scholar] [CrossRef]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Tufro, A.; Veron, D. Vegf and podocytes in diabetic nephropathy. Semin. Nephrol. 2012, 32, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces t helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Li, Y.; Li, M. The potential role of IL-33/ST2 signaling in fibrotic diseases. J. Leukoc. Biol. 2015, 98, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Tonacci, A.; Quattrocchi, P.; Gangemi, S. IL33/ST2 axis in diabetic kidney disease: A literature review. Medicina 2019, 55, 50. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Wang, Y.; Niu, Z.; Wang, C.; Wang, R.; Zhang, Z.; Chen, T.; Wang, X.M.; Li, Q.; Lee, V.W.S.; et al. Potentiating tissue-resident type 2 innate lymphoid cells by IL-33 to prevent renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2018, 29, 961–976. [Google Scholar] [CrossRef]
- Bao, Y.S.; Na, S.P.; Zhang, P.; Jia, X.B.; Liu, R.C.; Yu, C.Y.; Mu, S.H.; Xie, R.J. Characterization of interleukin-33 and soluble ST2 in serum and their association with disease severity in patients with chronic kidney disease. J. Clin. Immunol. 2012, 32, 587–594. [Google Scholar] [CrossRef]
- Wilding, J.P. The role of the kidneys in glucose homeostasis in type 2 diabetes: Clinical implications and therapeutic significance through sodium glucose co-transporter 2 inhibitors. Metab. Clin. Exp. 2014, 63, 1228–1237. [Google Scholar] [CrossRef]
- Vallon, V.; Thomson, S.C. Targeting renal glucose reabsorption to treat hyperglycaemia: The pleiotropic effects of SGLT2 inhibition. Diabetologia 2017, 60, 215–225. [Google Scholar] [CrossRef]
- Sano, M.; Takei, M.; Shiraishi, Y.; Suzuki, Y. Increased hematocrit during sodium-glucose cotransporter 2 inhibitor therapy indicates recovery of tubulointerstitial function in diabetic kidneys. J. Clin. Med. Res. 2016, 8, 844–847. [Google Scholar] [CrossRef]
- Dekkers, C.C.J.; Gansevoort, R.T.; Heerspink, H.J.L. New diabetes therapies and diabetic kidney disease progression: The role of SGLT-2 inhibitors. Curr. Diabetes Rep. 2018, 18, 27. [Google Scholar] [CrossRef] [PubMed]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Bottinger, E.P. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.A.; Wu, V.C.; Wang, C.Y. Autophagy in chronic kidney diseases. Cells 2019, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, A.; Yasuda, M.; Kume, S.; Yamahara, K.; Nakazawa, J.; Chin-Kanasaki, M.; Araki, H.; Araki, S.; Koya, D.; Asanuma, K.; et al. Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy. Diabetes 2016, 65, 755–767. [Google Scholar] [CrossRef]
- Lenoir, O.; Jasiek, M.; Henique, C.; Guyonnet, L.; Hartleben, B.; Bork, T.; Chipont, A.; Flosseau, K.; Bensaada, I.; Schmitt, A.; et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 2015, 11, 1130–1145. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Xu, L.; Shi, Y.; Zhuang, S. Podocyte autophagy: A potential therapeutic target to prevent the progression of diabetic nephropathy. J. Diabetes Res. 2017, 2017, 3560238. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Zhu, J.; Chen, X.; Zha, D.; Singhal, P.C.; Ding, G. High glucose induces autophagy in podocytes. Exp. Cell Res. 2013, 319, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Kim, J.K.; Kim, S.I.; Na, H.J.; Jun, S.Y.; Lee, S.J.; Choi, M.E. TGF-β1 protects against mesangial cell apoptosis via induction of autophagy. J. Biol. Chem. 2010, 285, 37909–37919. [Google Scholar] [CrossRef]
- Yasuda-Yamahara, M.; Kume, S.; Tagawa, A.; Maegawa, H.; Uzu, T. Emerging role of podocyte autophagy in the progression of diabetic nephropathy. Autophagy 2015, 11, 2385–2386. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Choi, M.E. Autophagy in diabetic nephropathy. J. Endocrinol. 2015, 224, R15–R30. [Google Scholar] [CrossRef]
- Yamahara, K.; Kume, S.; Koya, D.; Tanaka, Y.; Morita, Y.; Chin-Kanasaki, M.; Araki, H.; Isshiki, K.; Araki, S.; Haneda, M.; et al. Obesity-mediated autophagy insufficiency exacerbates proteinuria-induced tubulointerstitial lesions. J. Am. Soc. Nephrol. 2013, 24, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Macisaac, R.J.; Ekinci, E.I.; Jerums, G. Markers of and risk factors for the development and progression of diabetic kidney disease. Am. J. Kidney Dis. 2014, 63, S39–S62. [Google Scholar] [CrossRef] [PubMed]
- Sasagawa, T.; Suzuki, K.; Shiota, T.; Kondo, T.; Okita, M. The significance of plasma lysophospholipids in patients with renal failure on hemodialysis. J. Nutr. Sci. Vitaminol. 1998, 44, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Grove, K.J.; Voziyan, P.A.; Spraggins, J.M.; Wang, S.; Paueksakon, P.; Harris, R.C.; Hudson, B.G.; Caprioli, R.M. Diabetic nephropathy induces alterations in the glomerular and tubule lipid profiles. J. Lipid Res. 2014, 55, 1375–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, N.; Chun, J.; Duffield, J.S.; Lagares, D.; Wada, T.; Luster, A.D.; Tager, A.M. Lysophosphatidic acid signaling through its receptor initiates profibrotic epithelial cell fibroblast communication mediated by epithelial cell derived connective tissue growth factor. Kidney Int. 2017, 91, 628–641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.H.; Chen, H.; Vaziri, N.D.; Mao, J.R.; Zhang, L.; Bai, X.; Zhao, Y.Y. Metabolomic signatures of chronic kidney disease of diverse etiologies in the rats and humans. J. Proteome Res. 2016, 15, 3802–3812. [Google Scholar] [CrossRef] [PubMed]
- Saulnier-Blache, J.S.; Feigerlova, E.; Halimi, J.M.; Gourdy, P.; Roussel, R.; Guerci, B.; Dupuy, A.; Bertrand-Michel, J.; Bascands, J.L.; Hadjadj, S.; et al. Urinary lysophopholipids are increased in diabetic patients with nephropathy. J. Diabetes Its Complicat. 2017, 31, 1103–1108. [Google Scholar] [CrossRef]
- Michalczyk, A.; Dolegowska, B.; Heryc, R.; Chlubek, D.; Safranow, K. Associations between plasma lysophospholipids concentrations, chronic kidney disease and the type of renal replacement therapy. Lipids Health Disease 2019, 18, 85. [Google Scholar] [CrossRef] [Green Version]
- Mirzoyan, K.; Baiotto, A.; Dupuy, A.; Marsal, D.; Denis, C.; Vinel, C.; Sicard, P.; Bertrand-Michel, J.; Bascands, J.L.; Schanstra, J.P.; et al. Increased urinary lysophosphatidic acid in mouse with subtotal nephrectomy: Potential involvement in chronic kidney disease. J. Phys. Biochem. 2016, 72, 803–812. [Google Scholar] [CrossRef]
- Li, H.Y.; Oh, Y.S.; Choi, J.W.; Jung, J.Y.; Jun, H.S. Blocking lysophosphatidic acid receptor 1 signaling inhibits diabetic nephropathy in db/db mice. Kidney Int. 2017, 91, 1362–1373. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.Z.; Wang, X.; Yang, H.; Fogo, A.B.; Murphy, B.J.; Kaltenbach, R.; Cheng, P.; Zinker, B.; Harris, R.C. Lysophosphatidic acid receptor antagonism protects against diabetic nephropathy in a type 2 diabetic model. J. Am. Soc. Nephrol. 2017, 28, 3300–3311. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Sarker, M.K.; Choi, H.; Shin, D.; Kim, D.; Jun, H.S. Lysophosphatidic acid receptor 1 inhibitor, am095, attenuates diabetic nephropathy in mice by downregulation of TLR4/NF-κB signaling and NADPH oxidase. Biochim. Biophys. Acta. Mol. Basis Dis. 2019, 1865, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Diao, H.; Aplin, J.D.; Xiao, S.; Chun, J.; Li, Z.; Chen, S.; Ye, X. Altered spatiotemporal expression of collagen types i, iii, iv, and vi in LPAR3-deficient peri-implantation mouse uterus. Biol. Reprod. 2011, 84, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Canaud, G.; Bienaime, F.; Viau, A.; Treins, C.; Baron, W.; Nguyen, C.; Burtin, M.; Berissi, S.; Giannakakis, K.; Muda, A.O.; et al. Akt2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat. Med. 2013, 19, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Li, H.Y.; Lee, J.H.; Oh, Y.S.; Jun, H.S. Lysophosphatidic acid increases mesangial cell proliferation in models of diabetic nephropathy via RAC1/MAPK/KLF5 signaling. Exp. Mol. Med. 2019, 51, 18. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; He, P.; No, Y.R.; Yun, C.C. Kruppel-like factor 5 incorporates into the β-catenin/TCF complex in response to LPA in colon cancer cells. Cell. Signal. 2015, 27, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zha, Y.; Zeng, X.Z.; Dong, R.; Wang, Q.H.; Wang, D.T. Role of the wnt/β-catenin signaling pathway in inducing apoptosis and renal fibrosis in 5/6-nephrectomized rats. Mol. Med. Rep. 2017, 15, 3575–3582. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.J.; Zhou, D.; Zhou, L.; Liu, Y. Wnt/β-catenin signaling and kidney fibrosis. Kidney Int. Suppl. 2014, 4, 84–90. [Google Scholar] [CrossRef]
- Manigrasso, M.B.; Juranek, J.; Ramasamy, R.; Schmidt, A.M. Unlocking the biology of rage in diabetic microvascular complications. Trends Endocrinol. Metab. 2014, 25, 15–22. [Google Scholar] [CrossRef]
- Zaslavsky, A.; Singh, L.S.; Tan, H.; Ding, H.; Liang, Z.; Xu, Y. Homo- and hetero-dimerization of LPA/S1P receptors, OGR1 and GPR4. Biochimi. Biophys. Acta 2006, 1761, 1200–1212. [Google Scholar] [CrossRef]
- Yaghobian, D.; Don, A.S.; Yaghobian, S.; Chen, X.; Pollock, C.A.; Saad, S. Increased sphingosine 1-phosphate mediates inflammation and fibrosis in tubular injury in diabetic nephropathy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, S.; Takahashi-Fujigasaki, J.; Kanazawa, Y.; Matoba, K.; Kawanami, D.; Yokota, T.; Iwamoto, T.; Tajima, N.; Manome, Y.; Utsunomiya, K. Sphingosine-1-phosphate induces differentiation of cultured renal tubular epithelial cells under rho kinase activation via the S1P2 receptor. Clin. Exp. Nephrol. 2014, 18, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Liu, W.; Lan, T.; Xie, X.; Peng, J.; Huang, J.; Wang, S.; Shen, X.; Liu, P.; Huang, H. Berberine reduces fibronectin expression by suppressing the S1P-S1P2 receptor pathway in experimental diabetic nephropathy models. PLoS ONE 2012, 7, e43874. [Google Scholar] [CrossRef]
- Hutter, S.; van Haaften, W.T.; Hunerwadel, A.; Baebler, K.; Herfarth, N.; Raselli, T.; Mamie, C.; Misselwitz, B.; Rogler, G.; Weder, B.; et al. Intestinal activation of ph-sensing receptor OGR1 [GPR68] contributes to fibrogenesis. J. Crohn Colitis 2018, 12, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Colin-Santana, C.C.; Avendano-Vazquez, S.E.; Alcantara-Hernandez, R.; Garcia-Sainz, J.A. Egf and angiotensin ii modulate lysophosphatidic acid LPA(1) receptor function and phosphorylation state. Biochim. Biophys. Acta 2011, 1810, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Ma, L.; Li, Y.; Wang, F.; Zheng, G.Y.; Sun, Z.; Jiang, F.; Chen, Y.; Liu, H.; Dang, A.; et al. Genetic and functional evidence supports lpar1 as a susceptibility gene for hypertension. Hypertension 2015, 66, 641–646. [Google Scholar] [CrossRef]
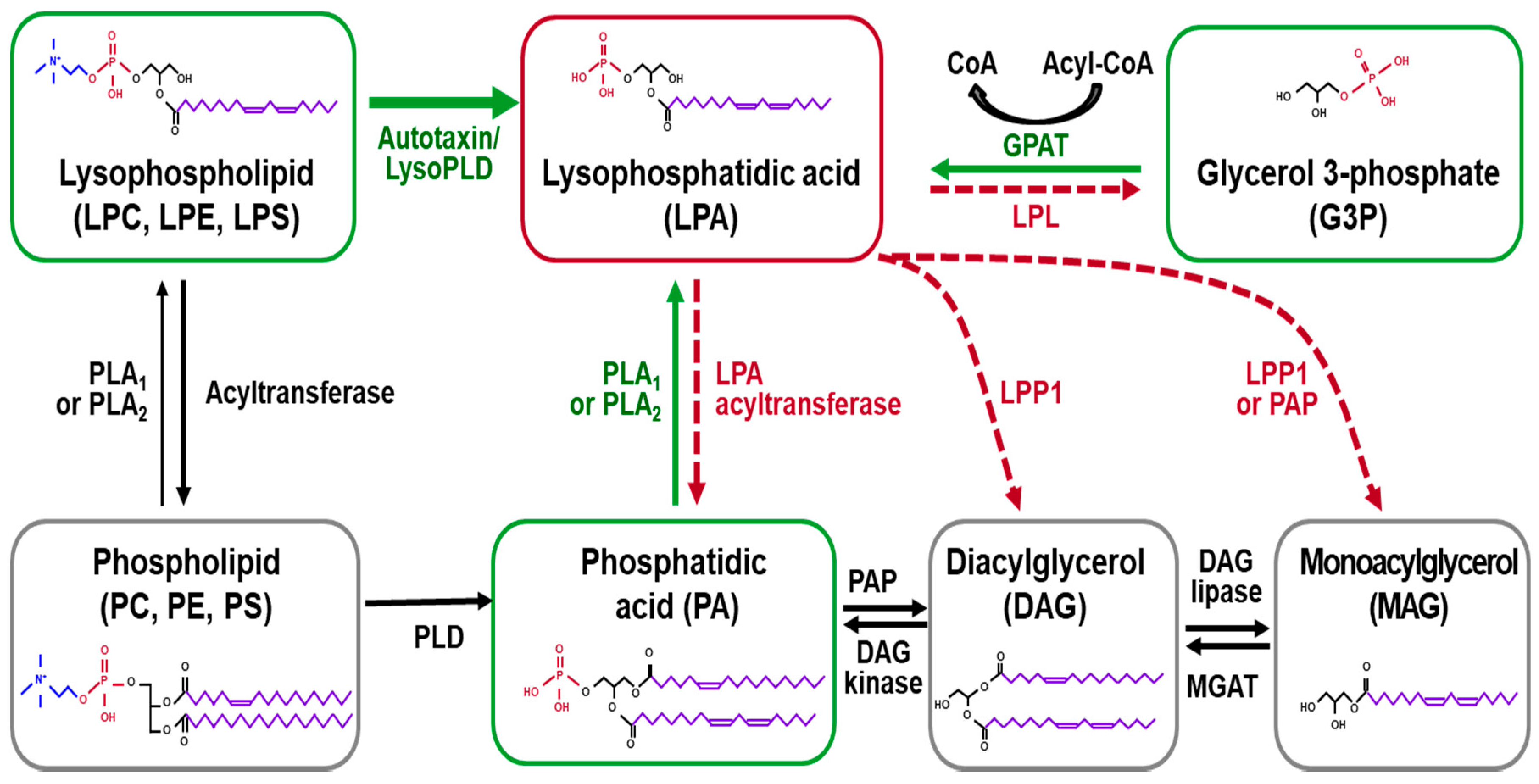
Figure 1.
The enzymatic pathways of lysophosphatidic acid (LPA) synthesis and degradation. LPA can be synthesized from different precursors, including lysophospholipids, phosphatidic acid, and glycerol 3-phosphate. The enzymes and pathways involved in LPA production are indicated using green text and a green solid line, respectively. LPA is converted into either monoacylglycerol or phosphatidic acid. The enzymes and pathways involved in LPA conversion are indicated using red text and a red dotted line, respectively. Lysophosphatidylcholine (LPC), lysophosphatidylethanolamine (LPE), lysophosphatidylserine (LPS), lysophospholipase D (Lyso PLD), lysophospholipase (LPL), glycerol 3-phosphate acyltransferase (GPAT), phospholipase C (PLC), phospholipase A1 or A2 (PLA1 and PLA2), diacylglycerol (DAG), monoacylglycerol (MAG), MAG acyltransferase (MGAT), lipid phosphate phosphatase 1 or 2 (LPP1 and LPP2), phosphatidate phosphatase (PAP), phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), and phospholipase D (PLD).
Figure 1.
The enzymatic pathways of lysophosphatidic acid (LPA) synthesis and degradation. LPA can be synthesized from different precursors, including lysophospholipids, phosphatidic acid, and glycerol 3-phosphate. The enzymes and pathways involved in LPA production are indicated using green text and a green solid line, respectively. LPA is converted into either monoacylglycerol or phosphatidic acid. The enzymes and pathways involved in LPA conversion are indicated using red text and a red dotted line, respectively. Lysophosphatidylcholine (LPC), lysophosphatidylethanolamine (LPE), lysophosphatidylserine (LPS), lysophospholipase D (Lyso PLD), lysophospholipase (LPL), glycerol 3-phosphate acyltransferase (GPAT), phospholipase C (PLC), phospholipase A1 or A2 (PLA1 and PLA2), diacylglycerol (DAG), monoacylglycerol (MAG), MAG acyltransferase (MGAT), lipid phosphate phosphatase 1 or 2 (LPP1 and LPP2), phosphatidate phosphatase (PAP), phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), and phospholipase D (PLD).

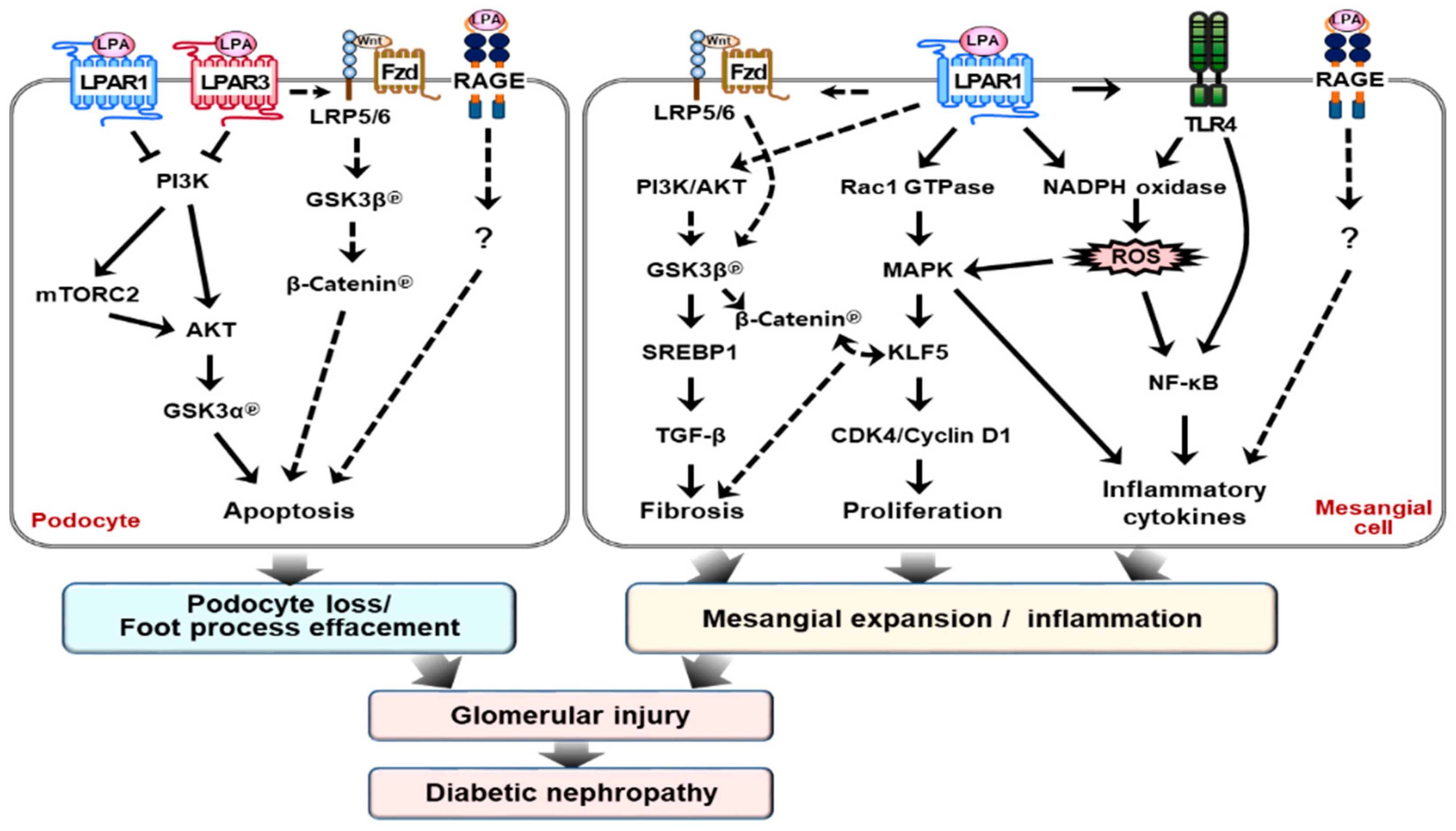
Figure 2.
LPA signaling pathways. LPA can induce multiple cellular effects via binding to specific GPCRs, including LPAR1–LPAR6, as well as binding to non-GPCRs, such as transient receptor potential vanilloid 1 (TRPV1), receptor for advanced glycation end products (RAGE), and intracellular peroxisome proliferator-activated receptor gamma (PPARγ). After binding to receptors, LPA activates downstream intracellular signaling pathways, thereby resulting in various physiological and pathophysiological responses, as described in detail in the text.
Figure 2.
LPA signaling pathways. LPA can induce multiple cellular effects via binding to specific GPCRs, including LPAR1–LPAR6, as well as binding to non-GPCRs, such as transient receptor potential vanilloid 1 (TRPV1), receptor for advanced glycation end products (RAGE), and intracellular peroxisome proliferator-activated receptor gamma (PPARγ). After binding to receptors, LPA activates downstream intracellular signaling pathways, thereby resulting in various physiological and pathophysiological responses, as described in detail in the text.

Figure 3.
Schematic representation of LPA signaling in diabetic nephropathy models.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, J.H.; Kim, D.; Oh, Y.S.; Jun, H.-S. Lysophosphatidic Acid Signaling in Diabetic Nephropathy. Int. J. Mol. Sci. 2019, 20, 2850. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112850
AMA Style
Lee JH, Kim D, Oh YS, Jun H-S. Lysophosphatidic Acid Signaling in Diabetic Nephropathy. International Journal of Molecular Sciences. 2019; 20(11):2850. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112850
Chicago/Turabian StyleLee, Jong Han, Donghee Kim, Yoon Sin Oh, and Hee-Sook Jun. 2019. "Lysophosphatidic Acid Signaling in Diabetic Nephropathy" International Journal of Molecular Sciences 20, no. 11: 2850. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112850
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.