Trimethylamine N-Oxide Does Not Impact Viability, ROS Production, and Mitochondrial Membrane Potential of Adult Rat Cardiomyocytes

Abstract
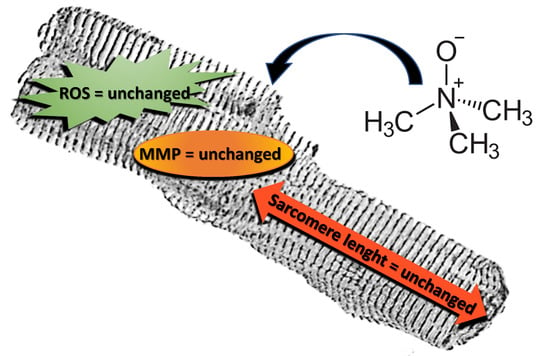
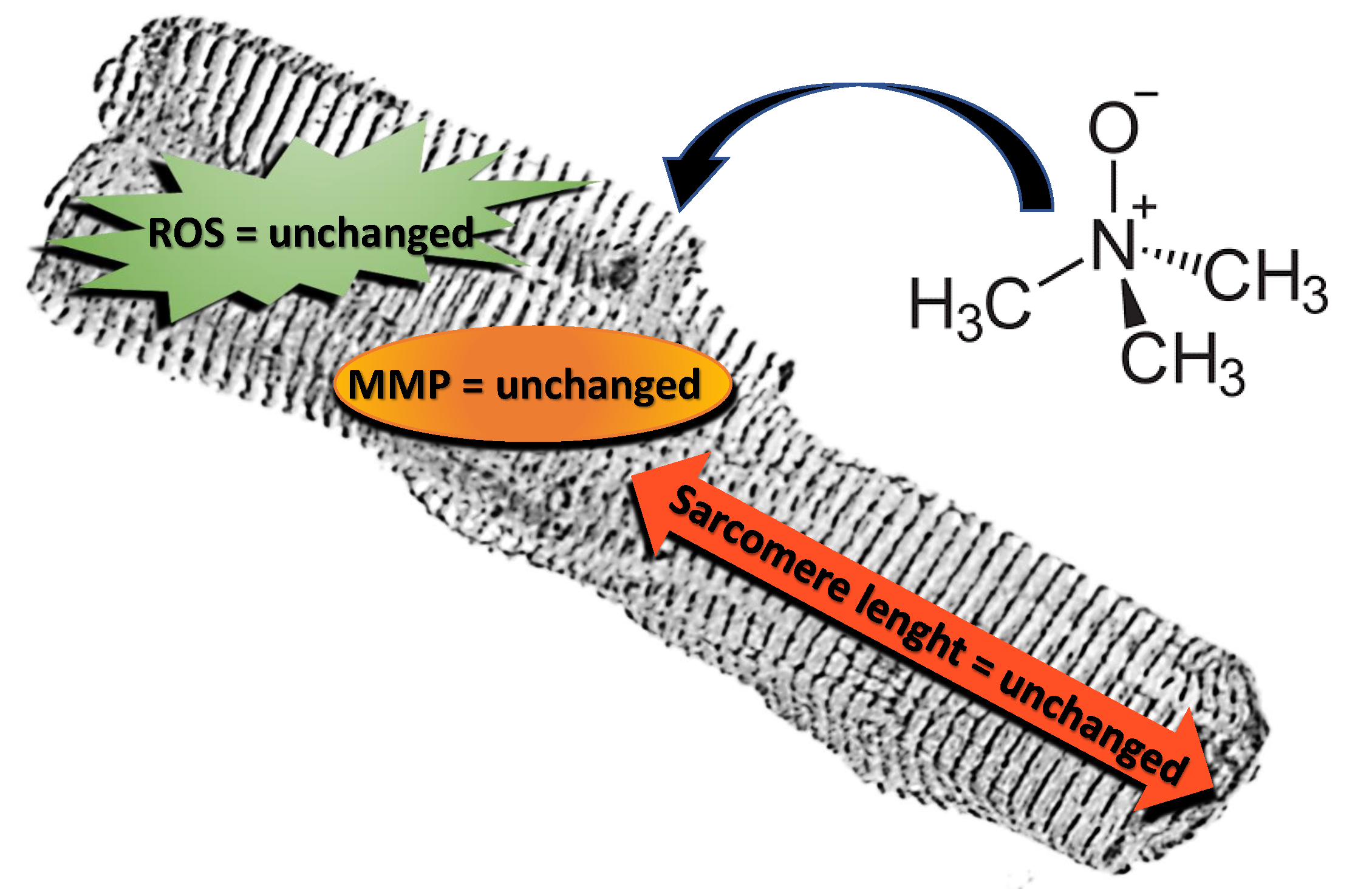
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. TMAO and Cell Viability
2.2. TMAO and Sarcomere Length
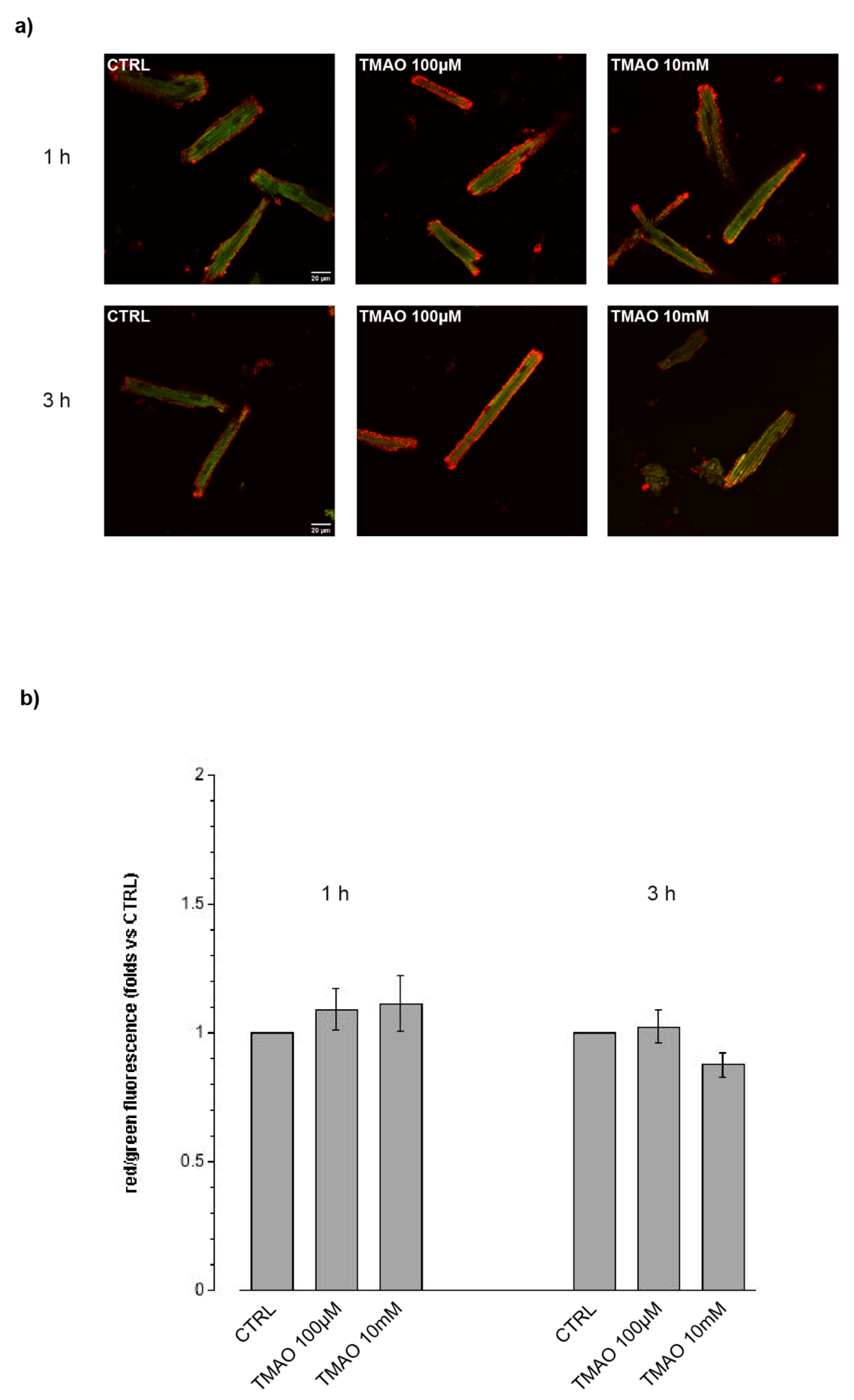
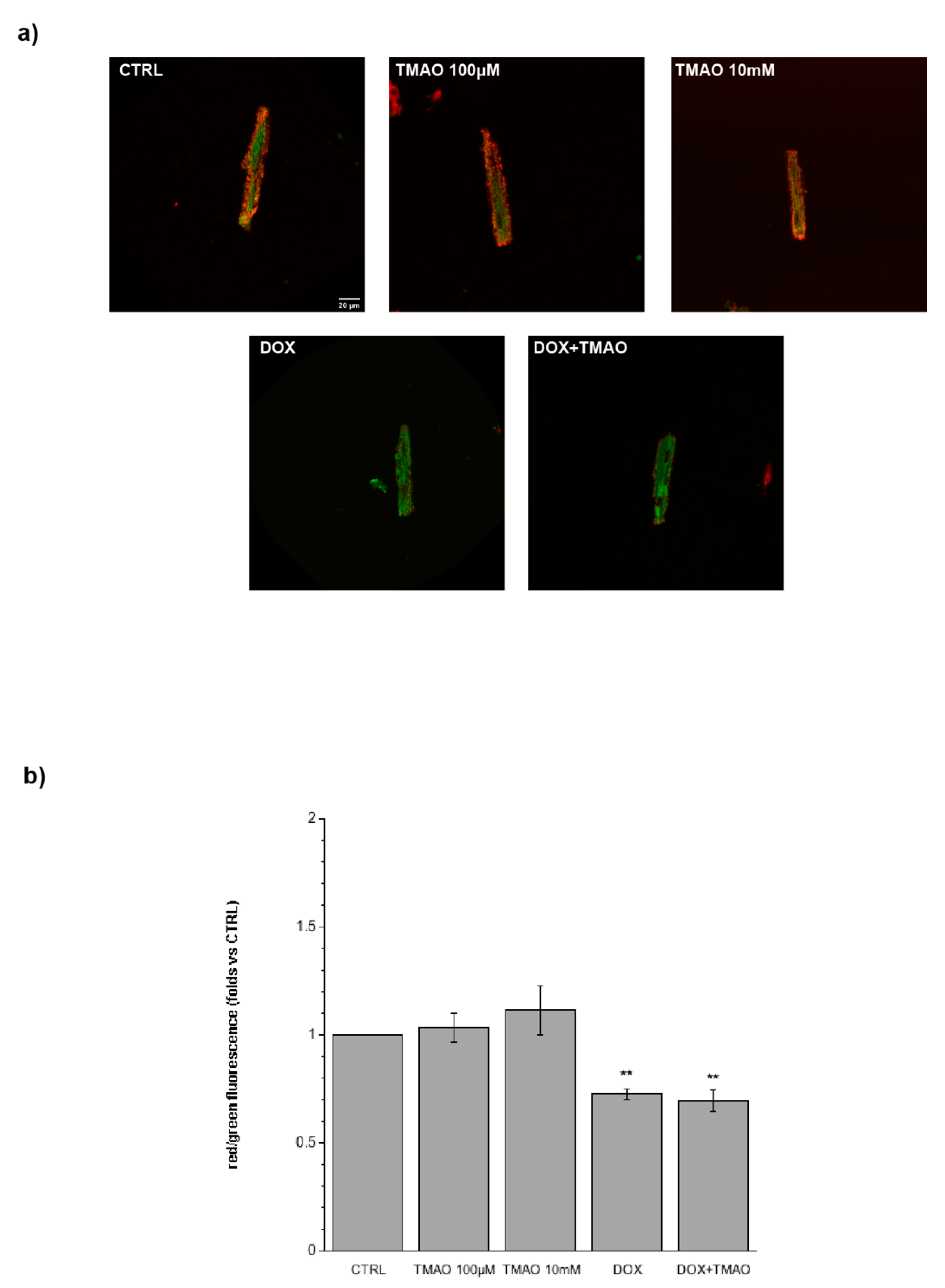
2.3. TMAO and Intracellular Reactive Oxygen Species (ROS)
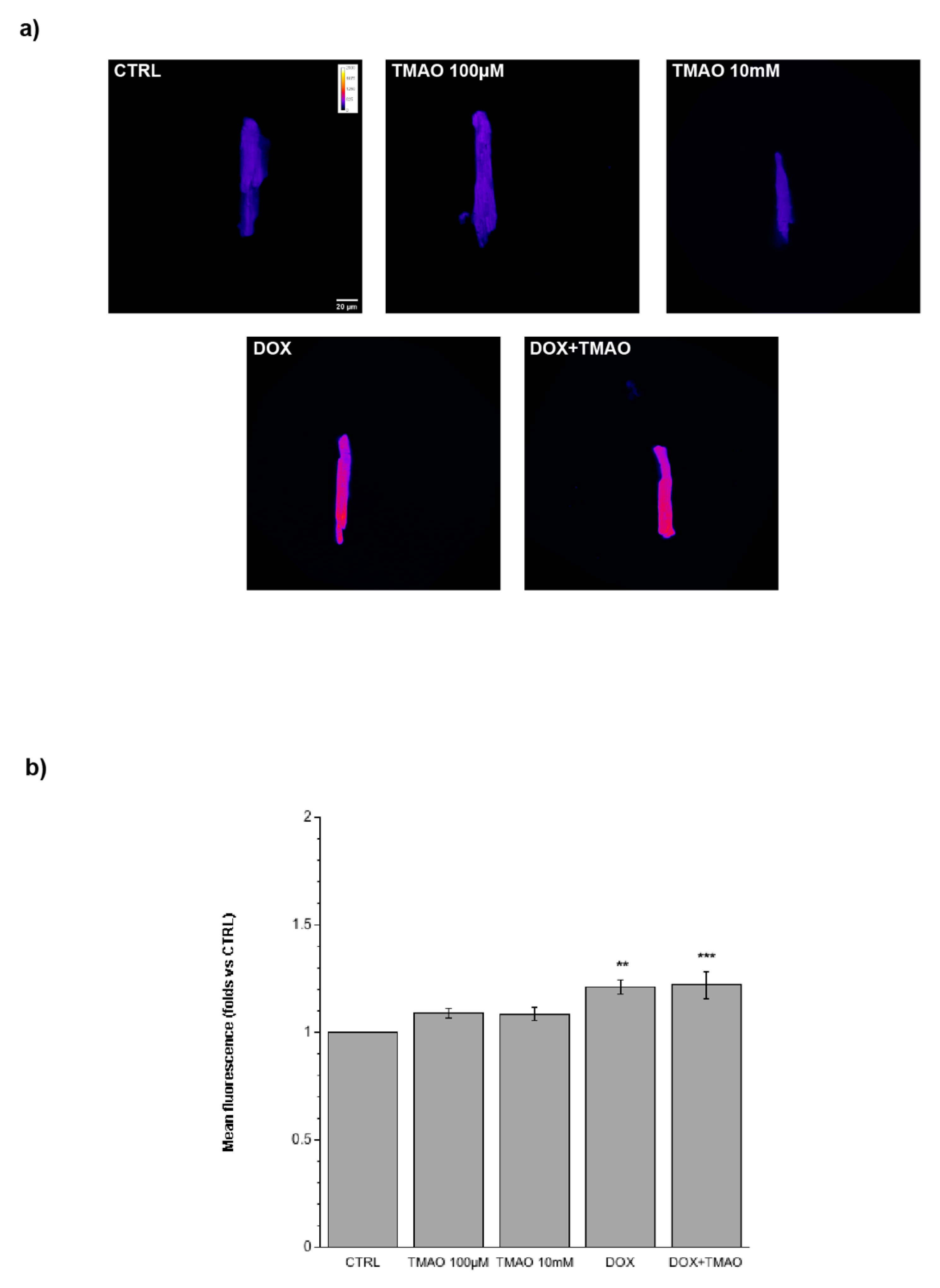
2.4. TMAO and Mitochondrial Membrane Potential
3. Discussion
4. Materials and Methods
4.1. Animal Care and Sacrifice
4.2. Solutions and Drugs
4.3. Adult Rat Ventricular Cell Isolation
4.4. Cell Viability
4.5. Evaluation of Sarcomere Length
4.6. Intracellular Reactive Oxygen Species (ROS) Measurement
4.7. Mitochondrial Membrane Potential Measurement
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TMAO | Trimethylamine N-oxide |
| PI | Propidium iodide |
| ROS | Reactive oxygen species |
| DCF-DA | 2′-7′-Dichlorofluorescein diacetate |
| JC-1 | 5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide |
References
- Ufnal, M.; Zadlo, A.; Ostaszewski, R. TMAO: A small molecule of great expectations. Nutrition 2015, 31, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Yancey, P.H. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J. Exp. Biol. 2005, 208, 2819–2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, Y.; Li, D.; Zhao, M.; Liu, C.; Liu, J.; Zeng, A.; Shi, X.; Cheng, S.; Pan, B.; Zheng, L.; et al. Gut flora-dependent metabolite Trimethylamine-N-oxide accelerates endothelial cell senescence and vascular aging through oxidative stress. Free Radic. Biol. Med. 2018, 116, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, Y.; Gua, C.; Li, X. Elevated Circulating Trimethylamine N-Oxide Levels Contribute to Endothelial Dysfunction in Aged Rats through Vascular Inflammation and Oxidative Stress. Front. Physiol. 2017, 8, 350. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, Z.; Sun, T.; Huang, H.; Chen, S.; Chen, L.; Luo, C.; Yang, W.; Yang, X.; Yoa, P.; Cheng, J.; et al. Association between microbiota-dependent metabolite trimethylamine-N-oxide and type 2 diabetes. Am. J. Clin. Nutr. 2017, 106, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Makrecka-Kuka, M.; Volska, K.; Antone, U.; Vilskersts, R.; Grinberga, S.; Bandere, D.; Liepinsh, E.; Dambrova, M. Trimethylamine N-oxide impairs pyruvate and fatty acid oxidation in cardiac mitochondria. Toxicol. Lett. 2017, 267, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Savi, M.; Bocchi, L.; Bresciani, L.; Falco, A.; Quaini, F.; Mena, P.; Brighenti, F.; Crozier, A.; Stilli, D.; Del Rio, D. Trimethylamine-N-Oxide (TMAO)-Induced Impairment of Cardiomyocyte Function and the Protective Role of Urolithin B-Glucuronide. Molecules 2018, 23, 549. [Google Scholar] [CrossRef]
- Arduini, A.; Zammit, V.A.; Bonomini, M. Identification of trimethylamine N-oxide (TMAO)-producer phenotype is interesting, but is it helpful? Gut 2019. [Google Scholar] [CrossRef]
- Nowiński, A.; Ufnal, M. Trimethylamine N-oxide: A harmful, protective or diagnostic marker in lifestyle diseases? Nutrition 2018, 46, 7–12. [Google Scholar] [CrossRef]
- Dumas, M.-E.; Maibaum, E.C.; Teague, C.; Ueshima, H.; Zhou, B.; Lindon, J.C.; Nicholson, J.K.; Stamler, J.; Elliott, P.; Chan, Q.; et al. Assessment of analytical reproducibility of 1H NMR spectroscopy based metabonomics for large-scale epidemiological research: The INTERMAP Study. Anal. Chem. 2006, 78, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- Collins, H.L.; Drazul-Schrader, D.; Sulpizio, A.C.; Koster, P.D.; Williamson, Y.; Adelman, S.J.; Owen, K.; Sanli, T.; Bellamine, A. L-Carnitine intake and high trimethylamine N-oxide plasma levels correlate with low aortic lesions in ApoE−/− transgenic mice expressing CETP. Atherosclerosis 2016, 244, 9–37. [Google Scholar] [CrossRef] [PubMed]
- Lupachyk, S.; Watcho, P.; Stavniichuk, R.; Shevalye, H.; Obrosova, I.G. Endoplasmic Reticulum Stress Plays a Key Role in the Pathogenesis of Diabetic Peripheral Neuropathy. Diabetes 2013, 62, 944–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisel, S.H.; Warrier, M. Trimethylamine N-Oxide, the Microbiome, and Heart and Kidney Disease. Annu. Rev. Nutr. 2017, 37, 157–181. [Google Scholar] [CrossRef] [PubMed]
- Sarvazyan, N. Visualization of doxorubicin-induced oxidative stress in isolated cardiac myocytes. Am. J. Physiol. 1996, 271, H2079–H2085. [Google Scholar] [CrossRef] [Green Version]
- Zununi Vahed, S.; Barzegari, A.; Zuluaga, M.; Letoumeur, D.; Pavon-Djavid, G. Myocardial infarction and gut microbiota: An incidental connection. Pharmacol. Res. 2018, 129, 308–317. [Google Scholar] [CrossRef]
- Cho, C.E.; Taesuwan, S.; Malysheva, O.V.; Bender, E.; Tulchinsky, N.F.; Yan, J.; Sutter, J.L.; Caudill, M.A. Trimethylamine-N-oxide (TMAO) response to animal source foods varies among healthy young men and is influenced by their gut microbiota composition: A randomized controlled trial. Mol. Nutr. Food Res. 2017, 61, 1600324. [Google Scholar] [CrossRef]
- Ufnal, M.; Jazwiec, R.; Dadlez, M.; Drapala, A.; Sikora, M.; Skrzypecki, J. Trimethylamine-N-oxide: A carnitine-derived metabolite that prolongs the hypertensive effect of angiotensin II in rats. Can. J. Cardiol. 2014, 30, 1700–1705. [Google Scholar] [CrossRef]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef]
- Dambrova, M.; Latkovskis, G.; Kuka, J.; Strele, I.; Konrade, I.; Grinberga, S.; Hartmane, D.; Pugovics, O.; Erglis, A.; Liepinsh, E. Diabetes is Associated with Higher Trimethylamine N-oxide Plasma Levels. Exp. Clin. Endocrinol. Diabetes 2016, 124, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Mueller, D.M.; Allenspach, M.; Othman, A.; Saely, C.H.; Muendlein, A.; Vonbank, A.; Drexel, H.; von Eckardstein, A. Plasma levels of trimethylamine-N-oxide are confounded by impaired kidney function and poor metabolic control. Atherosclerosis 2015, 243, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Liao, S.-X.; He, Y.; Wang, S.; Xia, G.-H.; Liu, F.-T.; Zhu, J.-J.; You, C.; Chen, Q.; Zhou, L.; et al. Dysbiosis of Gut Microbiota with Reduced Trimethylamine-N-Oxide Level in Patients With Large-Artery Atherosclerotic Stroke or Transient Ischemic Attack. J. Am. Heart Assoc. 2015, 4, e002699. [Google Scholar] [CrossRef] [PubMed]
- Huc, T.; Drapala, A.; Gawrys, M.; Konop, M.; Bielinska, K.; Zaorska, E.; Samborowska, E.; Wyczalkowska-Tomasik, A.; Paczek, L.; Dadlez, M.; et al. Chronic, low-dose TMAO treatment reduces diastolic dysfunction and heart fibrosis in hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1805–H1820. [Google Scholar] [CrossRef] [PubMed]
- Gallo, M.P.; Femminò, S.; Antoniotti, S.; Querio, G.; Alloatti, G.; Levi, R. Catestatin induces glucose uptake and GLUT4 trafficking in adult rat cardiomyocytes. Biomed. Res. Int. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lenky, C.C.; McEntyre, C.J.; Lever, M. Measurement of marine osmolytes in mammalian serum by liquid chromatography-tandem mass spectrometry. Anal. Biochem. 2012, 420, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Somero, G.N. From dogfish to dogs: Trimethylamines protect proteins from urea. Am. J. Physiol. 1986, 1, 9–12. [Google Scholar] [CrossRef]
- Lagranha, C.L.; Deschamps, A.; Aponte, A.; Steenbergen, C.; Murphy, E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection on females. Circ. Res. 2010, 106, 1981–1991. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Querio, G.; Antoniotti, S.; Levi, R.; Gallo, M.P. Trimethylamine N-Oxide Does Not Impact Viability, ROS Production, and Mitochondrial Membrane Potential of Adult Rat Cardiomyocytes. Int. J. Mol. Sci. 2019, 20, 3045. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20123045
Querio G, Antoniotti S, Levi R, Gallo MP. Trimethylamine N-Oxide Does Not Impact Viability, ROS Production, and Mitochondrial Membrane Potential of Adult Rat Cardiomyocytes. International Journal of Molecular Sciences. 2019; 20(12):3045. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20123045
Chicago/Turabian StyleQuerio, Giulia, Susanna Antoniotti, Renzo Levi, and Maria Pia Gallo. 2019. "Trimethylamine N-Oxide Does Not Impact Viability, ROS Production, and Mitochondrial Membrane Potential of Adult Rat Cardiomyocytes" International Journal of Molecular Sciences 20, no. 12: 3045. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20123045