Potential of Transcript Editing Across Mitogenomes of Early Land Plants Shows Novel and Familiar Trends


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Characteristics of Newly Sequenced Liverwort Mitogenomes
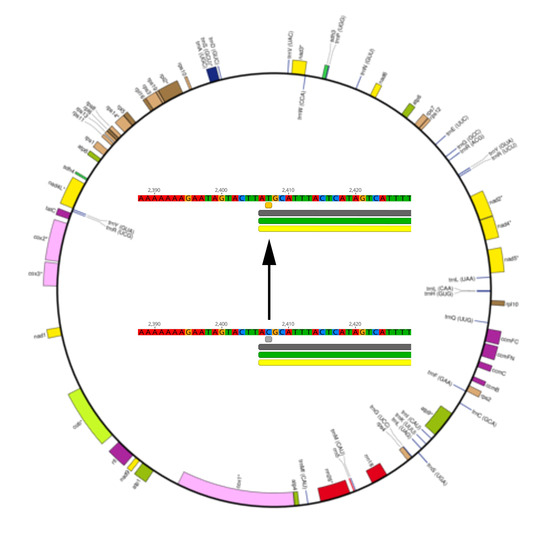
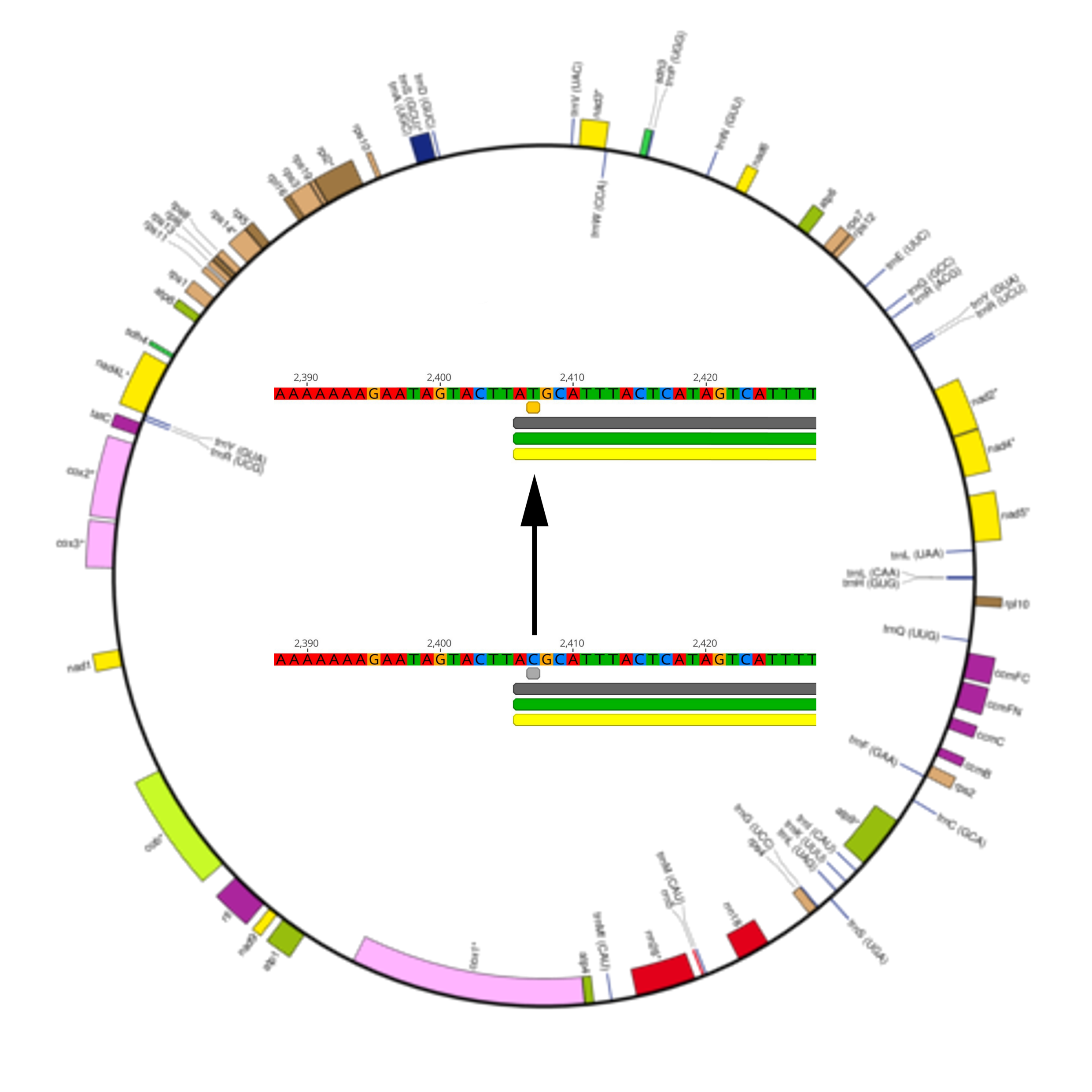
2.2. RNA Editing Predictions in Liverwort Mitochondria
2.3. RNA Editing Predictions in Moss Mitochondria
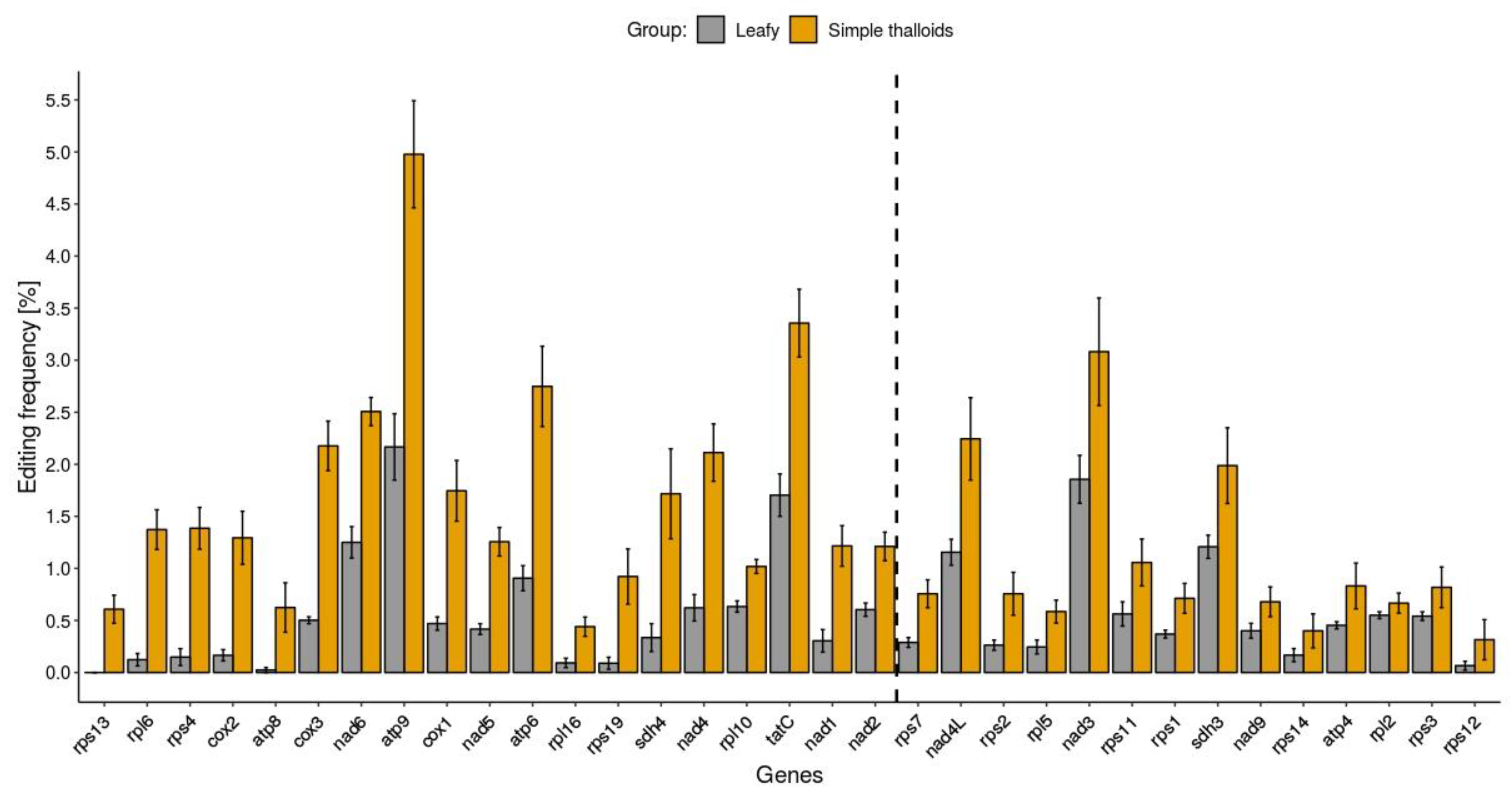
2.4. RNA Editing Across Mitochondrial Genes of Bryophytes
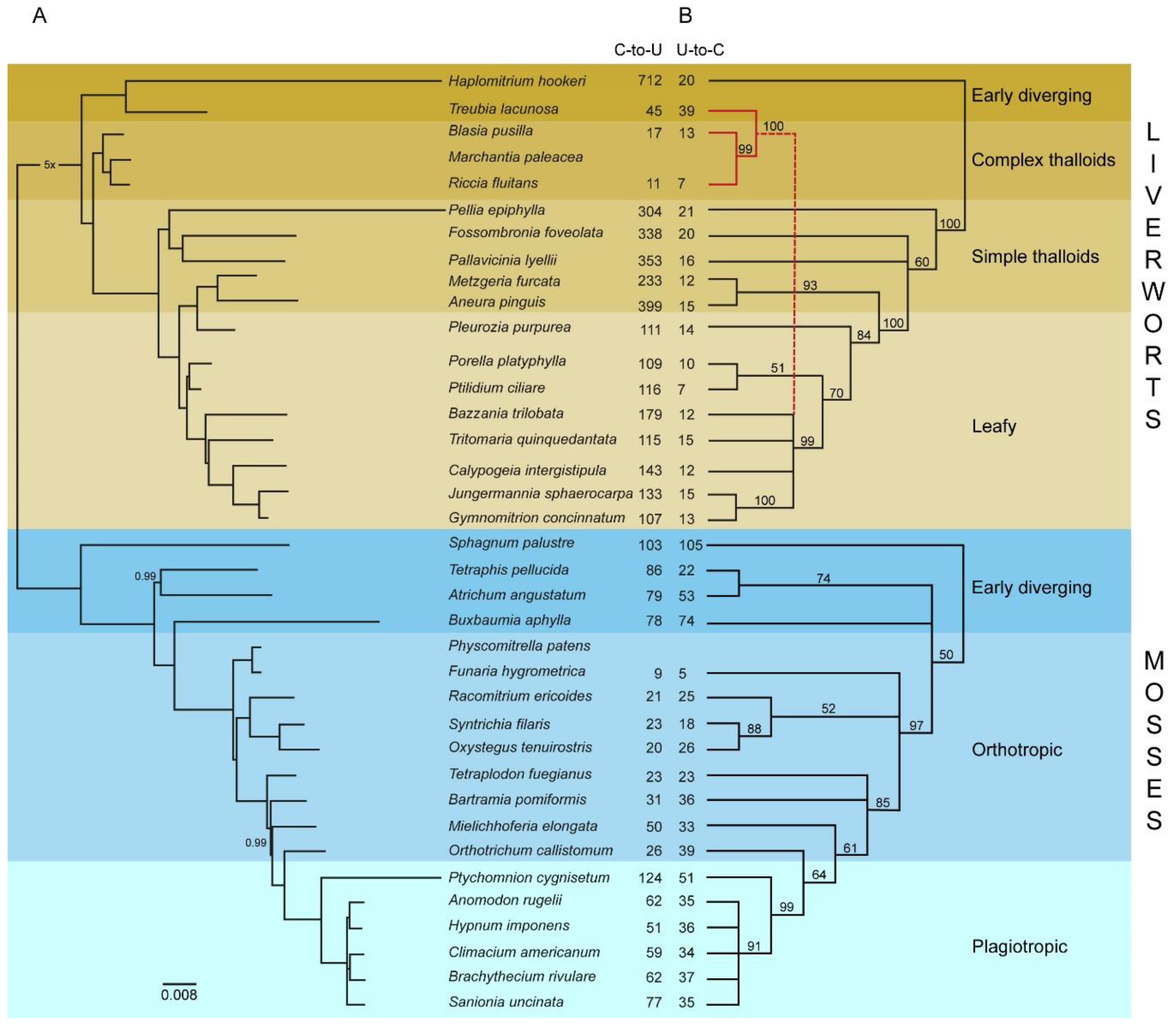
2.5. Phylogenetic Relationships Based on Protein-Coding Mitochondrial Genes
3. Materials and Methods
3.1. Material
3.2. Mitochondrial Genome Assembly
3.3. Prediction and Verification of RNA Editing Sites
3.4. Phylogenetic Analyses
3.5. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- An, N.A.; Ding, W.; Yang, X.; Peng, J.; He, B.Z.; Shen, Q.S.; Lu, F.; He, A.; Zhang, E.Y.; Tan, B.C.M.; et al. Evolutionarily significant A-to-I RNA editing events orginated through G-to-A mutations in primates. Genome Biol. 2019, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Teichert, I. Adenosine to inosine mRNA editing in fungi and how it may relate to fungal pathogenesis. PLoS Pathog. 2018, 14, e1007231. [Google Scholar] [CrossRef] [PubMed]
- Rüdinger, M.; Fritz-Laylin, L.; Polsakiewicz, M.; Knoop, V. Plant-type mitochondrial RNA editing in the protist Naegleria gruberi. RNA 2011, 17, 2058–2062. [Google Scholar] [CrossRef]
- Knoop, V. When you can’t trust the DNA: RNA editing changes transcript sequences. Cell. Mol. Life Sci. 2011, 68, 567–586. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, M.; Sugita, M. RNA editing and its molecular mechanism in plant organelles. Genes 2017, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, J.J. The emerging role of RNA editing in plasticity. J. Exp. Biol. 2015, 218, 1812–1821. [Google Scholar] [CrossRef] [Green Version]
- Maier, R.M.; Zeltz, P.; Kössel, H.; Bonnard, G.; Gualberto, J.M.; Grienenberger, J.M. RNA editing in plant mitochondrial and chloroplasts. Plant Mol. Biol. 1996, 32, 343–365. [Google Scholar] [CrossRef]
- Steinhauser, S.; Beckert, S.; Capesius, I.; Malek, O.; Knoop, V. Plant mitochondrial RNA editing. J. Mol. Evol. 1999, 48, 303–312. [Google Scholar] [CrossRef]
- Ruwe, H.; Castandet, B.; Schmitz-Linneweber, C.; Stern, D.B. Arabidopsis chloroplast quantitative editotype. FEBS Lett. 2013, 587, 1429–1433. [Google Scholar] [CrossRef]
- Meng, Y.; Chen, D.; Jin, Y.; Mao, C.; Wu, P.; Chen, M. RNA editing of nuclear transcripts in Arabidopsis thaliana. BMC Genom. 2010, 11, S12. [Google Scholar] [CrossRef]
- Börner, G.V.; Mörl, M.; Wissinger, B.; Brennicke, A.; Schmelzer, C. RNA editing of a group II intron in Oenotheraas a prerequisite for splicing. Mol. Gen. Genet. 1995, 246, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Castandet, B.; Choury, D.; Bégu, D.; Jordana, X.; Araya, A. Intron RNA editing is essential for splicing in plant mitochondria. Nucleic Acids Res. 2010, 38, 7112–7121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, K.; Hammani, K.; Tanz, S.K.; Peng, L.; Fukao, Y.; Myouga, F.; Motohashi, R.; Shinozaki, K.; Small, I.; Shikanai, T. The pentatricopeptide repeat protein OTP82 is required for RNA editing of plastid ndhB and ndhG transcripts. Plant J. 2010, 61, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Kudla, J.; Bock, R. RNA editing in an untranslated region of the Gingko chloroplast genome. Gene 1999, 234, 81–86. [Google Scholar] [CrossRef]
- Bentolila, S.; Oh, J.; Hanson, M.R.; Bukowski, R. Comprehensive high-resolution analysis of the role of an Arabidopsis gene family in RNA editing. PLoS Genet. 2013, 9, e1003584. [Google Scholar] [CrossRef] [PubMed]
- Rüdinger, M.; Volkmar, U.; Lenz, H.; Groth-Malonek, M.; Knoop, V. Nuclear DYW-Type PPR gene families diversify with increasing RNA editing frequencies in liverwort and moss mitochondria. J. Mol. Evol. 2012, 74, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Small, I. The evolution of RNA editing and pentatricopeptide repeat genes. New Phytol. 2011, 191, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Knie, N.; Grewe, F.; Fischer, S.; Knoop, V. Reverse U-to-C editing exceeds C-to-U RNA editing in some ferns–a monilophyte-wide comparison of chloroplast and mitochondrial RNA editing suggests independent evolution of the two processes in both organelles. BMC Evol. Biol. 2016, 16, 134. [Google Scholar] [CrossRef]
- Edera, A.A.; Gandini, C.L.; Sanchez-Puerta, M.V. Towards a comprehensive picture of C-to-U RNA editing sites in angiosperm mitochondria. Plant Mol. Biol. 2018, 97, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Myszczyński, K.; Górski, P.; Ślipiko, M.; Sawicki, J. Sequencing of organellar genomes of Gymnomitrion concinnatum (Jungermanniales) revealed the first exception in the structure and gene order of evolutionary stable liverworts mitogenomes. BMC Plant Biol. 2018, 18, 321. [Google Scholar] [CrossRef] [PubMed]
- Rüdinger, M.; Funk, H.T.; Rensing, S.A.; Maier, U.G.; Knoop, V. RNA editing: Only eleven sites are present in the Physcomitrella patens mitochondrial transcriptome and a universal nomenclature proposal. Mol. Genet. Genom. 2009, 281, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Bell, N.E.; Boore, J.L.; Brent, D.M.; Hyvönen, J. Organellar genomes of the four-toothed moss, Tetraphis pellucida. BMC Genom. 2014, 15, 383. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.L.; Li, L.B.; Wang, B.; Chen, Z.D.; Knoop, V.; Groth-Malonek, M.; Dombrovska, O.; Lee, J.; Kent, L.; Rest, J.; et al. The deepest divergences in land plants inferred from phylogenomic evidence. Proc. Natl. Acad. Sci. USA 2006, 103, 15511–15516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, C.J.; Li, B.; Foster, P.G.; Embley, T.M.; Civáň, P. Conflicting phylogenies for early land plants are caused by composition biases among synonymous substitutions. Syst. Biol. 2014, 63, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Wickett, N.J.; Mirarab, S.; Nguyen, N.; Warnow, T.; Carpenter, E.; Matasci, N.; Ayyampalayam, S.; Barker, M.S.; Gitzendanner, G.B.; Ruhfel, B.R. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. USA 2014, 111, e4859–e4868. [Google Scholar] [CrossRef]
- Puttick, M.N.; Morris, J.L.; Williams, T.A.; Cox, C.J.; Edwards, D.; Kenrick, P.; Pressel, S.; Wellman, C.H.; Schneider, H.; Pisani, D.; et al. The interrelationships of land plants and the nature of the ancestral embryophyte. Curr. Biol. 2018, 28, 1–13. [Google Scholar] [CrossRef]
- Liu, Y.; Johnson, M.G.; Cox, C.J.; Medina, R.; Devos, N.; Vanderpoorten, A.; Hedenas, L.; Bell, N.E.; Shevock, J.R.; Aguero, B. Resolution of the ordinal phylogeny of mosses using targeted exons from organellar and nuclear genomes. Nat. Commun. 2019, 10, 1485. [Google Scholar] [CrossRef]
- Liu, Y.; Medina, R.; Goffinet, B. 350 my of mitochondrial genome stasis in mosses, an early land plant lineage. Mol. Biol. Evol. 2014, 31, 2586–2591. [Google Scholar] [CrossRef]
- Forrest, L.L.; Davis, E.C.; Long, D.; Crandall-Stotler, B.J.; Clark, A.; Hollingsworth, M.L. Unraveling the evolutionary history of the liverworts (Marchantiophyta): Multiple taxa, genomes and analysis. Bryologist 2006, 109, 303–334. [Google Scholar] [CrossRef]
- Groth-Malonek, M.; Wahrmund, U.; Polsakiewicz, M.; Knoop, V. Evolution of a pseudogene: Exclusive survival of a functional mitochondrial nad7 gene supports Haplomitrium as the earliest liverwort lineage and proposes a secondary loss of RNA editing in Marchantiidae. Mol. Biol. Evol. 2007, 24, 1068–1074. [Google Scholar] [CrossRef]
- Rüdinger, M.; Polsakiewicz, M.; Knoop, V. Organellar RNA editing and plant-specific extensions of pentatricopeptide repeat proteins in jungermanniid but not in marchantiid liverworts. Mol. Biol. Evol. 2008, 25, 1405–1414. [Google Scholar] [CrossRef]
- Sugita, M.; Ichinose, M.; Ide, M.; Sugita, C. Architecture of the PPR gene family in the moss Physcomitrella patens. RNA Biol. 2013, 10, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, N.F.; Christoff, A.P.; da Fonseca, G.C.; Kulcheski, F.R.; Margis, R. Unveiling chloroplast RNA editing events using next generation small RNA sequencing data. Front. Plant Sci. 2017, 8, 1686. [Google Scholar] [CrossRef] [PubMed]
- Lenz, H.; Rüdinger, M.; Volkmar, U.; Fischer, S.; Herres, S.; Grewe, F.; Knoop, V. Introducing the plant RNA editing prediction and analysis computer tool PREPACT and an update on RNA editing site nomenclature. Curr. Genet. 2010, 56, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Lenz, H.; Hein, A.; Knoop, V. Plant organelle RNA editing and its specificity factors: Enhancements of analyses and new database features in PREPACT 3.0. BMC Bioinform. 2018, 19, 255. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, K. Transfer RNA genes in the mitochondrial genome from a liverwort, Marchantia polymorpha: The absence of chloroplast-like tRNAs. Nucleic Acids Res. 1992, 20, 3773–3777. [Google Scholar]
- Wang, B.; Xue, J.; Li, L.; Liu, Y.; Qiu, Y.L. The complete mitochondrial genome sequence of the liverwort Pleurozia purpurea reveals extremely conservative mitochondrial genome evolution in liverworts. Curr. Genet. 2009, 55, 601–609. [Google Scholar] [CrossRef]
- Liu, Y.; Xue, J.Y.; Wang, B.; Li, L.; Qiu, Y.L. The mitochondrial genomes of the early land plants Treubia lacunosa and Anomodon rugelii: Dynamic and conservative evolution. PLoS ONE 2011, 6, 10. [Google Scholar] [CrossRef]
- Myszczyński, K.; Bączkiewicz, A.; Buczkowska, K.; Ślipiko, M.; Szczecińska, M.; Sawicki, J. The extraordinary variation of the organellar genomes of the Aneura pinguis revealed advanced cryptic speciation of the early land plants. Sci. Rep. 2017, 7, 9804. [Google Scholar] [CrossRef]
- Ślipiko, M.; Myszczyński, K.; Buczkowska-Chmielewska, K.; Bączkiewicz, A.; Szczecińska, M.; Sawicki, J. Comparative analysis of four Calypogeia species revealed unexpected change in evolutionarily-stable liverwort mitogenomes. Genes 2017, 8, 395. [Google Scholar] [CrossRef]
- Takenaka, M.; Verbitskiy, D.; van der Merwe, J.A.; Zehrmann, A.; Brennicke, A. The process of RNA editing in plant mitochondria. Mitochondrion 2008, 8, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, M.; Zehrmann, A.; Verbitskiy, D.; Hartel, B.; Brennicke, A. RNA editing in plants and its evolution. Annu. Rev. Genet. 2013, 47, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Hepburn, N.J.; Schmidt, D.W.; Mower, J.P. Loss of two introns from the Magnolia tripetala mitochondrial cox2 gene implicates horizontal gene transfer and gene conversion as a novel mechanism of intron loss. Mol. Biol. Evol. 2012, 29, 3111–3120. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, A.; Ross, G.; Graham, S.W.; Barrett, C.F.; Davis, J.I.; Seberg, O.; Petersen, G. Localized retroprocessing as a model of intron loss in the plant mitochondrial genome. Genome Biol. Evol. 2016, 8, 2176–2189. [Google Scholar] [CrossRef] [PubMed]
- Geiss, K.T.; Abbas, G.M.; Makaroff, C.A. Intron loss from the nadh sehydrogenase subunit 4 gene of lettuce mitochondrial DNA—Evidence for homologous recombination of a cDNA intermediate. Mol. Gen. Genet. 1994, 243, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Itchoda, N.; Nishizawa, S.; Nagano, H.; Kubo, T.; Mikami, T. The sugar beet mitochondrial nad4 gene. An intron loss and its phylogenetic implication in the Caryophyllales. Theor. Appl. Genet. 2012, 104, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; MacQueen, A.H.; Alverson, A.J.; Palmer, J.D.; Taylor, D.R. Extensive loss of RNA editing sites in rapidly evolving Silene mitochondrial genomes: Selection vs. retroprocessing as the driving force. Genetics 2010, 185, 1369–1380. [Google Scholar] [CrossRef]
- Grewe, F.; Herres, S.; Viehöver, P.; Polsakiewicz, M.; Weisshaar, B.; Knoop, V. A unique transcriptome: 1782 positions of RNA editing alter 1406 codon identities in mitochondrial mRNAs of the lycophyte Isoëtes engelmannii. Nucleic Acids Res. 2011, 39, 2890–2902. [Google Scholar] [CrossRef]
- Rudinger, M.; Szovenyi, P.; Rensing, S.A.; Knoop, V. Assigning DYW-type PPR proteins to RNA editing sites in the funariid mosses Physcomitrella patens and Funaria hygrometrica. Plant J. 2011, 67, 370–380. [Google Scholar] [CrossRef]
- Matasci, N.; Hung, L.H.; Yan, Z.; Carpenter, E.J.; Wickett, N.J.; Mirarab, S.; Nguyen, N.; Warnow, T.; Ayyampalayam, S.; Barker, M.; et al. Data access for the 1,000 plants (1 KP) project. GigaScience 2014, 3, 17. [Google Scholar] [CrossRef]
- Licht, K.; Jantsch, M.F. Rapid and dynamic transcriptome regulation by RNA editing and RNA modifications. J. Cell Biol. 2016, 213, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.H.; Waqas, M.; Lee, I.J. Silicon regulates antioxidant activities of crop plants under abiotic-induced oxidative stress: A review. Front. Plant Sci. 2017, 8, 510. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Andrade, J.; Ramirez, V.; Lopez, A.; Vera, P. Mediated plastid RNA editing in plant immunity. PLoS Pathog. 2013, 10, e1003713. [Google Scholar] [CrossRef] [PubMed]
- Bakir, Y.; Eldem, V.; Zararsiz, G.; Unver, T. Global transcriptome analysis reveals differences in gene expression patterns between nonhyperhydric and hyperhydric peach leaves. Plant Genome 2016, 9, 2. [Google Scholar] [CrossRef]
- Zhao, X.; Huang, J.; Chory, J. GUN1 interacts with MORF2 to regulate plastid RNA editing during retrograde signaling. Proc. Natl. Acad. Sci. USA 2019, 20, 10162–10167. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.H.; Danan-Gotthold, M.; Ben-Izhak, M.; Rechavi, G.; Cohen, C.J.; Louzoun, Y.; Levanon, E.Y. Increased RNA editing may provide a soruce of autoantigens in systemic lupus erythematosus. Cell Rep. 2018, 23, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Diao, L.; Yu, S.; Xu, X.; Li, J.; Zhang, R.; Yang, Y.; Werner, H.M.J.; Eterovic, A.K.; Yuan, Y. The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell 2015, 28, 515–528. [Google Scholar] [CrossRef]
- Sapiro, A.; Shmueli, A.; Henry, G.L.; Li, Q.; Shalit, T.; Yaron, O.; Paas, Y.; Li, J.B.; Shohat-Ophir, G. Illuminating spatial A-to-I RNA editing signatures within the Drosophila brain. Proc. Natl. Acad. Sci. USA 2019, 116, 2318–2327. [Google Scholar] [CrossRef]
- Chen, T.C.; Liu, Y.C.; Wang, X.; Wu, C.H.; Huang, C.H.; Chang, C.C. Whole plastid transcriptomes reveal abundant RNA editing sites and differential editing status in Phalaenopsis aphrodite subsp. formosana. Bot. Stud. 2017, 58, 38. [Google Scholar] [CrossRef]
- Mower, J.P.; Palmer, J.D. Patterns of partial RNA editing in mitochondrial genes of Beta vulgaris. Mol. Genet. Genom. 2007, 276, 285–293. [Google Scholar] [CrossRef]
- Ye, N.; Wang, X.; Li, J.; Bi, C.; Xu, Y.; Wu, D.; Ye, Q. Assembly and comparative analysis of complete mitochondrial genome sequences of an economic plant Salix suchowensis. PeerJ 2017, 29, e3148. [Google Scholar] [CrossRef] [PubMed]
- Brenner, W.G.; Mader, M.; Muller, N.A.; Hoenicka, H.; Schroeder, H.; Zorn, I.; Fladung, M.; Kersten, B. High level conservation of mitochondrial RNA editing sites among fours Populus species. G3 2019, 7, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Renzaglia, K. Phylogeny and diversification of bryophytes. Am. J. Bot. 2004, 91, 1557–1581. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.J.; Goffinet, B.; Wickett, N.J.; Boles, S.B.; Shaw, A.J. Moss diversity: A molecular phylogenetic analysis of genera. Phytotaxa 2010, 9, 175–195. [Google Scholar] [CrossRef]
- Rosato, M.; Kovařík, A.; Garilleti, R.; Rosselló, J.A. Conserved Organisation of 45S rDNA Sites and rDNA Gene Copy Number among Major Clades of Early Land Plants. PLoS ONE 2016, 11, e0162544. [Google Scholar] [CrossRef] [PubMed]
- Flores, J.R.; Catalano, S.A.; Munoz, J.; Suarez, G.M. Combined phylogenetic analysis of the subclass Marchantiidae (Marchantiophyta): Towards a robustly diagnosed classification. Cladistics 2017, 34, 517–541. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of ogranelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, 4. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Pohlert, T. The Pairwise Multiple Comparison of Mean Ranks Package (PMCMR). Available online: https://cran.r-project.org/web/packages/PMCMR/index.html (accessed on 20 February 2019).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Accession Number | Genome Size (bp) | Mean Coverage | GC Content (%) | Total Number of Genes | Protein Coding Genes | rRNA | tRNA | Genes with Introns | Total Number of Introns |
|---|---|---|---|---|---|---|---|---|---|---|
| Fossombronia foveolata | MK749462 | 175,222 | 80.5 | 45.2 | 73 | 42 | 3 | 27 | 14 | 25 |
| Haplomitrium hookeri | MK749465 | 174,448 | 44.4 | 47.6 | 74 | 43 | 3 | 27 | 16 | 30 |
| Jungermannia sphaerocarpa | MK749463 | 156,050 | 64.8 | 44.8 | 72 | 42 | 3 | 27 | 14 | 25 |
| Metzgeria furcata | MK749464 | 171,891 | 99.2 | 46.7 | 74 | 42 | 3 | 28 | 15 | 29 |
| Porella platyphylla | MK749461 | 179,081 | 31.9 | 44.7 | 74 | 42 | 3 | 28 | 15 | 29 |
| Ptilidium ciliare | MK749460 | 179,562 | 233.0 | 44.8 | 74 | 42 | 3 | 28 | 15 | 29 |
| Riccia fluitans | MK749459 | 185,621 | 202.8 | 42.4 | 74 | 42 | 3 | 28 | 16 | 30 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Myszczyński, K.; Ślipiko, M.; Sawicki, J. Potential of Transcript Editing Across Mitogenomes of Early Land Plants Shows Novel and Familiar Trends. Int. J. Mol. Sci. 2019, 20, 2963. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20122963
Myszczyński K, Ślipiko M, Sawicki J. Potential of Transcript Editing Across Mitogenomes of Early Land Plants Shows Novel and Familiar Trends. International Journal of Molecular Sciences. 2019; 20(12):2963. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20122963
Chicago/Turabian StyleMyszczyński, Kamil, Monika Ślipiko, and Jakub Sawicki. 2019. "Potential of Transcript Editing Across Mitogenomes of Early Land Plants Shows Novel and Familiar Trends" International Journal of Molecular Sciences 20, no. 12: 2963. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20122963