Long Noncoding Competing Endogenous RNA Networks in Age-Associated Cardiovascular Diseases


1
Molecular Cardiology Laboratory, IRCCS Policlinico San Donato, San Donato Milanese, 20097 Milan, Italy
2
Laboratory of Epigenetics, Istituti Clinici Scientifici Maugeri IRCCS, 27100 Pavia, Italy
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(12), 3079; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20123079
Submission received: 29 May 2019
/
Revised: 17 June 2019
/
Accepted: 21 June 2019
/
Published: 24 June 2019
(This article belongs to the Special Issue RNAs in Cardiovascular Diseases-CardioRNA EU COST Action)
Abstract
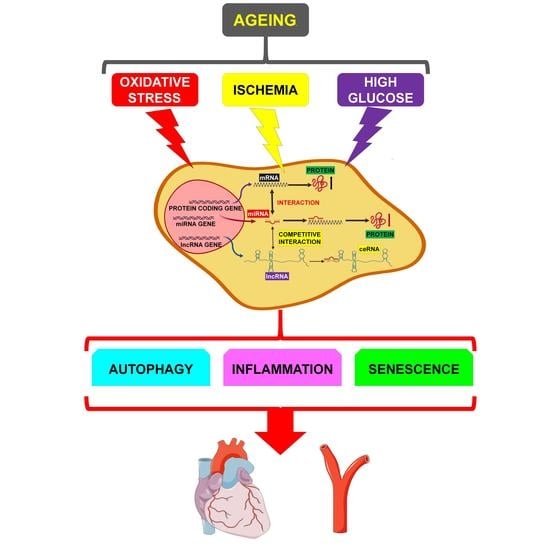
:Cardiovascular diseases (CVDs) are the most serious health problem in the world, displaying high rates of morbidity and mortality. One of the main risk factors for CVDs is age. Indeed, several mechanisms are at play during aging, determining the functional decline of the cardiovascular system. Aging cells and tissues are characterized by diminished autophagy, causing the accumulation of damaged proteins and mitochondria, as well as by increased levels of oxidative stress, apoptosis, senescence and inflammation. These processes can induce a rapid deterioration of cellular quality-control systems. However, the molecular mechanisms of age-associated CVDs are only partially known, hampering the development of novel therapeutic strategies. Evidence has emerged indicating that noncoding RNAs (ncRNAs), such as long ncRNAs (lncRNAs) and micro RNAs (miRNAs), are implicated in most patho-physiological mechanisms. Specifically, lncRNAs can bind miRNAs and act as competing endogenous-RNAs (ceRNAs), therefore modulating the levels of the mRNAs targeted by the sponged miRNA. These complex lncRNA/miRNA/mRNA networks, by regulating autophagy, apoptosis, necrosis, senescence and inflammation, play a crucial role in the development of age-dependent CVDs. In this review, the emerging knowledge on lncRNA/miRNA/mRNA networks will be summarized and the way in which they influence age-related CVDs development will be discussed.
1. Introduction
Better healthcare and living conditions have contributed to an increase in people’s longevity, which has resulted in a higher prevalence of age-related debilitating and life-threatening diseases such as cardiovascular diseases (CVDs), cancer and neurodegeneration.
CVDs share with infectious diseases the worst rate of morbidity and mortality, with now more than 30% of all deaths worldwide according to the World Health Organization [1], and it is expected that, as the world population ages, this situation will worsen [2,3].
Indeed, age represents a major independent risk factor for cardiovascular-related morbidity and mortality [4,5]. Aging is characterized by a progressive decline in numerous physiological processes, worsening the outcome of other diseases, such as diabetes mellitus, hypertension and coronary disease [6,7,8]. Increased CVD prevalence is also associated with frailty, a condition of increased vulnerability to stressors [9]. In view of this, it is conceivable to define unsuccessful cardiovascular aging a disease per se [10]. Thus, preserving a well-functioning cardiovascular system, which delivers oxygenated blood to all body tissues, is a prerequisite for the good health of all organs and for longevity.
An ever-increasing number of evidence indicates that noncoding RNAs (ncRNAs), such as long ncRNAs (lncRNAs) and micro RNAs (miRNAs) are implicated in most pathophysiological mechanisms. By inhibiting post-transcriptionally both protein coding- (mRNAs) and noncoding-genes, miRNAs regulate gene expression [11,12]. LncRNAs can exert sponging-like effects on both miRNAs and mRNAs, and have both beneficial and detrimental effects. Moreover, the lncRNA family can also regulate molecular processes by acting as host transcripts for miRNAs [13,14].
There are several pieces of evidence reporting that ncRNAs are causally linked to the development of age-associated CVDs by regulating inflammation, cell proliferation, apoptosis, senescence and autophagy. Herein, we will review and summarize the mechanistic, functional, and pathological role of the lncRNAs/miRNAs/mRNA networks in these events and their specific involvement in age-related CVDs.
2. Aging and Cardiovascular System Deterioration
Several studies suggest that cardiomyocyte apoptosis and vascular stiffness are associated with aging-induced structural and functional alterations, but the underlying mechanisms are yet to be fully understood. Moreover, the health of the arterial and cardiac systems are mutually correlated because the age-dependent increase of arterial stiffness induces, as compensatory mechanisms, myocardial hypertrophy and fibrosis, which in turn lead to the reduced cardiac output, frequently observed in the elderly.
Considering these facts, we will here summarize the main age-related pathophysiological processes, both in the vascular system and in the heart.
2.1. Vascular Functional Impairment
Endothelial dysfunction and generalized central arterial stiffness are two major vascular modifications caused by aging [7].
Atherosclerosis is a chronic silent inflammatory disease, which can evolve to plaque rupture and thrombosis [15]. The Framingham Heart Study has demonstrated that endothelial dysfunction is independent of clinical disease, as it is also observed in healthy old adults, but creates a pro-atherogenic environment, facilitating the formation of plaques [16]. If other atherosclerotic risk factors are also present (e.g., cigarette smoking, hypertension, high levels of serum cholesterol and its fractions, low levels of HDL and diabetes mellitus), these alterations can lead to the enlargement and rupture of an atherosclerotic stable plaque.
ECs (Endothelial Cells), VSMCs (Vascular Smooth Muscle Cells), and macrophages are the primary cells contributing to the formation of atherosclerotic lesions. The atherosclerotic plaque results from the infiltration of circulating monocytes in the sub-endothelial space, their differentiation into macrophages, followed by the internalization of modified lipoproteins with transformation in foam cells [17,18].
Another typical age-related vascular alteration is the increase of arterial stiffness. Many studies have demonstrated, throughout the vascular tree, a gradual, age-related impairment of arterial compliance, diffuse intimal and medial thickening and reduced distensibility of central arteries, which result in decreased vascular dilatation and elevated systolic pressure [19,20]. These changes represent major risk factors for the development of atherosclerosis, hypertension, stroke, and arterial fibrillation [7]. In particular, the loss of aorta distensibility associated with ageing is a very complex process involving several mechanisms. One is the inversion of the elastin/collagen ratio. This is due to both increased collagen deposition and depletion of elastin, mediated by the activation of the matrix enzymes produced by the inflammatory cells in the aorta [21]. Another mechanism is the non-enzymatic glycosylation of collagen present in the arterial wall, which results in the cross-linking of adjacent proteins [21,22]. The fragmentation and calcification of elastic fibers, the increased deposition of collagen and collagen cross-linking, amyloid deposition in the medial layer, and migration/proliferation of VSMC are frequent histologic findings in aged vessels (Figure 1A). In addition, in the elderly, increased levels of markers of both EC oxidative stress and inflammation have been reported [23,24,25]. Initially, the cardiovascular system is able to counteract the stress by moderately increasing the adrenergic signaling. When the stress becomes chronic, however, these responses are strengthened by the activation of the renin-angiotensin-aldosterone and endothelin signaling mechanisms. The activation of these signaling pathways induces an exaggerated chronic inflammatory response and, consequently, more oxidative stress and age-associated structural and functional arterial remodeling [26,27]. Thus, the mechanisms accompanying “physiologic” arterial aging become, with chronic stress, “pathophysiologic”. Moreover, epigenetic control mechanisms are also critically involved in atherosclerosis plaque development and vulnerability [28,29,30].
2.2. Cardiac Function Impairment
There is a variety of natural and pathological insults to the myocardium that, throughout life, causes attrition of cardiomyocytes, including ethanol or drugs abuses, increased food intake, viral myocarditis and myocardial infarctions.
The aging process of the cardiac system is characterized by a reduced peak cardiac output and left ventricular (LV) diastolic function, an altered response to catecholamine, an incomplete relaxation during early diastolic filling, and increased myocardial stiffness [10,19]. Moreover, the reduction in cardiac output stimulates a compensation mechanism by increasing muscle mass that leads to cardiac hypertrophy and LV wall thickening [31,32]. This mechanism enhances cardiac output at the beginning, but reduces the cardiac function as hypertrophy increases [33]. In addition, there is an asymmetric growth of the interventricular septum leading to a change in the heart shape [34,35] (Figure 1B).
Increased apoptosis and necrosis are frequent histologic findings in the myocardium of old animals and humans [36,37]. Moreover, cardiomyocytes, as well as other post-mitotic cells, are more likely, during aging, to accumulate the granular pigment ‘lipofuscin’, which is composed of oxidized lipids, cross-linked proteins and oligosaccharides, and is considered as a marker of cellular aging [38].
A typical process associated with ageing is the decline in the number of cardiomyocytes, and this process is more pronounced in males than females [39]. Moreover, the replacement of cardiomyocytes is a very rare event [40,41], due to the withdrawal of cardiomyocytes from the cell cycle after birth [42]. This cell number decline leads to an adaptive hypertrophy of the remaining cardiomyocyte and, eventually, to a reduction of cardiac capacity [31,32].
A decrease in autophagy efficiency has been found to be associated with the ageing process, determining the abnormal deposition of intracellular protein aggregates, enhanced ROS production, decreased ATP production, and cell death [43,44].
Several neurodegenerative diseases, such as Alzheimer’s, Huntington’s and Parkinson’s diseases, are characterized by amyloidosis, which is the accumulation of misfolded proteins as intracellular or extracellular aggregates. Some cardiomyopathies share these features with neurodegenerative diseases. Recent evidence indicates that not only the accumulation of β-amyloid fibrils in extracellular plaques, but also the soluble intermediates of fibril formation seem to be toxic [45]. Indeed, soluble oligomers have been found in animal models of HF as well as in human HF [46,47,48,49,50,51]. Another amyloid-protein, the transthyretin (TTR), is a tetrameric protein synthesized mostly by the liver. This protein can be deposited as insoluble fibrils into the heart as consequence of protein misfolding due to gene mutations or as an ageing-related phenomenon, such as the age-dependent increased oxidative stress [52,53,54]. The pathogenitic role of senile amyloidosis in dilated cardiomyopathies is also demonstrated by the accumulation of wt TTR in patients with heart failure with preserved ejection fraction (HFpEF) [55,56].
3. The Noncoding RNAs Network
3.1. microRNAs
miRNAs are small non-coding RNA sequences, ~22 nucleotide-long, that, by partially interacting with target mRNAs, lower their translation and/or stability [12]. The primary miRNA transcript, the pri-miRNA, is longer than the mature form and, after cleavage by the microprocessor complex (containing the ribonuclease Drosha), generates the miRNA precursor (pre-miRNA) that is exported to the cytoplasm [57]. After translocation to the cytoplasm, the pre-miRNA is cleaved by Dicer to generate a ~22-bp-long duplex RNA. Only one strand of the duplex represents the mature miRNA, and it is loaded onto the RNA-induced silencing complex (RISC), which contains Argonaute (Ago) proteins [58]. This complex can target a family of target mRNAs by partial hybridization, frequently at the miRNA ‘seed’ region (nucleotides 2–7). In this way, in most circumstances, this complex acts as a negative regulator of gene expression. Worth noting is that more than one miRNAs can recognize the same mRNA generating a complex miRNA/mRNA network. miRNAs mediate the regulation of gene expression in virtually all aspects of cell biology, and dysregulation of miRNAs has been causally linked to several age-dependent CVDs [13].
3.2. Long Noncoding RNAs
The threshold of 200 nucleotides usually separates short from long ncRNAs. The biogenesis and turnover of most lncRNA are closely similar to those of the mRNAs coding for proteins. As for mRNAs, lncRNA transcription is mediated by promoter elements, transcription factors and histone modifications [59].
According to their genomic localization and biogenesis, lncRNAs can be classified as: (1) intergenic lncRNA (lincRNA), transcribed independently of protein-coding genes; (2) antisense lncRNAs (lncRNA-AS), when they are expressed from the opposite strand of mRNAs; (3) pseudogene-encoded lncRNAs, that are transcribed from vestigial genes that lost their protein-coding potential; (4) intronic lncRNAs, when they are present in introns of coding genes; (5) promoter-associated lncRNAs, if they are transcribed from the promoter regions of coding mRNAs [13,60].
The lncRNA can function through different mechanisms of action: (1) Epigenetic regulation, when they act as scaffolds, bridges, and tethers of factors that regulate the state of the chromatin; (2) Transcriptional regulation, when they modulate the rates of RNA polymerase II initiation and elongation; (3) Nuclear compartmentalization, to maintain nuclear structures; (4) Post-transcriptional gene regulation by both basepairing with mRNAs, or acting as cofactors or competitors of RNA-binding proteins; (5) Competing endogenous RNAs (ceRNAs), when lncRNAs can function as decoys or sponges for miRNAs [61,62]. In particular, lncRNAs acting as ceRNAs harbor miRNA response elements for binding miRNAs and generally display increased expression and/or stability compared to other lncRNAs. The presence of one or multiple binding site and the high level of the ceRNA make it able to sequester the target miRNA, thus titrating the miRNA-RISC away from the mRNAs it regulates [63,64]. Thus, by acting as ceRNAs, lncRNAs are at the center of a large-scale regulatory network across the transcriptome, greatly expanding the complexity of gene expression regulation (Figure 2).
There are clearly several complications of the system and miRNA sponging may become biologically relevant only for a small subset of ceRNAs/miRNAs whose cellular concentration and target abundance meet a narrow range of values [65]. Other relevant factors are the affinity of the competitive binding to the miRNA for the lncRNA and the mRNA, as well as the miRNA ability to induce the degradation of the bound RNA (both lncRNA and mRNA). It should also be considered that more than one miRNA has multiple target mRNAs and possibly more than one ceRNA-lncRNA may be part of the same network, yielding an extremely complicated system that may provide stability to the structure.
4. miRNA/lncRNA/mRNA Network Modulation by Age-Related Mechanisms
The altered signaling observed during the aging of the cardiovascular system involves several mechanisms, such as a reduced response to acute stress, reduced autophagy, higher levels of markers of chronic stress, including reactive oxygen species (ROS) and inflammation, increased cell death and necrosis, with reduced cell replacement [68,69].
These cellular events, in general, have detrimental consequences and eventually can determine the development of age-associated pathologies. As an example, the accumulation of ROS sensitizes the heart to the renin-angiotensin-aldosterone system, inducing apoptosis and increasing the propensity for adverse cardiac remodeling, diastolic dysfunction and heart failure [70].
In the following paragraphs, we will review the role of autophagy, inflammation and senescence in the modulation of the lncRNA/miRNA/mRNA networks and their involvement in age-dependent CVDs (summarized in Table 1).
4.1. Autophagy Impairment
Autophagy is a fundamental process regulating cellular quality control. The term “autophagy” means “self-eating” in Greek, and it is an intracytoplasmic degradation process of organelles such as mitochondria, endoplasmic reticulum and peroxisomes, as well as intracellular pathogens [92]. Autophagy leads to the dynamic recycling of protein aggregates, thus providing both energy and building material for new protein and membrane production [70]. In particular, the autophagy related protein (ATG) complexes coordinate protein degradation by the formation of the autophagosome, which, in turn, fuses with lysosomes to generate autolysosome. Inflammation, oxidized lipoprotein, ER stress and ROS production are some of the factors that induce the autophagic process [93]. A decrease in the efficiency of autophagy determines the abnormal deposition of intracellular protein aggregates, enhanced ROS production, decreased ATP production, and cell death [43,44].
Several data demonstrate that physiological autophagy is a protective mechanism that serves to maintain normal cardiovascular function [70]. Actually, impaired autophagy is associated with CVDs development [68,69].
In particular, autophagy, by degrading the damaged intracellular organelles, promotes the survival of the cellular components of the plaque and reduces the accumulation of ROS [68]. This activity stabilizes the plaque, thus preventing its rupture and the consequent detrimental effects, such as arterial occlusion, acute coronary syndrome, myocardial infarction, and stroke [69]. Moreover, autophagy, by degrading the accumulated dysregulated proteins, decreases the cardiac mass, counteracting ventricular hypertrophy [94]. Autophagy is also induced in ischemia and reperfusion (I/R) [95]. Indeed, during I/R, the oxygen supply limitation to the heart activates the autophagic process as a homeostatic mechanism to protect the myocardium from further ischemia [96].
It is noteworthy that, in spite of its well-known cardiovascular protective effects, dysregulated autophagy can also have detrimental effects. Actually, pressure overload triggers autophagy, which in turn leads to cell death, thus worsening heart failure, and plaque destabilization [69].
Autophagy can play a negative role also in I/R. In fact, following I/R injury, a burst of oxidative stress occurs, which results in enhanced cardiomyocyte autophagy followed by cell apoptosis [97].
The factors determining whether autophagy will be adaptive or detrimental are still unknown, but the extent and duration of autophagy seem to be important [98]. The involvement of the lncRNA/miRNA/mRNA network in autophagy is reported in the following sub-paragraphs and summarized in Figure 3.
4.1.1. APF/miR-188-3p/ATG7
Wang and collaborators investigated the role of the network APF/miR-188-3p/ATG7 in the regulation of the I/R-induced autophagy [71]. In particular, the expression of the lncRNA APF (also named AH079427) increases in cardiomyocytes treated with anoxia/reperfusion, as well as in vivo after I/R [71]. The exogenous expression of APF promotes both autophagy and cell death and these effects are mediated by sequestration of miR-188-3p and induction of the pro-autophagic gene ATG7. Accordingly, in vivo, the silencing of APF leads to decreased autophagy and reduction of myocardial infarction size and thus to an amelioration of myocardial functions.
4.1.2. AK088388/miR-30a/Beclin-1 and LC3-II
LncRNA AK088388 has been recently found to modulate autophagy in cardiomyocytes functioning as an endogenous RNA sponge of miR-30a under Hypoxia/Reperfusion conditions [72]. The inhibition of AK088388 is linked to increased levels of miR-30a and attenuation of the expression of Beclin-1 and LC3- II. This effect leads to a reduction in cardiomyocyte damage and autophagy [72].
4.1.3. AK139328/miR-204-3p/ATGs
AK139328 is another lncRNA induced by I/R alone or in combination with diabetes (DM/IR) [73]. Interestingly, the inhibition in vivo of AK139328 in DM/IR reduces LV enlargement, fibrosis and myocardial infarct size, improving the heart function [73]. Accordingly, AK139328 silencing in DM/IR derived cardiomyocytes attenuates autophagy and apoptosis [73]. Mechanistically, AK139328 sponges miR-204, an anti-autophagy miRNA also important for metabolic recovery after I/R [73]. Indeed, the simultaneous silencing of AK139328 and overexpression of miR-204 relieves the hypoxia/reperfusion injury through inhibition of the autophagic cascade [73].
4.1.4. BACE1-AS/miRNAs/BACE1
The abnormal deposition of intracellular protein aggregates such as amyloid has been found to be associated with a decreased efficiency in autophagy with ageing [43,44]. Recently, the overexpression of the lncRNA beta-secretase 1-antisense RNA (BACE1-AS) in post-ischemic HF has been related to both the accumulation of BACE1 mRNA, which encodes for the enzyme responsible for β–amyloid, and the cardiac deposition of β–amyloid [51]. Indeed, the dysregulation of the network formed by BACE1-AS, BACE1 and β–amyloid has been related to the cell toxic effect of β–amyloid [51]. Different miRNA-mediated mechanisms have been proposed through which BACE1-AS mediates the stabilization of BACE1 mRNA. Indeed, BACE1-AS can act by masking the binding site for miR-485-5p on BACE1 mRNA [74]. Moreover, BACE1-AS shares with BACE1 the miRNA-responsive-elements for miR-29, miR-107, miR-124, miR-485 and miR-761, acting as a ceRNA preventing BACE1 targeting [75].
4.1.5. Galont/miR-338/ATG5
“GATA1 activated lncRNA” or Galont is induced by anoxia/reperfusion in neonatal cardiomyocytes [76], and its overexpression triggers both autophagic and apoptotic responses. miR-338 is a Galont direct target and its expression is repressed in anoxia/reperfusion [76]. Indeed, Galont overexpression in vitro reduces both autophagy and apoptosis induced by anoxia/reperfusion through the inhibition of miR-338 target ATG5 [76].
4.1.6. GAS5/miR-26a/ATGs
GAS5 is an lncRNA that has been found to be increased in atherosclerosis and its silencing reduces the apoptosis of macrophages and ECs after treatment with oxidized low density lipoproteins (ox-LDL) [99]. Very recently, Liang et al. [77] investigated the mechanisms underpinning these effects. The expression of GAS5 and miR-26a, which interacts with the lncRNA, are up- and down-regulated, respectively, in both plasma samples from patients suffering from aortic stenosis and in aortic ECs treated with ox-LDL. The treatment of ECs with ox-LDL determines the increase of cell apoptosis and the decrease of autophagy. GAS5 silencing and treatment with ox-LDL leads to increased expression of miR-26a and of autophagic markers [77]. These data suggest a novel regulatory mechanism for ox-LDL-induced impaired autophagy flux in ECs, with GAS5/miR-26a axis that might provide potential targets in the process of atherosclerosis.
4.1.7. UCA1/miR-128/HSP70
In both mouse cardiomyocyte H9c2 cells under Hypoxia/Reperfusion or rat hearts undergoing I/R, the expression of miR-128 has been found to be increased, while the level of its target heat shock protein 70 (HSP70) was decreased [79]. The post-conditioning by morphine treatment leads to the increase of lncRNA urothelial carcinoma-associated 1 (UCA1) level, which, by sponging miR-128, results in the increase of HSP70 and the attenuation of cell autophagy and cardiac injury [79].
4.1.8. TGFB2-OT1/miR-4459/ATG13
The lncRNA TGFB2-OT1 (TGFβ2 overlapping transcript 1 or FLJ11812) is located in the 3′UTR of TGFβ2 (transforming growth factor, β2). Ge et al. reported that TGFB2-OT1 can sequester miR-4459, thus increasing the level of its target ATG13 (autophagy related 13) and promoting autophagy [78]. In particular, the treatment of ECs with the small molecule 3BDO (3-benzyl-5-((2-nitrophenoxy) methyl)–dihydrofuran-2(3H)-one) inhibits both rapamycin-induced autophagy and the expression of TGFB2-OT1 [78], and activates the MTOR signaling [100]. MTOR activation phosphorylates TIA1 (TIA1 cytotoxic granule-associated RNA binding protein), which, in this state, represses the expression of TGFB2-OT1 [71]. The reduced expression of TGFB2-OT1, which competes with miR-4459 for binding to ATG13, leads to an inhibition of autophagy [78]. Thus, the modulation of TGFB2-OT1/miR-4459/ATG13 axis could represent a potential therapeutic approach for autophagy regulation in CVDs.
4.2. “Inflammageing”
An hallmark of ageing is also represented by the presence, even in the absence of risk factors and clinically active diseases, of high levels of pro-inflammatory markers in cells and tissues of older individuals, a condition that is called “inflammageing” [101,102].
It is not yet known whether high levels of tissue and circulating pro-inflammatory molecules might contribute to associated pathological conditions or whether they represent only reactive markers of the underlying pathology. Nevertheless, clear evidence indicates that inflammageing is a risk factor for CVDs [103,104].
Inflammation plays a crucial role in fighting infections or extraneous molecules, but, despite this protective role, sustained and prolonged inflammation might be detrimental to health. Actually, during inflammation, ECs are affected by an early damage, which, in turn, might be pro-atherogenetic. Atherosclerotic plaques can in turn produce additional pro-inflammatory molecules, amplifying the role of inflammation in atherogenesis [69,105,106].
Noncoding RNAs, including miRNAs and lncRNAs are emerging regulators of the inflammatory signalling. Several ncRNAs have been so far identified to be involved in the regulation of NF-κB signalling and inflammation [107,108,109].
Moreover, another layer of complexity added to vascular ageing is represented by the link between oxidative stress and inflammation, both leading to endothelial dysfunction [110]. Indeed, an increase in inflammatory cytokines and chemokines leads to infiltration of T cells and macrophages, but also to the internalization of oxidized lipoproteins (ox-LDL) with transformation in foam cells, both contributing to tissue injury [17,18].
Thus, the role of lncRNA/miRNA/mRNA network in cardiovascular physio-pathology will be discussed in light of these processes and findings will be summarized in Figure 4.
4.2.1. TGFB2-OT1/miR-4459, miR-3960 and miR-4488/CERS1, NAT8L, ATG13 and LARP
Autophagy and inflammation may play a coordinated role in the progression of cardiovascular events [111,112]. In particular, autophagy regulates the inflammation cascade by both clearing mitochondrial ROS and inhibiting NF-kB activation, thus preventing further injury to the atherosclerotic plaque [113,114]. In the previous section, the role of TGFB2-OT1/miRNAs network in autophagy has been discussed [78]. Works from the same research group that identified this function has showed that TGFB2-OT1/miRNA interaction plays a role in inflammation too [87]. Indeed, Huang and colleagues [87] have shown that the expression of TGFB2-OT1 is induced in ECs treated with LPS (lipopolysaccharide) or ox-LDL and that this increase is mediated by NUPR1 which, in turn, induces TIA1, responsible for TGFB2-OT1 processing from 3′UTR of TGFB2 [78]. TGFB2-OT1 acts as a ceRNA of miR-4459 [78], as well as of miR-3960 and miR-4488 [87]. The sponging of these miRNAs, in turn, augments the protein levels of their targets CERS1 (ceramide synthase 1), NAT8L (N-acetyltransferase 8-like [GCN5-related, putative]), ATG13 and LARP1 (La ribonucleoprotein domain family, member 1). LARP1 further increases the levels of SQSTM1 (Sequestosome 1), ATG3 and ATG7. Worth noting is that CERS1 and NAT8L regulate autophagy by modulating mitochondrial function. ATG3, ATG7 and ATG13 also regulate autophagy, while increased SQSTM1 levels activate RELA and CASP1, inducing the release of the inflammatory cytokines. Collectively, these data indicate that ECs autophagy and inflammation are both promoted by TGFB2-OT1 expression.
4.2.2. GAS5/miR-26a/HMGB1
The saturated fatty acid palmitic acid (PA) induces an inflammatory phenotype in cardiomyocytes, characterized by increased release of pro-inflammatory cytokines and oxidants, leading to cellular hypertrophy and apoptosis [115,116]. The expression of GAS5 is increased in PA-treated mouse cardiomyocytes (H9c2 cells), and silencing in vitro experiments have demonstrated that GAS5 is involved in both the release of inflammatory mediators and cellular injury [80]. In vitro assays have validated the bioinformatics prediction of GAS5 as interactor and of high mobility group box 1 (HMGB1) as target of miR-26a, suggesting that the network GAS5/miR-26a/HMGB1 may have a role in the mechanism of myocardial lipotoxic injury [80].
4.2.3. GAS5/miR-221/MMPs
In ox-LDL-stimulated macrophages, the expression of GAS5 is increased and its elevation aggravates ox-LDL-induced inflammation by inducing IL-6, TNF-α and IL-1β, while its silencing reversed ox-LDL-induced inflammation [81]. GAS5 expression is also increased in atherosclerotic plaques of aortic stenosis patients [81]. The pro-inflammatory effects of GAS5 on ox-LDL-stimulated macrophages are mediated by the sponging miR-221 and the consequent elevation of MMP-2 and MMP-9.
4.2.4. H19/let-7/PERIOSTIN
Cao et al., [82] have showed that H19 expression is increased in the serum of patients with atherosclerosis and in the ox-LDL-treated human umbilical vein endothelial cells (HUVECs). The effects of H19 silencing reverses the ox-LDL-mediated cell toxicity, and the secretion of inflammatory mediators and reactive oxygen species (ROS). The inflammatory and oxidative roles of H19 are mediated by periostin overexpression due to H19 sponging of let-7a [82].
4.2.5. HOTAIR/miR-34a/SIRT1
Very recently, the importance of HOTAIR has been evaluated in H9c2 rat cardiomyocytes exposed to high glucose (HG) and in a mouse model of experimental diabetes, showing that HOTAIR functions as a ceRNA to upregulate SIRT1 by sponging miR-34a [83].
HOTAIR is a lncRNA located at the antisense strand of the HOXC gene locus [117], and its modulation has been identified to be associated with CVDs [118,119,120].
In cardiomyocytes treated with HG, the silencing of HOTAIR induces a higher expression of inflammatory cytokines, ROS and apoptosis compared to cells treated with HG only [83]. Moreover, in diabetic mice, the cardiac tissue infiltration of inflammatory cells was reduced by HOTAIR overexpression, indicating an anti-inflammatory role of HOTAIR [83]. HOTAIR silencing de-represses miR-34a, reducing the expression of miR-34a target SIRT1 [83], a well-known anti-ageing factor, repressor of inflammation and oxidative-stress [121,122]. Indeed, in diabetic mice, SIRT1 knockout increased the leukocytes infiltration, the oxidative stress and, thus, the cardiac performance [83].
4.2.6. MALAT-1/miR-155/SOCS1
Several evidence indicates that the lncRNA MALAT-1 plays a vascular protective role. In particular: (1) MALAT1 regulates the migration of vascular ECs as well as vascular growth, both in vitro and in vivo, (2) it protects ECs against ox-LDL-induced dysfunction and (3) its expression is upregulated in response to high glucose treatment [123,124,125]. The expression of this lncRNA has been found to be increased in human coronary artery ECs upon ox-LDL stimulation. The ox-LDL treatment of the ECs in combination with MALAT-1 silencing stimulates the release of inflammatory factors, while MALAT-1 overexpression reduces the release of cytokines and apoptosis [84]. Interactor of MALAT-1 is miR-155, which, in turn, is induced by ox-LDL [126], and a relevant target of miR-155 is SOCS1, involved in atherosclerosis inflammation [127]. Thus, the activation of the pathway MALAT-1/miR-155/SOCS1 by repressing JAK/STAT signaling and reducing the release of cytokines, relieves the inflammation of ECs mediated by ox-LDL [84].
4.2.7. RNCR3/miR-185-5p/KLF-2
The lncRNA RNCR3 has a protective effect against inflammation in the atherosclerotic lesions, and this function is mediated by a crosstalk between KLF2 and RNCR3 through the interaction with miR-185-5p [85].
Indeed, RNCR3 has been found to be upregulated in human and mouse atherosclerotic plaques; its knock-down in ApoE−/− mice and after high-fat diet increases atherosclerosis, cholesterol and triglycerides plasma levels and the release of inflammatory mediators, indicating an anti-atherosclerotic role of RNCR3 [85]. Mechanistically, RNCR3, by sponging miR-185- 5p, induces the expression of its target KLF-2, a transcriptional factor conferring an endothelial vasoprotective phenotype [128]. Of interest is the fact that the atherosclerotic segments of aortas from apoE−/− mice expressed higher levels of both RNCR3 and KLF2. Similar data were obtained comparing human aortic atherosclerotic plaques with surrounding normal aortic tissue, confirming the relevance of the mouse model.
Thus, in the atherosclerotic plaque, the upregulation of the RNCR3/KLF-2 axis, by reducing inflammation, lipid accumulation and atherosclerosis extent, might be protective against CVDs.
4.2.8. RP5-833A20.1/miR-382/NFIA
The lncRNA RP5-833A20.1 is located in the second intron of the Nuclear Factor IA (NFIA) sequence, and its transcription direction is opposite to that of NFIA. Microarray analysis of human macrophage–derived foam cells shows that RP5-833A20.1 is upregulated, while NFIA expression is downregulated [86]. A similar modulation has been demonstrated when macrophages are exposed to both oxidized and acetylated low-density lipoprotein (ox-/Ac-LDL). The down-regulation of NFIA is RP5-833A20.1-dependent and this effect is mediated by the sponging of miR-382-5p, which targets NFIA. Moreover, the network RP5-833A20.1/miR-382/NFIA is also involved in the regulation of lipid accumulation and inflammation. Accordingly, in vivo over-expression of NFIA in apoE−/− mice reduces the atherosclerotic plaque formation [86]. Therefore, NFIA might be considered as a potential target to treat atherosclerotic vascular disease.
4.3. Cellular Senescence
There is a complex and not yet fully understood connection between oxidative stress and cell senescence. Senescence was originally observed as a mechanism of permanent withdrawal from the cell cycle after a reproducible number of cell divisions [129]. Senescence is a mechanism induced by both endogenous and exogenous stimuli, which mediate replicative senescence or stress-induced premature senescence (SIPS), respectively [130,131]. The best characterized endogenous stimulus is telomere erosion. Telomeres consist of tandem repeats of the TTAGGG sequence and are involved in the maintenance of genomic and cellular stability and replication [132]. Several mechanisms can shorten the telomere, such as cell division, aging, and ROS accumulation [131,133]. Consequently, cardiovascular risk factors such as smoking, high cholesterol levels and obesity can promote telomere attrition [134]. Among the exogenous stimuli inducing SIPS, oxidative stress is a prominent one.
Two distinct and partially intersecting pathways can mediate replicative senescence and SIPS. Among the pathways activated by senescence are the p53/p21 (replicative senescence) and the p16Ink4a/retinoblastoma (SIPS) protein pathways, both contributing to cell growth arrest [135,136].
Senescence is associated with the senescence-associated secretory phenotype (SASP), which consists in the release of a large number of pro-inflammatory cytokines and growth factors. These factors cause an increased burden of tissue inflammation and oxidative stress that shorten further the telomeres, thus aggravating the senescence process [137]. It is fair to assume that telomere shortening and cellular senescence are involved in aging and in the development of CVDs. Indeed, experiments conducted in the INK-ATTAC mouse model show that elimination of senescent cells can delay ageing-associated disorders [138].
There are several ncRNAs involved in the senescence phenotype at transcriptional, post-transcriptional, and post-translational levels (for detailed reviews see: [139,140]). Hereafter, the involvement of the lncRNA/miRNA/mRNA network in senescence is discussed and data summarized in Figure 5.
4.3.1. GAS5/miR-223/NAMPT
Recently, the network GAS5/miR-223/nicotinamide phosphoribosyltransferase (NAMPT) has been associated to the modulation of cellular senescence of endothelial progenitor cells (EPCs) [88]. The lncRNA GAS5, by sponging miR-223 in EPCs at late passages, downregulates miR-223 and subsequently de-represses the expression of miR-223-target NAMPT, which induces cell proliferation and inhibits cellular senescence by acting on PI3K/AKT signaling [88].
4.3.2. H19/miR-29b-3p/cIAP1
H19 has been demonstrated to relieve the hypoxia/post-conditioning (H/Post)-associated injury in cardiac cells through miR-29b-3p and its target cIAP1 (Cellular Inhibitor of Apoptosis Protein 1) [89]. D-Galactose derives from the digestion of lactose and, being a reducing sugar, reacts with free amines of amino acids in proteins forming advanced glycation products. In this way, the oversupply of D-galactose could generate advanced glycation products that induce oxidative-stress [141]. Zhang and colleagues have shown that, in neonatal cardiomyocytes, the induction of senescence by D-Galactose reduces H19 levels [89]. Moreover, during H/Post, both the expression of H19 and the cell number increase, but not in senescence cells. Mechanistically, during H/post, the sponging of miR-29b-3p and the increase of miR-29-3p target cIAP1 mediate the anti-apoptotic effect of H19 [89].
The ischemic post-conditioning is, normally, a cardio-protective mechanism since it reduces I/R injury [142,143]. Data from Zhang and colleagues, suggest that, in the aged heart, this protective effect can be lost, since the senescence process lowers the levels of H19, decreasing the antiapoptotic potential of post-conditioning [89].
4.3.3. lncRNA-ES3/miR-34c-5p/BMF
Vascular calcification is a well-known major risk factor for the development of cardiovascular diseases [144,145] and this process increases with ageing. While the underpinning mechanisms that are responsible for calcification still remain elusive, it has been shown that the osteoblastic program is activated in senescent VSMCs [145]. Recently, Lin et al., [90], have found that the expression of miR-34c-5p is reduced in human aorta VSMCs with the senescence/calcification phenotype induced by the hyperglycemic stimulus. Moreover, the overexpression of miR-34c-5p reduces the level of the senescence markers p16 and p21, suggesting that this miRNA is important in counteracting the senescence phenotype [90]. The authors have also demonstrated that the lncRNA-ES3 (LIN00458) is induced in VSMCs cultured in high-glucose and that lncRNA-ES3 interacts with miR-34c-5p repressing its expression and increasing the expression of miR-34c-target BMF (Bcl-2 modifying factor). Both lncRNA-ES3 and BMF overexpression induce the senescence/calcification phenotype, suggesting a role for the lncRNA-ES3/miR-34c-5p/BMF axis in vascular ageing.
4.3.4. MEG3/miR-128/Girdin
MEG3 is a well characterized lncRNA which controls vascularization and angiogenesis, EC proliferation, and senescence [146,147,148]. Girdin, also known as GIV, is involved in the signaling of G protein-coupled receptors, such as VEGF-R [149]. In particular, Lan and colleagues found that both MEG3 and Girdin are downregulated in vessels derived from aged humans or mice [91]. Moreover, reduced levels of MEG3 and Girdin have been found in senescent ECs [91]. By contrast, miR-128, which is associated with senescence [150], displayes an opposite pattern of expression [91]. In keeping with bioinformatics prediction, luciferase assay and RNA pulldown experiments indicate that there is an interplay between MEG3, miR-128 and Girdin, where decreased expression of MEG3 decreases the sponging of miR-128, which can reduce the expression of Girdin [91]. In particular, MEG3 silencing in ECs reduces platelets phagocytosis, membrane fluidity and increases lipoprotein oxidation, ROS accumulation and telomere shortening [91]. Altogether, these effects are characteristics of endothelial senescence and are associated with the onset of the atherosclerotic process.
5. Conclusions and Future Perspectives
CeRNA networks are very complex and are influenced by a variety of parameters. As described in the previous sections, the affinity of the miRNA binding site, the ability of the miRNA to induce the degradation of the bound RNA, the relative abundance of direct players are some of the elements that contribute to the complexity of lncRNA/miRNA/mRNA networks, determining the biological outcome.
In spite of all these complications, evidence of the existence and biological relevance of these networks is now clear in virtually all physiological and physio-pathological situations.
In this review, we described the lncRNAs harboring miRNA binding sites and functioning as molecular decoys or sponges by sequestering miRNAs away from other transcripts. However, other types of ncRNA interaction are also relevant for age-related CVDs. Indeed, miRNA sequences may be embedded into lncRNAs that, in this way, can act as progenitors or reservoirs of miRNAs [14,151]. An example is H19, involved in several CVDs and deregulated under cell stress conditions [119,148,152,153], which is the precursor of miR-675 [154]. In particular, the lncRNA H19 via miR-675 regulates the expression of miR-675 target VDAC1 [155]. This pathway modulates mitochondrial apoptosis induced by high-glucose and attenuates hyperglycemia-mediated oxidative stress in myocardial tissue [155]. Another target of miR-675, USP10, has been found to be involved in the modulation of the anti-senescence actions of melatonin on H2O2-treated C-kit+ cardiac progenitor cells (CPCs) [156].
A particular class of ncRNAs acting as ceRNAs is constituted by circular RNAs (circRNAs), covalently closed RNA circles [157,158]. The prototype is constituted by ciRS-7/CDR1as, expressed mostly in the brain, serving as miR-7 sponge and containing more than 70 binding sites [63,159]. Likewise, sry, a testis-specific circRNA, harbors 16 binding sites for miR-138 [159]. While these seem to be extreme cases, many circRNAs harboring only one or two bindings sites have been reported to act as miRNA decoys or sponges, also in the cardiovascular system [157]. It is expected that circRNA ceRNAs may play a role in autophagy, apoptosis, necrosis, senescence and inflammation regulation in CVDs as well.
In view of the fact that both miRNAs and lncRNAs influence mRNA function in CVDs, a clear understanding of how lncRNAs and miRNA interact may be instrumental for developing innovative therapeutic strategies. Currently, significant effort has been made to develop techniques for in vitro and in vivo manipulation of miRNA and lncRNA levels and also to translate these results to a clinical scenario.
In this respect, one element seems to be of great importance: while miRNAs are in most cases highly conserved, typically, lncRNAs display poor conservation across species, showing conserved “patterns” of bases surrounded by large less conserved sequences [14,160]. This could represent a significant hurdle to the transferability of findings in animal models of disease to humans, since some of the lncRNAs/miRNA networks are based on non-conserved lncRNA sequences.
Another important element is lncRNA annotation. While greatly improved, the process of lncRNA annotation is far from being completed and much work is still needed before an annotation accuracy compared to that of coding RNAs is obtained.
In spite of all these difficulties, many lncRNAs/miRNA/mRNA interactions have been identified, and the field is well poised to reveal more insights into how these networks are regulated and function to impact age-related CVDs. The ultimate goal is translating this knowledge to treat human CVDs [161,162]. Generally, there are two strategies for therapeutic targeting of noncoding RNAs: (1) restoring the function of noncoding RNAs that are insufficiently expressed; (2) blocking the actions of noncoding RNAs that are aberrantly overexpressed.
Both these approaches are applicable to miRNAs. Indeed, since miRNAs have a small size, and are often cytoplasmic localized, their function can be restored with synthetic miRNA mimics, miRNAs vectors and small molecules. On the other hand, their function can be blocked by a variety of strategies such as LNA anti-miR, miR-sponges, and antagomiRs.
For long noncoding RNAs, the overexpression and silencing approaches are in many aspects similar to those of coding mRNA. Unlike mature mRNAs, certain lncRNA are mostly nuclear and can be efficiently targeted by GapmeRs.
Author Contributions
S.G., C.G. and F.M. structured the text and context. S.G. designed tables and figures and reviewed the literature. All the authors approved the final version of the manuscript.
Funding
The support of Ministero della Salute (Ricerca Corrente, 5 × 1000) and of COST action (cardioRNA CA17129) is acknowledged.
Acknowledgments
Figures were partly generated by using Servier Medical Art.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design, execution, interpretation, or writing of the study.
References
- World Health Organization (WHO). Cardiovascular Diseases (CVDs) Fact Sheet; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Paneni, F.; Diaz Canestro, C.; Libby, P.; Luscher, T.F.; Camici, G.G. The Aging Cardiovascular System: Understanding it at the Cellular and Clinical Levels. J. Am. Coll. Cardiol. 2017, 69, 1952–1967. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Albert, N.M.; Allen, L.A.; Bluemke, D.A.; Butler, J.; Fonarow, G.C.; Ikonomidis, J.S.; Khavjou, O.; Konstam, M.A.; Maddox, T.M.; et al. Forecasting the Impact of Heart Failure in the United States: A Policy Statement from the American Heart Association. Circ. Heart Fail. 2013, 6, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G. Cardiovascular Regulatory Mechanisms in Advanced Age. Physiol. Rev. 1993, 73, 413–467. [Google Scholar] [CrossRef] [PubMed]
- Najjar, S.S.; Scuteri, A.; Lakatta, E.G. Arterial Aging: Is it an Immutable Cardiovascular Risk Factor? Hypertension 2005, 46, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.; Doblhammer, G.; Rau, R.; Vaupel, J.W. Ageing Populations: The Challenges Ahead. Lancet 2009, 374, 1196–1208. [Google Scholar] [CrossRef]
- Lakatta, E.G. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises: Part III: Cellular and Molecular Clues to Heart and Arterial Aging. Circulation 2003, 107, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.; Zipes, D.; Libby, P.; Bonow, R. Cardiovascular Disease in the Elderly. In Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine; Anonymous, Ed.; Saunders Elsevier: Philadelphia, PA, USA, 2007; pp. 1923–1953. [Google Scholar]
- Afilalo, J.; Karunananthan, S.; Eisenberg, M.J.; Alexander, K.P.; Bergman, H. Role of Frailty in Patients with Cardiovascular Disease. Am. J. Cardiol. 2009, 103, 1616–1621. [Google Scholar] [CrossRef]
- Lakatta, E.G. So! what’s Aging? is Cardiovascular Aging a Disease? J. Mol. Cell. Cardiol. 2015, 83, 1–13. [Google Scholar] [CrossRef]
- Greco, S.; Gorospe, M.; Martelli, F. Noncoding RNA in Age-Related Cardiovascular Diseases. J. Mol. Cell. Cardiol. 2015, 83, 142–155. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA Translation and Stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [Green Version]
- Greco, S.; Salgado Somoza, A.; Devaux, Y.; Martelli, F. Long Noncoding RNAs and Cardiac Disease. Antioxid. Redox Signal. 2018, 29, 880–901. [Google Scholar] [CrossRef] [PubMed]
- Zampetaki, A.; Albrecht, A.; Steinhofel, K. Long Non-Coding RNA Structure and Function: Is there a Link? Front. Physiol. 2018, 9, 1201. [Google Scholar] [CrossRef] [PubMed]
- Fuster, V.; Badimon, L.; Badimon, J.J.; Chesebro, J.H. The Pathogenesis of Coronary Artery Disease and the Acute Coronary Syndromes (2). N. Engl. J. Med. 1992, 326, 310–318. [Google Scholar] [PubMed]
- Mitchell, G.F.; Parise, H.; Benjamin, E.J.; Larson, M.G.; Keyes, M.J.; Vita, J.A.; Vasan, R.S.; Levy, D. Changes in Arterial Stiffness and Wave Reflection with Advancing Age in Healthy Men and Women: The Framingham Heart Study. Hypertension 2004, 43, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Thorp, E.B. Mechanisms of Failed Apoptotic Cell Clearance by Phagocyte Subsets in Cardiovascular Disease. Apoptosis 2010, 15, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Miller, Y.I.; Hedrick, C.C. Monocyte and Macrophage Dynamics during Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1506–1516. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Levy, D. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises: Part II: The Aging Heart in Health: Links to Heart Disease. Circulation 2003, 107, 346–354. [Google Scholar] [CrossRef]
- Lee, H.Y.; Oh, B.H. Aging and Arterial Stiffness. Circ. J. 2010, 74, 2257–2262. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, P.M. Hemodynamic Aging as the Consequence of Structural Changes Associated with Early Vascular Aging (EVA). Aging Dis. 2014, 5, 109–113. [Google Scholar]
- Aronson, D. Cross-Linking of Glycated Collagen in the Pathogenesis of Arterial and Myocardial Stiffening of Aging and Diabetes. J. Hypertens. 2003, 21, 3–12. [Google Scholar] [CrossRef]
- Donato, A.J.; Eskurza, I.; Silver, A.E.; Levy, A.S.; Pierce, G.L.; Gates, P.E.; Seals, D.R. Direct Evidence of Endothelial Oxidative Stress with Aging in Humans: Relation to Impaired Endothelium-Dependent Dilation and Upregulation of Nuclear Factor-kappaB. Circ. Res. 2007, 100, 1659–1666. [Google Scholar] [CrossRef] [PubMed]
- Krabbe, K.S.; Pedersen, M.; Bruunsgaard, H. Inflammatory Mediators in the Elderly. Exp. Gerontol. 2004, 39, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Manas, L.; El-Assar, M.; Vallejo, S.; Lopez-Doriga, P.; Solis, J.; Petidier, R.; Montes, M.; Nevado, J.; Castro, M.; Gomez-Guerrero, C.; et al. Endothelial Dysfunction in Aged Humans is Related with Oxidative Stress and Vascular Inflammation. Aging Cell 2009, 8, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Dulak-Lis, M.; Tsiropoulou, S.; Harvey, A.; Briones, A.M.; Touyz, R.M. Oxidative Stress and Human Hypertension: Vascular Mechanisms, Biomarkers, and Novel Therapies. Can. J. Cardiol. 2015, 31, 631–641. [Google Scholar] [CrossRef]
- Harvey, A.; Montezano, A.C.; Lopes, R.A.; Rios, F.; Touyz, R.M. Vascular Fibrosis in Aging and Hypertension: Molecular Mechanisms and Clinical Implications. Can. J. Cardiol. 2016, 32, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Pelisek, J.; Jin, Z.G. Atherosclerosis is an Epigenetic Disease. Trends Endocrinol. Metab. 2018, 29, 739–742. [Google Scholar] [CrossRef]
- Xu, S.; Kamato, D.; Little, P.J.; Nakagawa, S.; Pelisek, J.; Jin, Z.G. Targeting Epigenetics and Non-Coding RNAs in Atherosclerosis: From Mechanisms to Therapeutics. Pharmacol. Ther. 2019, 196, 15–43. [Google Scholar] [CrossRef]
- Zhang, W.; Song, M.; Qu, J.; Liu, G.H. Epigenetic Modifications in Cardiovascular Aging and Diseases. Circ. Res. 2018, 123, 773–786. [Google Scholar] [CrossRef]
- Sessions, A.O.; Engler, A.J. Mechanical Regulation of Cardiac Aging in Model Systems. Circ. Res. 2016, 118, 1553–1562. [Google Scholar] [CrossRef] [Green Version]
- Piek, A.; de Boer, R.A.; Sillje, H.H. The Fibrosis-Cell Death Axis in Heart Failure. Heart Fail. Rev. 2016, 21, 199–211. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Levy, D. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises: Part I: Aging Arteries: A “Set Up” for Vascular Disease. Circulation 2003, 107, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Yeon, S.B.; Salton, C.J.; Gona, P.; Chuang, M.L.; Blease, S.J.; Han, Y.; Tsao, C.W.; Danias, P.G.; Levy, D.; O’Donnell, C.J.; et al. Impact of Age, Sex, and Indexation Method on MR Left Ventricular Reference Values in the Framingham Heart Study Offspring Cohort. J. Magn. Reson. Imaging 2015, 41, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- Hees, P.S.; Fleg, J.L.; Lakatta, E.G.; Shapiro, E.P. Left Ventricular Remodeling with Age in Normal Men Versus Women: Novel Insights using Three-Dimensional Magnetic Resonance Imaging. Am. J. Cardiol. 2002, 90, 1231–1236. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac Fibrosis: Cell Biological Mechanisms, Molecular Pathways and Therapeutic Opportunities. Mol. Asp. Med. 2018, 65, 70–99. [Google Scholar] [CrossRef] [PubMed]
- Swynghedauw, B.; Besse, S.; Assayag, P.; Carre, F.; Chevalier, B.; Charlemagne, D.; Delcayre, C.; Hardouin, S.; Heymes, C.; Moalic, J.M. Molecular and Cellular Biology of the Senescent Hypertrophied and Failing Heart. Am. J. Cardiol. 1995, 76, 2D–7D. [Google Scholar] [CrossRef]
- Terman, A.; Brunk, U.T. Oxidative Stress, Accumulation of Biological ‘Garbage’, and Aging. Antioxid. Redox Signal. 2006, 8, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Fornes, P.; Costagliola, R.; Esposito, B.; Belmin, J.; Lecomte, D.; Tedgui, A. Age and Gender Effects on Cardiomyocyte Apoptosis in the Normal Human Heart. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2001, 56, M719–M723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eschenhagen, T.; Bolli, R.; Braun, T.; Field, L.J.; Fleischmann, B.K.; Frisen, J.; Giacca, M.; Hare, J.M.; Houser, S.; Lee, R.T.; et al. Cardiomyocyte Regeneration: A Consensus Statement. Circulation 2017, 136, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Graham, E.; Bergmann, O. Dating the Heart: Exploring Cardiomyocyte Renewal in Humans. Physiology (Bethesda) 2017, 32, 33–41. [Google Scholar] [CrossRef]
- Zacchigna, S.; Giacca, M. Extra- and Intracellular Factors Regulating Cardiomyocyte Proliferation in Postnatal Life. Cardiovasc. Res. 2014, 102, 312–320. [Google Scholar] [CrossRef]
- Gatica, D.; Chiong, M.; Lavandero, S.; Klionsky, D.J. Molecular Mechanisms of Autophagy in the Cardiovascular System. Circ. Res. 2015, 116, 456–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Zhang, Y. Targeting Autophagy in Aging and Aging-Related Cardiovascular Diseases. Trends Pharmacol. Sci. 2018, 39, 1064–1076. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble Pool of Abeta Amyloid as a Determinant of Severity of Neurodegeneration in Alzheimer’s Disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef]
- McLendon, P.M.; Robbins, J. Desmin-related cardiomyopathy: An unfolding story. J. Physiol. Heart Circ. Physiol. 2011, 301, H1220–H1228. [Google Scholar] [CrossRef] [PubMed]
- Agnetti, G.; Halperin, V.L.; Kirk, J.A.; Chakir, K.; Guo, Y.; Lund, L.; Nicolini, F.; Gherli, T.; Guarnieri, C.; Caldarera, C.M.; et al. Desmin Modifications Associate with Amyloid-Like Oligomers Deposition in Heart Failure. Cardiovasc. Res. 2014, 102, 24–34. [Google Scholar] [CrossRef] [PubMed]
- McLendon, P.M.; Robbins, J. Proteotoxicity and Cardiac Dysfunction. Circ. Res. 2015, 116, 1863–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianni, D.; Li, A.; Tesco, G.; McKay, K.M.; Moore, J.; Raygor, K.; Rota, M.; Gwathmey, J.K.; Dec, G.W.; Aretz, T.; et al. Protein Aggregates and Novel Presenilin Gene Variants in Idiopathic Dilated Cardiomyopathy. Circulation 2010, 121, 1216–1226. [Google Scholar] [CrossRef] [Green Version]
- Pattison, J.S.; Sanbe, A.; Maloyan, A.; Osinska, H.; Klevitsky, R.; Robbins, J. Cardiomyocyte Expression of a Polyglutamine Preamyloid Oligomer Causes Heart Failure. Circulation 2008, 117, 2743–2751. [Google Scholar] [CrossRef]
- Greco, S.; Zaccagnini, G.; Fuschi, P.; Voellenkle, C.; Carrara, M.; Sadeghi, I.; Bearzi, C.; Maimone, B.; Castelvecchio, S.; Stellos, K.; et al. Increased BACE1-AS Long Noncoding RNA and Beta-Amyloid Levels in Heart Failure. Cardiovasc. Res. 2017, 113, 453–463. [Google Scholar] [CrossRef]
- Gonzalez-Lopez, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; de Haro-Del Moral, F.J.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Wild-Type Transthyretin Amyloidosis as a Cause of Heart Failure with Preserved Ejection Fraction. Eur. Heart J. 2015, 36, 2585–2594. [Google Scholar] [CrossRef]
- Connors, L.H.; Sam, F.; Skinner, M.; Salinaro, F.; Sun, F.; Ruberg, F.L.; Berk, J.L.; Seldin, D.C. Heart Failure Resulting from Age-Related Cardiac Amyloid Disease Associated with Wild-Type Transthyretin: A Prospective, Observational Cohort Study. Circulation 2016, 133, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Merlini, G.; Quarta, C.C.; Riva, L.; Longhi, S.; Leone, O.; Salvi, F.; Ciliberti, P.; Pastorelli, F.; Biagini, E.; et al. Systemic Cardiac Amyloidoses: Disease Profiles and Clinical Courses of the 3 Main Types. Circulation 2009, 120, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.F.; Mirzoyev, S.A.; Edwards, W.D.; Dogan, A.; Grogan, D.R.; Dunlay, S.M.; Roger, V.L.; Gertz, M.A.; Dispenzieri, A.; Zeldenrust, S.R.; et al. Left Ventricular Amyloid Deposition in Patients with Heart Failure and Preserved Ejection Fraction. JACC Heart Fail. 2014, 2, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Michels da Silva, D.; Langer, H.; Graf, T. Inflammatory and Molecular Pathways in Heart Failure-Ischemia, HFpEF and Transthyretin Cardiac Amyloidosis. Int. J. Mol. Sci. 2019, 20, 2322. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Loedige, I.; Filipowicz, W. The Widespread Regulation of microRNA Biogenesis, Function and Decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Gorospe, M.; Abdelmohsen, K. MicroRegulators Come of Age in Senescence. Trends Genet. 2011, 27, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Amort, T.; Souliere, M.F.; Wille, A.; Jia, X.Y.; Fiegl, H.; Worle, H.; Micura, R.; Lusser, A. Long Non-Coding RNAs as Targets for Cytosine Methylation. RNA Biol. 2013, 10, 1003–1008. [Google Scholar] [CrossRef]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the Classification of Long Non-Coding RNAs. RNA Biol. 2013, 10, 925–933. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome Regulation by Long Noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Clark, M.B.; Mattick, J.S. Long Noncoding RNAs in Cell Biology. Semin. Cell Dev. Biol. 2011, 22, 366–376. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a Large Class of Animal RNAs with Regulatory Potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wee, L.M.; Flores-Jasso, C.F.; Salomon, W.E.; Zamore, P.D. Argonaute Divides its RNA Guide into Domains with Distinct Functions and RNA-Binding Properties. Cell 2012, 151, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Devaux, Y.; Zangrando, J.; Schroen, B.; Creemers, E.E.; Pedrazzini, T.; Chang, C.P.; Dorn, G.W., 2nd; Thum, T.; Heymans, S. Cardiolinc network. Long Noncoding RNAs in Cardiac Development and Ageing. Nat. Rev. Cardiol. 2015, 12, 415–425. [Google Scholar] [PubMed]
- Kim, J.; Kim, K.M.; Noh, J.H.; Yoon, J.H.; Abdelmohsen, K.; Gorospe, M. Long Noncoding RNAs in Diseases of Aging. Biochim. Biophys. Acta 2016, 1859, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; Roth, L.; Schrijvers, D.M.; De Meyer, G.R.Y.; Martinet, W. Defective Autophagy in Atherosclerosis: To Die or to Senesce? Oxid. Med. Cell. Longev. 2018, 2018, 7687083. [Google Scholar] [CrossRef]
- Libby, P. Mechanisms of Acute Coronary Syndromes and their Implications for Therapy. N. Engl. J. Med. 2013, 368, 2004–2013. [Google Scholar] [CrossRef]
- Mei, Y.; Thompson, M.D.; Cohen, R.A.; Tong, X. Autophagy and Oxidative Stress in Cardiovascular Diseases. Biochim. Biophys. Acta 2015, 1852, 243–251. [Google Scholar] [CrossRef]
- Wang, K.; Liu, C.Y.; Zhou, L.Y.; Wang, J.X.; Wang, M.; Zhao, B.; Zhao, W.K.; Xu, S.J.; Fan, L.H.; Zhang, X.J.; et al. APF lncRNA Regulates Autophagy and Myocardial Infarction by Targeting miR-188-3p. Nat. Commun. 2015, 6, 6779. [Google Scholar] [CrossRef]
- Wang, J.J.; Bie, Z.D.; Sun, C.F. Long Noncoding RNA AK088388 Regulates Autophagy through miR-30a to Affect Cardiomyocyte Injury. J. Cell. Biochem. 2019, 120, 10155–10163. [Google Scholar] [CrossRef]
- Yu, S.Y.; Dong, B.; Fang, Z.F.; Hu, X.Q.; Tang, L.; Zhou, S.H. Knockdown of lncRNA AK139328 Alleviates Myocardial Ischaemia/Reperfusion Injury in Diabetic Mice Via Modulating miR-204-3p and Inhibiting Autophagy. J. Cell. Mol. Med. 2018, 22, 4886–4898. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Zhang, M.; Huang, J.; Modarresi, F.; Van der Brug, M.P.; Nalls, M.A.; Cookson, M.R.; St-Laurent, G., 3rd; Wahlestedt, C. Evidence for Natural Antisense Transcript-Mediated Inhibition of microRNA Function. Genome Biol. 2010, 11, R56. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Ni, H.; Yu, Y.; Zhang, M.; Wu, M.; Wang, Q.; Wang, L.; Xu, S.; Xu, Z.; Xu, C.; et al. BACE1-AS Prevents BACE1 mRNA Degradation through the Sequestration of BACE1-Targeting miRNAs. J. Chem. Neuroanat. 2019, 98, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Yang, X.; Li, Q.; Guo, Z. GATA1 Activated lncRNA (Galont) Promotes Anoxia/Reoxygenation-Induced Autophagy and Cell Death in Cardiomyocytes by Sponging miR-338. J. Cell. Biochem. 2018, 119, 4161–4169. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Fan, T.; Liu, L.; Zhang, L. Knockdown of Growth-Arrest Specific Transcript 5 Restores Oxidized Low-Density Lipoprotein-Induced Impaired Autophagy Flux Via Upregulating miR-26a in Human Endothelial Cells. Eur. J. Pharmacol. 2019, 843, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Han, L.; Huang, S.; Peng, N.; Wang, P.; Jiang, Z.; Zhao, J.; Su, L.; Zhang, S.; Zhang, Y.; et al. Identification of a Novel MTOR Activator and Discovery of a Competing Endogenous RNA Regulating Autophagy in Vascular Endothelial Cells. Autophagy 2014, 10, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, R.; Niu, Q.; Wang, H.; Yang, Z.; Bao, Y. Morphine Postconditioning Alleviates Autophage in Ischemia-Reperfusion Induced Cardiac Injury through Up-Regulating lncRNA UCA1. Biomed. Pharmacother. 2018, 108, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Yue, Q.; Zhao, C.; Wang, Y.; Zhao, L.; Zhu, Q.; Li, G.; Wu, N.; Jia, D.; Ma, C. Downregulation of Growth Arrest specific Transcript 5 Alleviates Palmitic Acid induced Myocardial Inflammatory Injury through the miR26a/HMGB1/NFkappaB Axis. Mol. Med. Rep. 2018, 18, 5742–5750. [Google Scholar]
- Ye, J.; Wang, C.; Wang, D.; Yuan, H. LncRBA GSA5, Up-Regulated by Ox-LDL, Aggravates Inflammatory Response and MMP Expression in THP-1 Macrophages by Acting Like a Sponge for miR-221. Exp. Cell Res. 2018, 369, 348–355. [Google Scholar] [CrossRef]
- Cao, L.; Zhang, Z.; Li, Y.; Zhao, P.; Chen, Y. LncRNA H19/miR-Let-7 Axis Participates in the Regulation of Ox-LDL-Induced Endothelial Cell Injury Via Targeting Periostin. Int. Immunopharmacol. 2019, 72, 496–503. [Google Scholar] [CrossRef]
- Gao, L.; Wang, X.; Guo, S.; Xiao, L.; Liang, C.; Wang, Z.; Li, Y.; Liu, Y.; Yao, R.; Liu, Y.; et al. LncRNA HOTAIR Functions as a Competing Endogenous RNA to Upregulate SIRT1 by Sponging miR-34a in Diabetic Cardiomyopathy. J. Cell. Physiol. 2019, 234, 4944–4958. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sun, Y.; Zhong, L.; Xiao, Z.; Yang, M.; Chen, M.; Wang, C.; Xie, X.; Chen, X. The Suppression of Ox-LDL-Induced Inflammatory Cytokine Release and Apoptosis of HCAECs by Long Non-Coding RNA-MALAT1 Via Regulating microRNA-155/SOCS1 Pathway. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Shan, K.; Jiang, Q.; Wang, X.Q.; Wang, Y.N.; Yang, H.; Yao, M.D.; Liu, C.; Li, X.M.; Yao, J.; Liu, B.; et al. Role of Long Non-Coding RNA-RNCR3 in Atherosclerosis-Related Vascular Dysfunction. Cell Death Dis. 2016, 7, e2248. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.W.; Zhao, J.Y.; Li, S.F.; Huang, J.L.; Qiu, Y.R.; Ma, X.; Wu, S.G.; Chen, Z.P.; Hu, Y.R.; Yang, J.Y.; et al. RP5-833A20.1/miR-382-5p/NFIA-Dependent Signal Transduction Pathway Contributes to the Regulation of Cholesterol Homeostasis and Inflammatory Reaction. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Lu, W.; Ge, D.; Meng, N.; Li, Y.; Su, L.; Zhang, S.; Zhang, Y.; Zhao, B.; Miao, J. A New microRNA Signal Pathway Regulated by Long Noncoding RNA TGFB2-OT1 in Autophagy and Inflammation of Vascular Endothelial Cells. Autophagy 2015, 11, 2172–2183. [Google Scholar] [CrossRef]
- Yao, J.; Shi, Z.; Ma, X.; Xu, D.; Ming, G. lncRNA GAS5/miR-223/NAMPT Axis Modulates the Cell Proliferation and Senescence of Endothelial Progenitor Cells through PI3K/AKT Signaling. J. Cell. Biochem. 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cheng, L.; Xu, L.; Zhang, Y.; Yang, Y.; Fu, Q.; Mi, W.; Li, H. The Lncrna, H19 Mediates the Protective Effect of Hypoxia Postconditioning Against Hypoxia-Reoxygenation Injury to Senescent Cardiomyocytes by Targeting MicroRNA-29b-3p. Shock 2018. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zhan, J.K.; Zhong, J.Y.; Wang, Y.J.; Wang, Y.; Li, S.; He, J.Y.; Tan, P.; Chen, Y.Y.; Liu, X.B.; et al. lncRNA-ES3/miR-34c-5p/BMF Axis is Involved in Regulating High-Glucose-Induced Calcification/Senescence of VSMCs. Aging (Albany NY) 2019, 11, 523–535. [Google Scholar] [CrossRef]
- Lin, C.; Ear, J.; Midde, K.; Lopez-Sanchez, I.; Aznar, N.; Garcia-Marcos, M.; Kufareva, I.; Abagyan, R.; Ghosh, P. Structural Basis for Activation of Trimeric Gi Proteins by Multiple Growth Factor Receptors Via GIV/Girdin. Mol. Biol. Cell 2014, 25, 3654–3671. [Google Scholar] [CrossRef]
- De Duve, C.; Wattiaux, R. Functions of Lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef]
- Wohlgemuth, S.E.; Calvani, R.; Marzetti, E. The Interplay between Autophagy and Mitochondrial Dysfunction in Oxidative Stress-Induced Cardiac Aging and Pathology. J. Mol. Cell. Cardiol. 2014, 71, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Rifki, O.F.; Hill, J.A. Cardiac Autophagy: Good with the Bad. J. Cardiovasc. Pharmacol. 2012, 60, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Hamacher-Brady, A.; Brady, N.R.; Gottlieb, R.A. The Interplay between Pro-Death and Pro-Survival Signaling Pathways in Myocardial Ischemia/Reperfusion Injury: Apoptosis Meets Autophagy. Cardiovasc. Drugs Ther. 2006, 20, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Wang, Y.; Chen, Y.; Cao, F. The Role of the Autophagy in Myocardial Ischemia/Reperfusion Injury. Biochim. Biophys. Acta 2015, 1852, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Essick, E.E.; Sam, F. Oxidative Stress and Autophagy in Cardiac Disease, Neurological Disorders, Aging and Cancer. Oxid. Med. Cell. Longev. 2010, 3, 168–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Meyer, G.R.; De Keulenaer, G.W.; Martinet, W. Role of Autophagy in Heart Failure Associated with Aging. Heart Fail. Rev. 2010, 15, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, W.; Guo, Y.; Chen, W.; Zheng, P.; Zeng, J.; Tong, W. Exosomal lncRNA GAS5 Regulates the Apoptosis of Macrophages and Vascular Endothelial Cells in Atherosclerosis. PLoS ONE 2017, 12, e0185406. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Meng, N.; Zhao, B.; Zhao, J.; Zhang, Y.; Zhang, S.; Miao, J. Protective Effects of a Synthesized Butyrolactone Derivative Against Chloroquine-Induced Autophagic Vesicle Accumulation and the Disturbance of Mitochondrial Membrane Potential and Na+,K+-ATPase Activity in Vascular Endothelial Cells. Chem. Res. Toxicol. 2009, 22, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-Aging. an Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic Inflammation in Ageing, Cardiovascular Disease, and Frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Candales, A.; Hernandez Burgos, P.M.; Hernandez-Suarez, D.F.; Harris, D. Linking Chronic Inflammation with Cardiovascular Disease: From Normal Aging to the Metabolic Syndrome. J. Nat. Sci. 2017, 3, e341. [Google Scholar] [PubMed]
- Zuliani, G.; Morieri, M.L.; Volpato, S.; Maggio, M.; Cherubini, A.; Francesconi, D.; Bandinelli, S.; Paolisso, G.; Guralnik, J.M.; Ferrucci, L. Insulin Resistance and Systemic Inflammation, but Not Metabolic Syndrome Phenotype, Predict 9 Years Mortality in Older Adults. Atherosclerosis 2014, 235, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Geovanini, G.R.; Libby, P. Atherosclerosis and Inflammation: Overview and Updates. Clin. Sci. (Lond.) 2018, 132, 1243–1252. [Google Scholar] [CrossRef]
- Lucas, A.R.; Korol, R.; Pepine, C.J. Inflammation in Atherosclerosis: Some Thoughts about Acute Coronary Syndromes. Circulation 2006, 113, e728–e732. [Google Scholar] [CrossRef] [PubMed]
- Boldin, M.P.; Baltimore, D. MicroRNAs, New Effectors and Regulators of NF-kappaB. Immunol. Rev. 2012, 246, 205–220. [Google Scholar] [CrossRef]
- Chew, C.L.; Conos, S.A.; Unal, B.; Tergaonkar, V. Noncoding RNAs: Master Regulators of Inflammatory Signaling. Trends Mol. Med. 2018, 24, 66–84. [Google Scholar] [CrossRef] [PubMed]
- Magagula, L.; Gagliardi, M.; Naidoo, J.; Mhlanga, M. Lnc-Ing Inflammation to Disease. Biochem. Soc. Trans. 2017, 45, 953–962. [Google Scholar] [CrossRef]
- Guzik, T.J.; Touyz, R.M. Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef]
- Zhang, J.; Rane, G.; Dai, X.; Shanmugam, M.K.; Arfuso, F.; Samy, R.P.; Lai, M.K.; Kappei, D.; Kumar, A.P.; Sethi, G. Ageing and the Telomere Connection: An Intimate Relationship with Inflammation. Ageing Res. Rev. 2016, 25, 55–69. [Google Scholar] [CrossRef]
- Razani, B.; Feng, C.; Coleman, T.; Emanuel, R.; Wen, H.; Hwang, S.; Ting, J.P.; Virgin, H.W.; Kastan, M.B.; Semenkovich, C.F. Autophagy Links Inflammasomes to Atherosclerotic Progression. Cell. Metab. 2012, 15, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in Infection, Inflammation and Immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in Immunity and Inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Guardia, D.; Palomer, X.; Coll, T.; Serrano, L.; Rodriguez-Calvo, R.; Davidson, M.M.; Merlos, M.; El Kochairi, I.; Michalik, L.; Wahli, W.; et al. PPARbeta/Delta Activation Blocks Lipid-Induced Inflammatory Pathways in Mouse Heart and Human Cardiac Cells. Biochim. Biophys. Acta 2011, 1811, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.; Zhu, H.; Liang, Z.; Ma, X.; Li, S. GLP1 Protects Cardiomyocytes from Palmitate-Induced Apoptosis Via Akt/GSK3b/B-Catenin Pathway. J. Mol. Endocrinol. 2015, 55, 245–262. [Google Scholar] [CrossRef]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional Demarcation of Active and Silent Chromatin Domains in Human HOX Loci by Noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Liu, Y.; Guo, S.; Yao, R.; Wu, L.; Xiao, L.; Wang, Z.; Liu, Y.; Zhang, Y. Circulating Long Noncoding RNA HOTAIR is an Essential Mediator of Acute Myocardial Infarction. Cell. Physiol. Biochem. 2017, 44, 1497–1508. [Google Scholar]
- Greco, S.; Zaccagnini, G.; Perfetti, A.; Fuschi, P.; Valaperta, R.; Voellenkle, C.; Castelvecchio, S.; Gaetano, C.; Finato, N.; Beltrami, A.P.; et al. Long Noncoding RNA Dysregulation in Ischemic Heart Failure. J. Transl. Med. 2016, 14, 183. [Google Scholar] [CrossRef]
- Lai, Y.; He, S.; Ma, L.; Lin, H.; Ren, B.; Ma, J.; Zhu, X.; Zhuang, S. HOTAIR Functions as a Competing Endogenous RNA to Regulate PTEN Expression by Inhibiting miR-19 in Cardiac Hypertrophy. Mol. Cell. Biochem. 2017, 432, 179–187. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Q.; Zeng, Z.; Wu, J.; Zhang, Y.; Chen, Z. Sirt1 Inhibits Oxidative Stress in Vascular Endothelial Cells. Oxid. Med. Cell. Longev. 2017, 2017, 7543973. [Google Scholar] [CrossRef]
- Kitada, M.; Ogura, Y.; Koya, D. The Protective Role of Sirt1 in Vascular Tissue: Its Relationship to Vascular Aging and Atherosclerosis. Aging (Albany NY) 2016, 8, 2290–2307. [Google Scholar] [CrossRef]
- Michalik, K.M.; You, X.; Manavski, Y.; Doddaballapur, A.; Zornig, M.; Braun, T.; John, D.; Ponomareva, Y.; Chen, W.; Uchida, S.; et al. Long Noncoding RNA MALAT1 Regulates Endothelial Cell Function and Vessel Growth. Circ. Res. 2014, 114, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Jin, X.; Xiang, Y.; Chen, Y.; Shen, C.X.; Zhang, Y.C.; Li, Y.G. The lncRNA MALAT1 Protects the Endothelium Against Ox-LDL-Induced Dysfunction Via Upregulating the Expression of the miR-22-3p Target Genes CXCR2 and AKT. FEBS Lett. 2015, 589, 3189–3196. [Google Scholar] [CrossRef] [PubMed]
- Puthanveetil, P.; Chen, S.; Feng, B.; Gautam, A.; Chakrabarti, S. Long Non-Coding RNA MALAT1 Regulates Hyperglycaemia Induced Inflammatory Process in the Endothelial Cells. J. Cell. Mol. Med. 2015, 19, 1418–1425. [Google Scholar] [CrossRef]
- Tili, E.; Michaille, J.J.; Cimino, A.; Costinean, S.; Dumitru, C.D.; Adair, B.; Fabbri, M.; Alder, H.; Liu, C.G.; Calin, G.A.; et al. Modulation of miR-155 and miR-125b Levels Following Lipopolysaccharide/TNF-Alpha Stimulation and their Possible Roles in Regulating the Response to Endotoxin Shock. J. Immunol. 2007, 179, 5082–5089. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, L.; Liang, X.; Zhu, G. MicroRNA-155 Promotes Atherosclerosis Inflammation Via Targeting SOCS1. Cell. Physiol. Biochem. 2015, 36, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Dekker, R.J.; Boon, R.A.; Rondaij, M.G.; Kragt, A.; Volger, O.L.; Elderkamp, Y.W.; Meijers, J.C.; Voorberg, J.; Pannekoek, H.; Horrevoets, A.J. KLF2 Provokes a Gene Expression Pattern that Establishes Functional Quiescent Differentiation of the Endothelium. Blood 2006, 107, 4354–4363. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Weinberg, R.A. The Signals and Pathways Activating Cellular Senescence. Int. J. Biochem. Cell Biol. 2005, 37, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Sack, M.N.; Fyhrquist, F.Y.; Saijonmaa, O.J.; Fuster, V.; Kovacic, J.C. Basic Biology of Oxidative Stress and the Cardiovascular System: Part 1 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 196–211. [Google Scholar] [CrossRef] [PubMed]
- de Lange, T. Shelterin: The Protein Complex that Shapes and Safeguards Human Telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- von Zglinicki, T. Oxidative Stress Shortens Telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Carnevali, S.; Petruzzelli, S.; Longoni, B.; Vanacore, R.; Barale, R.; Cipollini, M.; Scatena, F.; Paggiaro, P.; Celi, A.; Giuntini, C. Cigarette Smoke Extract Induces Oxidative Stress and Apoptosis in Human Lung Fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L955–L963. [Google Scholar] [CrossRef] [PubMed]
- van Deursen, J.M. The Role of Senescent Cells in Ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Ji, S. Cellular Senescence: Molecular Mechanisms and Pathogenicity. J. Cell. Physiol. 2018, 233, 9121–9135. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-Positive Senescent Cells Delays Ageing-Associated Disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.C.; Abdelmohsen, K.; Gorospe, M. SASP Regulation by Noncoding RNA. Mech. Ageing Dev. 2017, 168, 37–43. [Google Scholar] [CrossRef]
- Grammatikakis, I.; Panda, A.C.; Abdelmohsen, K.; Gorospe, M. Long Noncoding RNAs(lncRNAs) and the Molecular Hallmarks of Aging. Aging (Albany NY) 2014, 6, 992–1009. [Google Scholar] [CrossRef]
- Song, X.; Bao, M.; Li, D.; Li, Y.M. Advanced Glycation in D-Galactose Induced Mouse Aging Model. Mech. Ageing Dev. 1999, 108, 239–251. [Google Scholar] [CrossRef]
- Donato, M.; Evelson, P.; Gelpi, R.J. Protecting the Heart from Ischemia/Reperfusion Injury: An Update on Remote Ischemic Preconditioning and Postconditioning. Curr. Opin. Cardiol. 2017, 32, 784–790. [Google Scholar] [CrossRef]
- Lefer, D.J.; Marban, E. Is Cardioprotection Dead? Circulation 2017, 136, 98–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rennenberg, R.J.; Kessels, A.G.; Schurgers, L.J.; van Engelshoven, J.M.; de Leeuw, P.W.; Kroon, A.A. Vascular Calcifications as a Marker of Increased Cardiovascular Risk: A Meta-Analysis. Vasc. Health Risk Manag. 2009, 5, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.G.; Matsubara, H.; Ikeda, K. Pathophysiology of Vascular Calcification: Pivotal Role of Cellular Senescence in Vascular Smooth Muscle Cells. Exp. Gerontol. 2010, 45, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.Z.; Tian, W.; Fu, H.T.; Li, C.P.; Liu, B. Long Noncoding RNA-MEG3 is Involved in Diabetes Mellitus-Related Microvascular Dysfunction. Biochem. Biophys. Res. Commun. 2016, 471, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Hofmann, P.; Michalik, K.M.; Lozano-Vidal, N.; Berghauser, D.; Fischer, A.; Knau, A.; Jae, N.; Schurmann, C.; Dimmeler, S. Long Noncoding RNA Meg3 Controls Endothelial Cell Aging and Function: Implications for Regenerative Angiogenesis. J. Am. Coll. Cardiol. 2016, 68, 2589–2591. [Google Scholar] [CrossRef] [PubMed]
- Simion, V.; Haemmig, S.; Feinberg, M.W. LncRNAs in Vascular Biology and Disease. Vasc. Pharmacol. 2018, 114, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Li, Y.J.; Li, D.J.; Li, P.; Wang, J.Y.; Diao, Y.P.; Ye, G.D.; Li, Y.F. Long Non-Coding RNA MEG3 Prevents Vascular Endothelial Cell Senescence by Impairing miR-128-Dependent Girdin Down-Regulation. Am. J. Physiol. Cell. Physiol. 2018, 316, C830–C843. [Google Scholar] [CrossRef] [PubMed]
- Noren Hooten, N.; Abdelmohsen, K.; Gorospe, M.; Ejiogu, N.; Zonderman, A.B.; Evans, M.K. microRNA Expression Patterns Reveal Differential Expression of Target Genes with Age. PLoS ONE 2010, 5, e10724. [Google Scholar] [CrossRef] [PubMed]
- Dykes, I.M.; Emanueli, C. Transcriptional and Post-Transcriptional Gene Regulation by Long Non-Coding RNA. Genom. Proteom. Bioinform. 2017, 15, 177–186. [Google Scholar] [CrossRef]
- Voellenkle, C.; Garcia-Manteiga, J.M.; Pedrotti, S.; Perfetti, A.; De Toma, I.; Da Silva, D.; Maimone, B.; Greco, S.; Fasanaro, P.; Creo, P.; et al. Implication of Long Noncoding RNAs in the Endothelial Cell Response to Hypoxia Revealed by RNA-Sequencing. Sci. Rep. 2016, 6, 24141. [Google Scholar] [CrossRef]
- Merryman, W.D.; Clark, C.R. Lnc-Ing NOTCH1 to Idiopathic Calcific Aortic Valve Disease. Circulation 2016, 134, 1863–1865. [Google Scholar] [CrossRef] [PubMed]
- Keniry, A.; Oxley, D.; Monnier, P.; Kyba, M.; Dandolo, L.; Smits, G.; Reik, W. The H19 lincRNA is a Developmental Reservoir of miR-675 that Suppresses Growth and Igf1r. Nat. Cell Biol. 2012, 14, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H.; Yao, B.; Xu, W.; Chen, J.; Zhou, X. lncRNA H19/miR-675 Axis Regulates Cardiomyocyte Apoptosis by Targeting VDAC1 in Diabetic Cardiomyopathy. Sci. Rep. 2016, 6, 36340. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Ma, W.; Bi, C.; Yang, F.; Zhang, L.; Han, Z.; Huang, Q.; Ding, F.; Li, Y.; Yan, G.; et al. Long Noncoding RNA H19 Mediates Melatonin Inhibition of Premature Senescence of C-Kit+ Cardiac Progenitor Cells by Promoting miR-675. J. Pineal Res. 2016, 61, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Carrara, M.; Fuschi, P.; Ivan, C.; Martelli, F. Circular RNAs: Methodological Challenges and Perspectives in Cardiovascular Diseases. J. Cell. Mol. Med. 2018, 22, 5176–5187. [Google Scholar] [CrossRef] [PubMed]
- Li, H.M.; Ma, X.L.; Li, H.G. Intriguing Circles: Conflicts and Controversies in Circular RNA Research. Wiley Interdiscip. Rev. RNA 2019, e1538. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA Circles Function as Efficient microRNA Sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Gandhi, S.; Ruehle, F.; Stoll, M. Evolutionary Patterns of Non-Coding RNA in Cardiovascular Biology. Non-Coding RNA 2019, 5, 15. [Google Scholar] [CrossRef]
- Bassett, A.R.; Akhtar, A.; Barlow, D.P.; Bird, A.P.; Brockdorff, N.; Duboule, D.; Ephrussi, A.; Ferguson-Smith, A.C.; Gingeras, T.R.; Haerty, W.; et al. Considerations when Investigating lncRNA Function in Vivo. Elife 2014, 3, e03058. [Google Scholar] [CrossRef]
- Boon, R.A.; Jae, N.; Holdt, L.; Dimmeler, S. Long Noncoding RNAs: From Clinical Genetics to Therapeutic Targets? J. Am. Coll. Cardiol. 2016, 67, 1214–1226. [Google Scholar] [CrossRef]
Figure 1.
Cardiovascular function deterioration by ageing. (A) Vascular function impairment by ageing. The loss of aorta distensibility with ageing is associated to: (1) increased collagen deposition; (2) the depletion of elastin; (3) the non-enzymatic glycosylation of collagen, which results in the cross-linking of adjacent proteins; (4) amyloid deposition in the medial layer; (5) migration/proliferation of VSMCs and (6) endothelial hyperplasia. The impaired distensibility is responsible for the development of hypertension and decreased coronary perfusion. (B) Cardiac function impairment by ageing. The ageing process of the cardiac system is characterized by a reduction of both peak cardiac-output and left ventricular (LV) diastolic function, an altered response to catecholamine, an incomplete relaxation during early diastolic filling, and increased myocardial stiffness. These responses are compensated by increased muscle mass that leads to cardiac hypertrophy and LV wall thickening. This compensatory mechanism enhances cardiac output at the beginning, but reduces the cardiac function as hypertrophy increases. Fibroblast activation to myofibroblast, increased apoptosis, collagen deposition and crosslinking, inflammatory cells migration and perivascular fibrosis are frequent histologic findings in the old myocardium.
Figure 1.
Cardiovascular function deterioration by ageing. (A) Vascular function impairment by ageing. The loss of aorta distensibility with ageing is associated to: (1) increased collagen deposition; (2) the depletion of elastin; (3) the non-enzymatic glycosylation of collagen, which results in the cross-linking of adjacent proteins; (4) amyloid deposition in the medial layer; (5) migration/proliferation of VSMCs and (6) endothelial hyperplasia. The impaired distensibility is responsible for the development of hypertension and decreased coronary perfusion. (B) Cardiac function impairment by ageing. The ageing process of the cardiac system is characterized by a reduction of both peak cardiac-output and left ventricular (LV) diastolic function, an altered response to catecholamine, an incomplete relaxation during early diastolic filling, and increased myocardial stiffness. These responses are compensated by increased muscle mass that leads to cardiac hypertrophy and LV wall thickening. This compensatory mechanism enhances cardiac output at the beginning, but reduces the cardiac function as hypertrophy increases. Fibroblast activation to myofibroblast, increased apoptosis, collagen deposition and crosslinking, inflammatory cells migration and perivascular fibrosis are frequent histologic findings in the old myocardium.


Figure 2.
Competitive interactions between lncRNAs, miRNAs and mRNAs. The presence of miRNA response elements (MREs) allows the binding of miRNAs, which results in the inhibition of mRNA expression and/or stability. In addition, lncRNAs acting as ceRNAs harbor MREs and function as “sponges” for the miRNAs, which are sequestered so that the miRNA-RISC is unable to regulate the expression of target mRNAs. Thus, by acting as ceRNAs, lncRNAs are competitors for the binding of miRNA to the mRNA targets.
Figure 2.
Competitive interactions between lncRNAs, miRNAs and mRNAs. The presence of miRNA response elements (MREs) allows the binding of miRNAs, which results in the inhibition of mRNA expression and/or stability. In addition, lncRNAs acting as ceRNAs harbor MREs and function as “sponges” for the miRNAs, which are sequestered so that the miRNA-RISC is unable to regulate the expression of target mRNAs. Thus, by acting as ceRNAs, lncRNAs are competitors for the binding of miRNA to the mRNA targets.

Figure 3.
LncRNA-miRNA-mRNA networks and autophagy. A decrease in autophagic function is a hallmark of ageing. A variety of stimuli, such as hypoxia or ischemia/reperfusion, oxidized lipoproteins (ox-LDL) or treatment with an MTOR pathway agonist (3BDO), can activate the interaction of lncRNA and miRNA, and the specific modulation of direct and/or indirect targets that, in turn, can affect positively or negatively the autophagic process.
Figure 3.
LncRNA-miRNA-mRNA networks and autophagy. A decrease in autophagic function is a hallmark of ageing. A variety of stimuli, such as hypoxia or ischemia/reperfusion, oxidized lipoproteins (ox-LDL) or treatment with an MTOR pathway agonist (3BDO), can activate the interaction of lncRNA and miRNA, and the specific modulation of direct and/or indirect targets that, in turn, can affect positively or negatively the autophagic process.

Figure 4.
LncRNA-miRNA-mRNA networks and inflammation. During ageing, there is an increased accumulation of inflammatory molecules as well as a burst in oxidative stress. A prolonged state of inflammation can be detrimental for cardiovascular tissue and it can predispose to atherogenesis. Indeed, an increase in inflammatory cytokines and chemokines leads to the internalization of oxidized lipoproteins (ox-LDL) with transformation in foam cells, both contributing to tissue injury. The figure summarizes in vitro experiments performed in endothelial cells (ECs), macrophages cells (M∅) and cardiomyocytes (CMs).
Figure 4.
LncRNA-miRNA-mRNA networks and inflammation. During ageing, there is an increased accumulation of inflammatory molecules as well as a burst in oxidative stress. A prolonged state of inflammation can be detrimental for cardiovascular tissue and it can predispose to atherogenesis. Indeed, an increase in inflammatory cytokines and chemokines leads to the internalization of oxidized lipoproteins (ox-LDL) with transformation in foam cells, both contributing to tissue injury. The figure summarizes in vitro experiments performed in endothelial cells (ECs), macrophages cells (M∅) and cardiomyocytes (CMs).

Figure 5.
LncRNA-miRNA-mRNA networks and senescence. Cellular senescence is associated with the release of a large number of pro-inflammatory cytokines and growth factors. These factors cause an increased burden of tissue inflammation and oxidative stress that contribute to telomere shortening. Moreover, telomere shortening and cellular senescence are involved in aging and in the development of CVDs. The figure shows that the senescence phenotype can be induced by: (1) ageing the endothelial cells by culturing until late passages (p9) (2) stimulating cardiomyocytes (CMs) with D-galactose and (3) treating vascular smooth muscle cells (VSMCs) with high glucose. The lncRNA-miRNA-mRNA networks triggered by these conditions stimulate or inhibit senescence according to the modulated targets.
Figure 5.
LncRNA-miRNA-mRNA networks and senescence. Cellular senescence is associated with the release of a large number of pro-inflammatory cytokines and growth factors. These factors cause an increased burden of tissue inflammation and oxidative stress that contribute to telomere shortening. Moreover, telomere shortening and cellular senescence are involved in aging and in the development of CVDs. The figure shows that the senescence phenotype can be induced by: (1) ageing the endothelial cells by culturing until late passages (p9) (2) stimulating cardiomyocytes (CMs) with D-galactose and (3) treating vascular smooth muscle cells (VSMCs) with high glucose. The lncRNA-miRNA-mRNA networks triggered by these conditions stimulate or inhibit senescence according to the modulated targets.


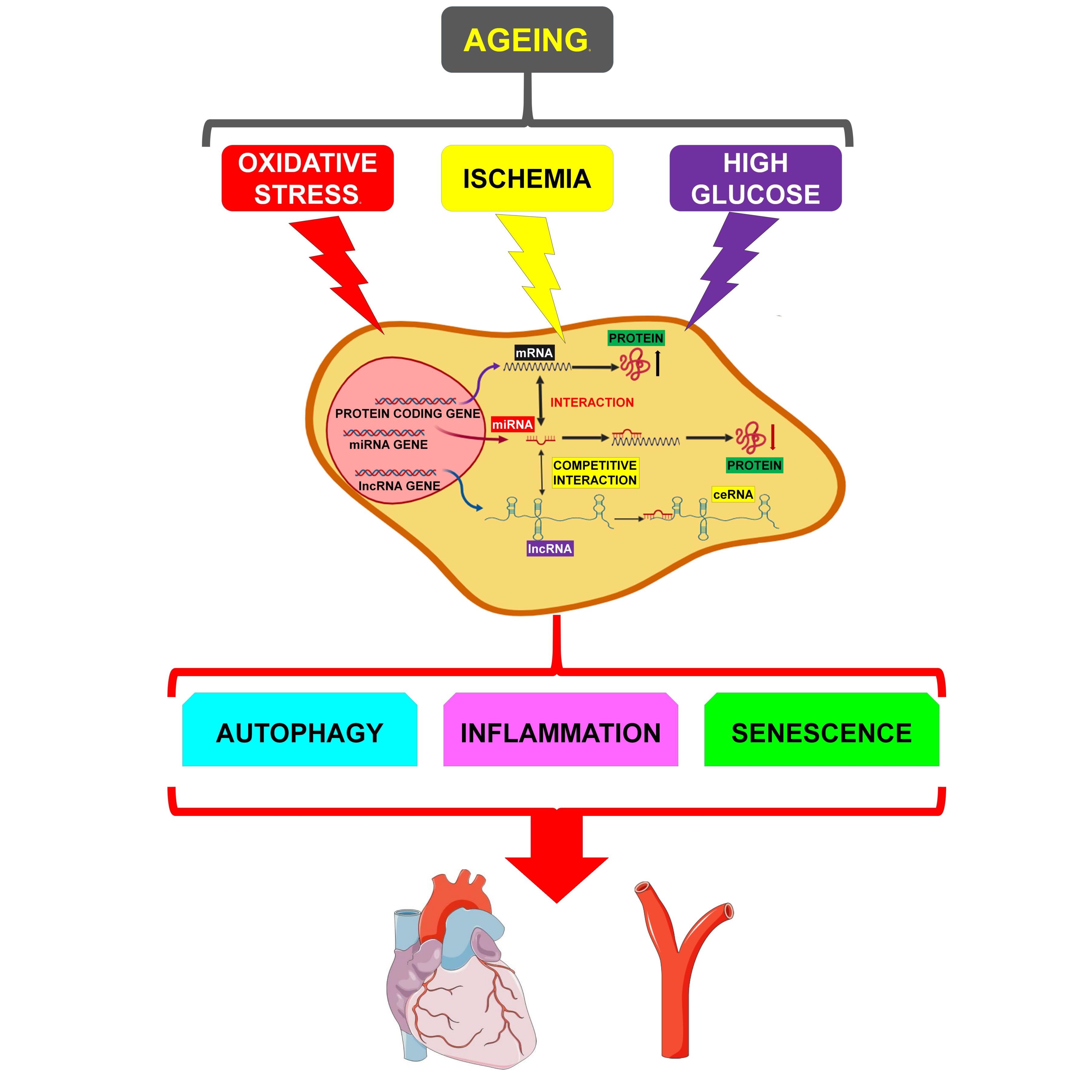{kind=link}
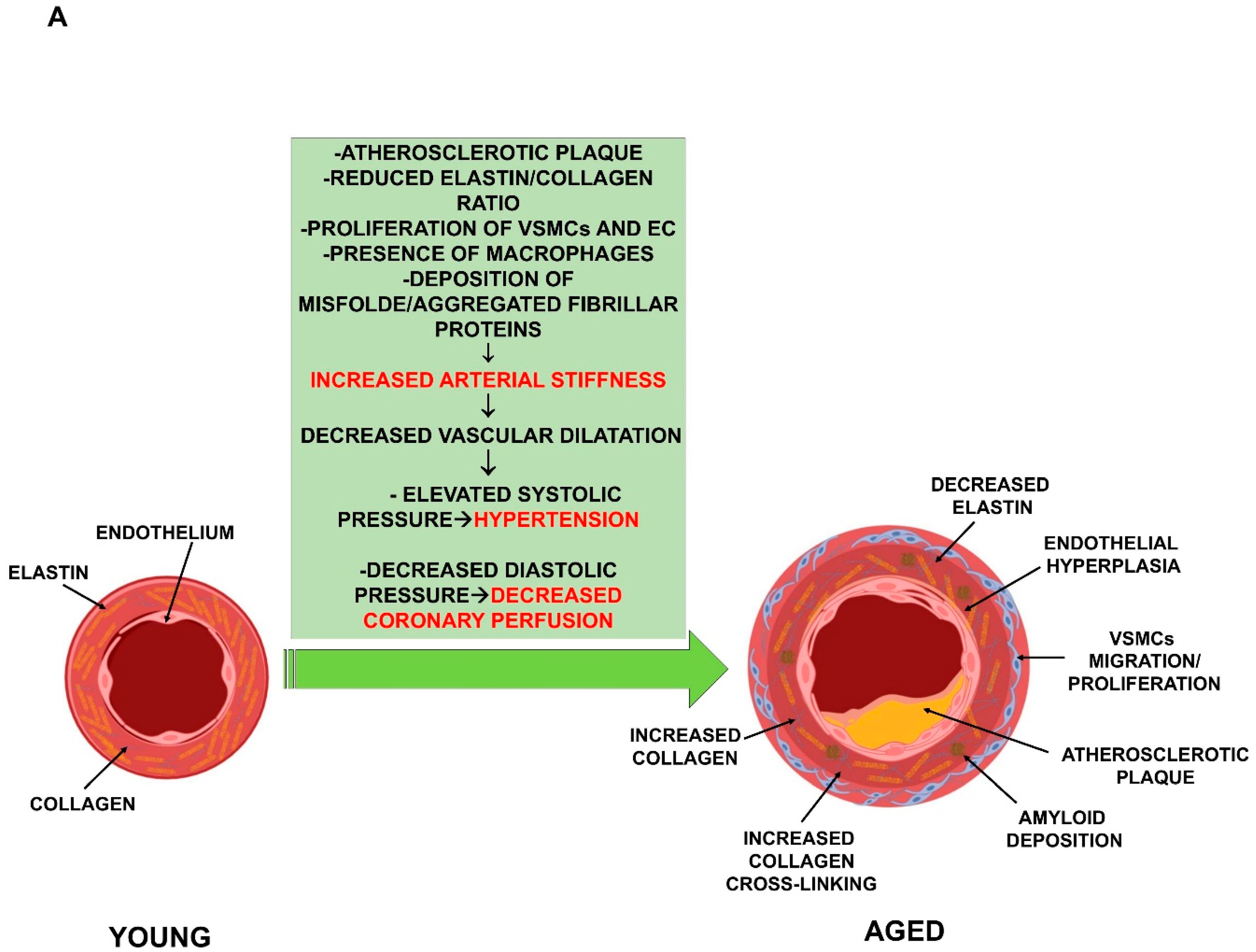{kind=link}
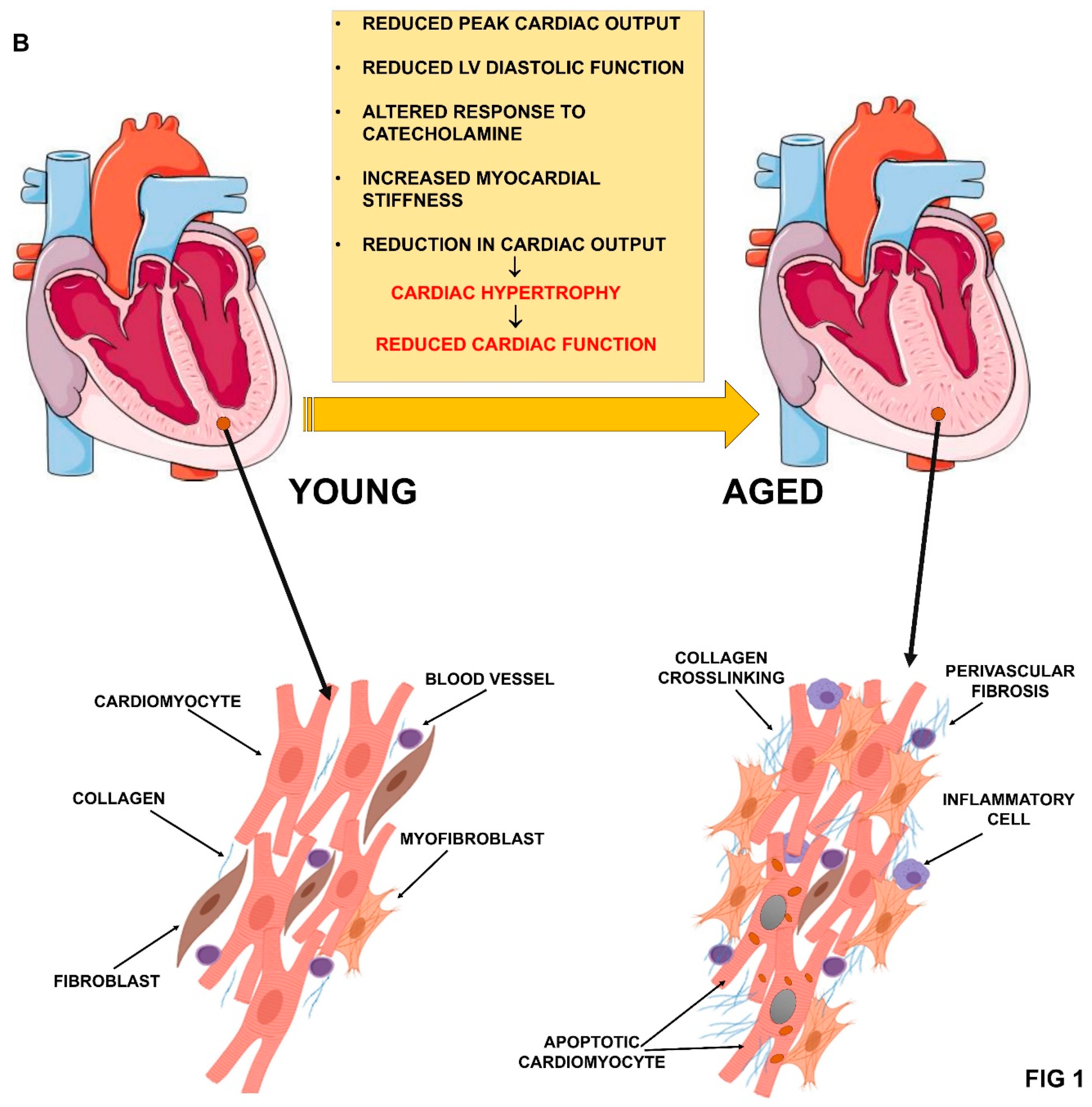{kind=link}
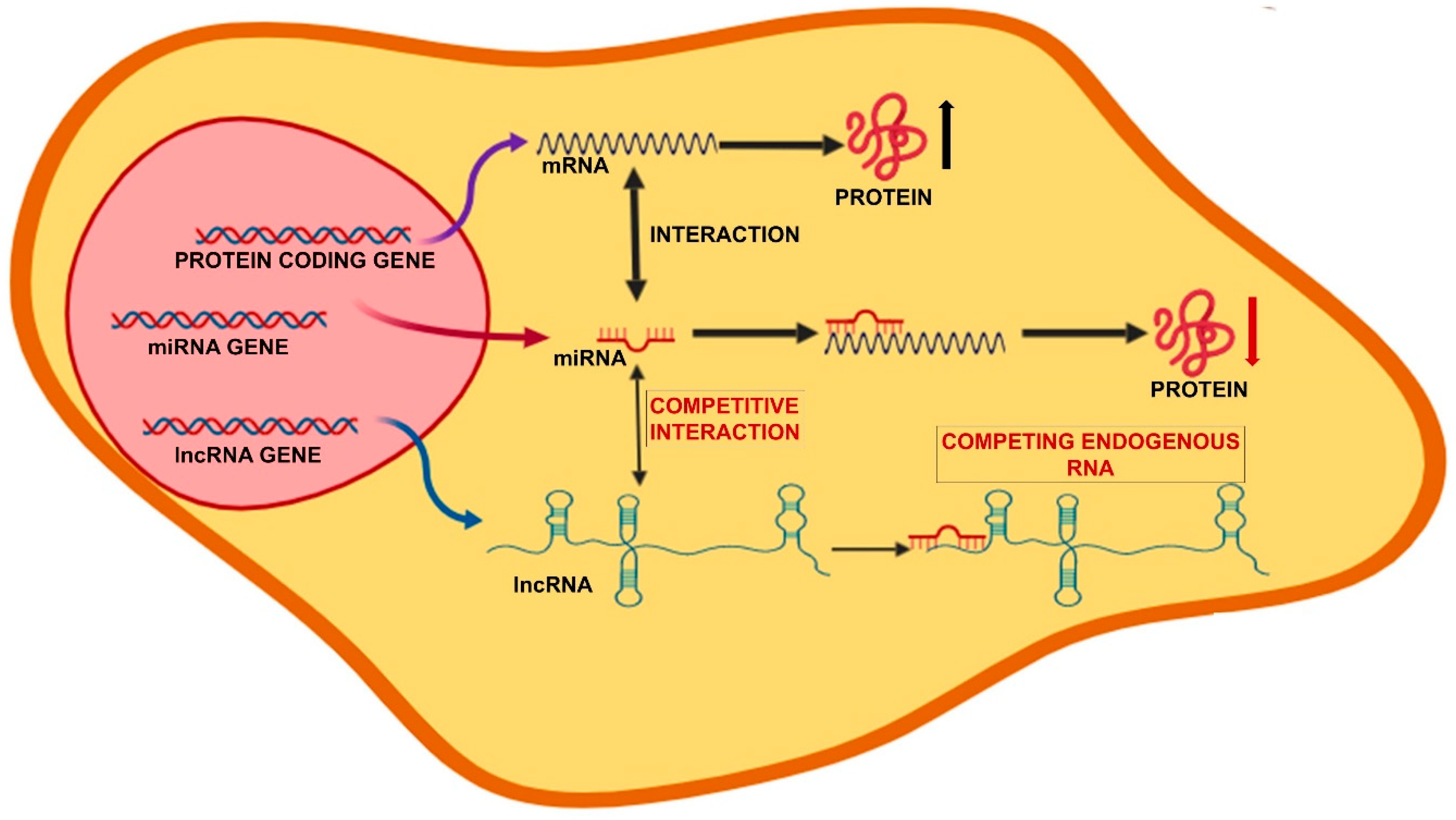{kind=link}
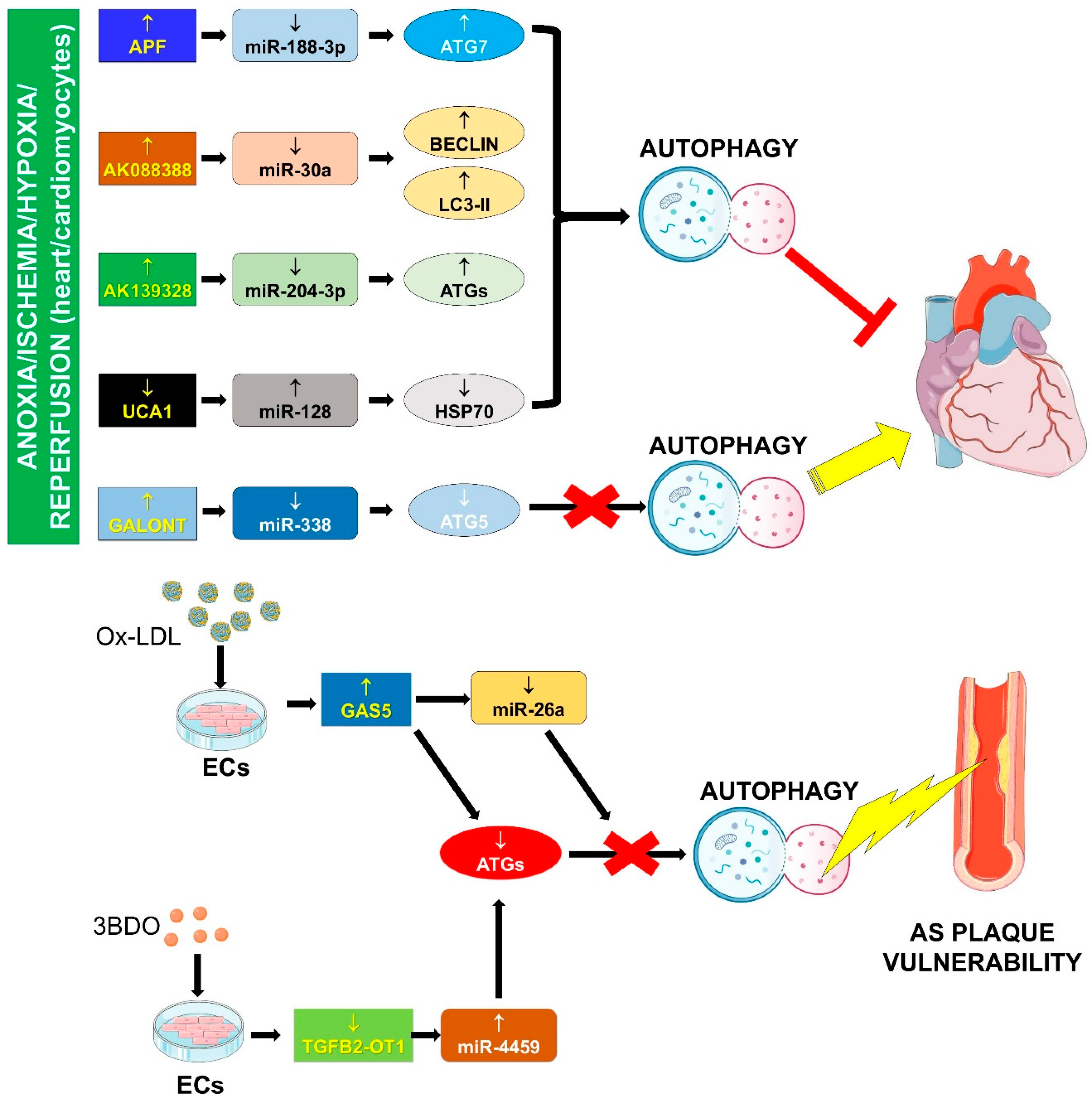{kind=link}
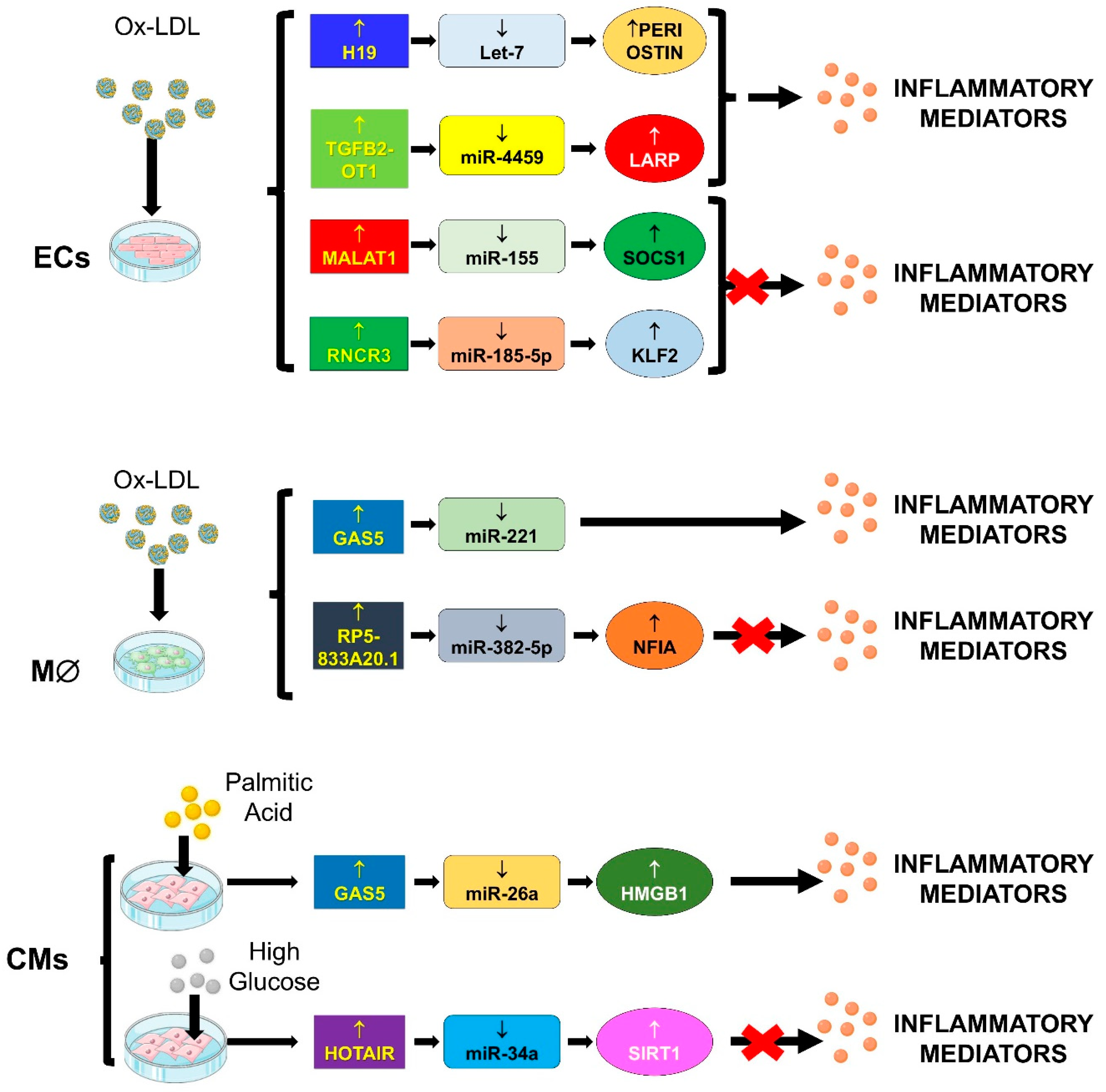{kind=link}
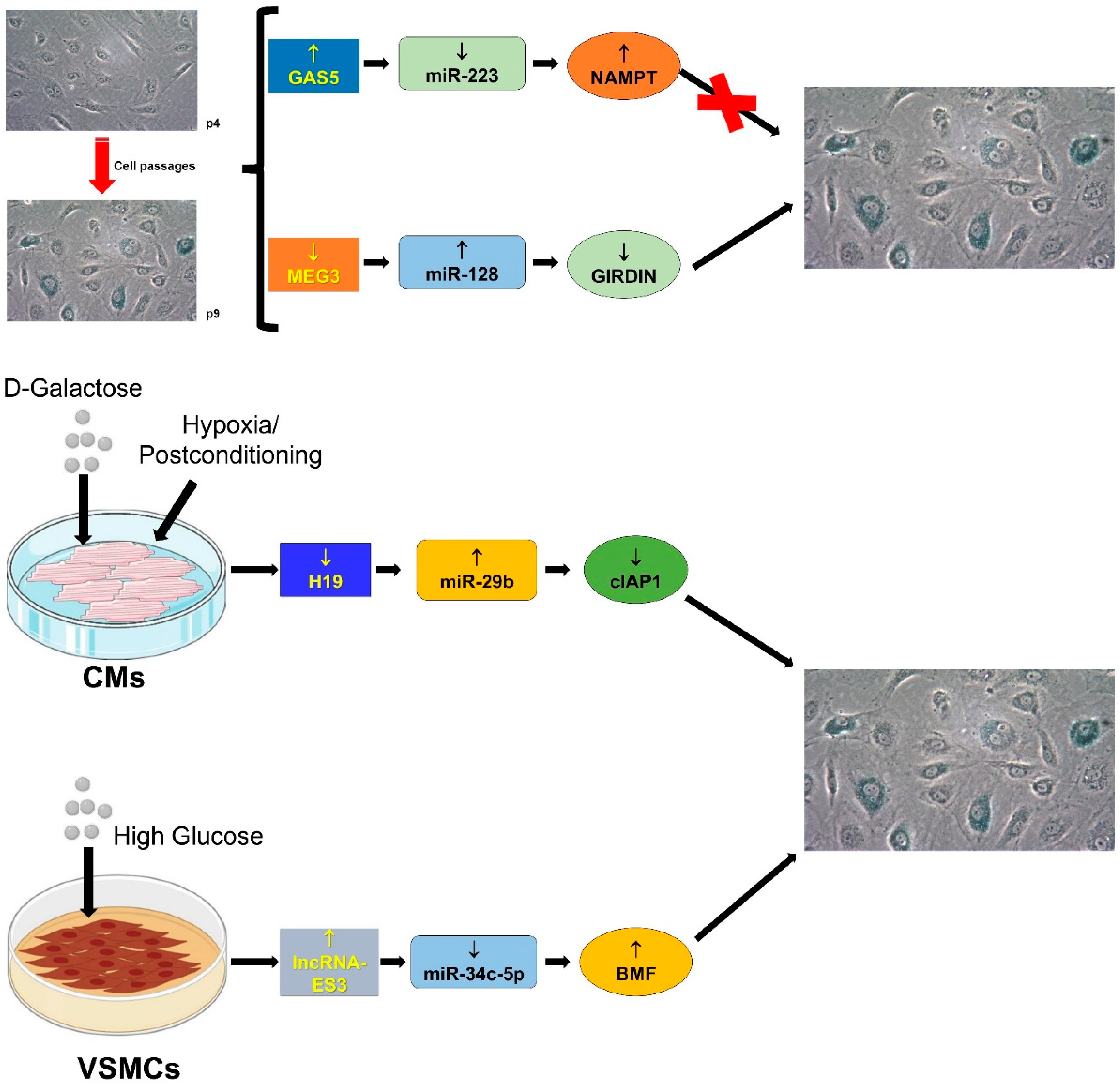{kind=link}
Table 1.
LncRNA/miRNA/mRNA networks and ageing mechanisms involved in cardiovascular diseases.
| Process | lncRNA | miRNA | Prediction Tool | miRNA Target (Direct/Indirect) | Pathology/Condition | Ref. |
|---|---|---|---|---|---|---|
| Autophagy | APF | miR-188-3p | RNAhybrid | ATG7 (direct) | -A/R in mouse CMs -I/R (mouse) | [71] |
| AK088388 | miR-30a | miRANDA TargetScan | Beclin-1 (direct) LC3-II (indirect) | -H/R in mouse CMs | [72] | |
| AK139328 | miR-204-3p | nd | ATG proteins (indirect) | -Diabetic mouse -I/R (mouse) | [73] | |
| BACE1-AS | miR-29/miR-107/miR-124/miR-485/miR-761 | miRANDA | BACE1 (direct) | -human DCM -HAOECs -mouse cardiomyocytes | [51,74,75] | |
| Galont | miR-338 | RNAhybrid | ATG5 (direct) | A/R cardiomyocytes (mouse) | [76] | |
| GAS5 | miR-26a | RNAhybrid | ATG proteins (indirect) | -ox-LDL stimulation of human ECs -plasma of AS patients | [77] | |
| TGFB2-OT1 | miR-4459 | Mirbase MicroInspector | ATG13 (direct) | -3BDO stimulation of human ECs | [78] | |
| UCA1 | miR-128 | nd | HSP70 (direct) | -I/R (mouse) -H/R in mouse CMs | [79] | |
| Inflammation | GAS5 | miR-26a | Starbase v2.0 LncBase Predicted v.2 tool | HMGB1 | -PA treated mouse CMs | [80] |
| GAS5 | miR-221 | nd | IL-1β, TNF-α, MMP-2, MMP-9 (indirect) | -Ox/LDL stimulation of THP-1 human cells -Human AS plaques | [81] | |
| H19 | let-7 | nd | Periostin | -Ox/LDL stimulation of human HUVEC | [82] | |
| HOTAIR | miR-34a | miRANDA | SIRT1 (direct) | -STZ mice -high glucose treated rat CMs | [83] | |
| MALAT-1 | miR-155 | TargetScan | SOCS1 (direct) | - ox-LDL stimulation of human HAOECs | [84] | |
| RNCR3 | miR-185-5p | TargetScan | KLF-2 (direct) | -Human aortic lesions -Aorta from apoE−/− mice - ox-LDL stimulation of human EC and VSMCs | [85] | |
| RP5-833A20.1 | miR-382-5p | miRBase, PicTar, TargetScan, RNAhybrid | NFIA (direct) | -Ox/Ac-LDL stimulation of THP-1 human cells -Aorta from apoE−/− mice | [86] | |
| TGFB2-OT1 | miR-4459 | MicroInspector | LARP/CERS1/ NAT8L/ ATG13 (direct) | -LPS/ox-LDL stimulation of human ECs | [87] | |
| Senescence | GAS5 | miR-223 | miRWalk miRANDA TargetScan microT-CDS | NAMPT (direct) | -Human EPCs (late passages) | [88] |
| H19 | miR-29b-3p | nd | cIAP1 (direct) | -D-galactose/H-Post treatment of CMs -I/Post (mouse) | [89] | |
| LncRNA-ES3 | miR-34c-5p | nd | BMF | -high glucose stimulated human aorta VSMCs | [90] | |
| MEG3 | miR-128 | nd | GIRDIN (direct) | -Coronary artery aged mice -HUVECs (late passages) | [91] |
Abbreviations: A/R = Anoxia/reperfusion; AS = aortic stenosis; CMs = cardiomyocytes; DCM = dilated cardiomyopathy; ECs = endothelial cells; EPCs = endothelial precursor cells; HAOECs = human aortic endothelial cells; H-Post = hypoxia-postconditioning; H/R = hypoxia/reperfusion; HUVECs = human umbilical endothelial cells; I-Post = ischemia-postconditioning; I/R = Ischemia/reperfusion; LPS = lipopolysaccharide; ox-LDL= oxidized low density lipoprotein; PA = palmitic acid; STZ = streptozotocin; VSMCs = vascular smooth muscle cells; 3BDO = 3-benzyl-5-((2-nitrophenoxy) methyl)-dihydrofuran-2(3H)-one.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Greco, S.; Gaetano, C.; Martelli, F. Long Noncoding Competing Endogenous RNA Networks in Age-Associated Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 3079. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20123079
AMA Style
Greco S, Gaetano C, Martelli F. Long Noncoding Competing Endogenous RNA Networks in Age-Associated Cardiovascular Diseases. International Journal of Molecular Sciences. 2019; 20(12):3079. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20123079
Chicago/Turabian StyleGreco, Simona, Carlo Gaetano, and Fabio Martelli. 2019. "Long Noncoding Competing Endogenous RNA Networks in Age-Associated Cardiovascular Diseases" International Journal of Molecular Sciences 20, no. 12: 3079. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20123079
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.