SerThr-PhosphoProteome of Brain from Aged PINK1-KO+A53T-SNCA Mice Reveals pT1928-MAP1B and pS3781-ANK2 Deficits, as Hub between Autophagy and Synapse Changes

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Strongest Downregulations Affect Microtubular Functions
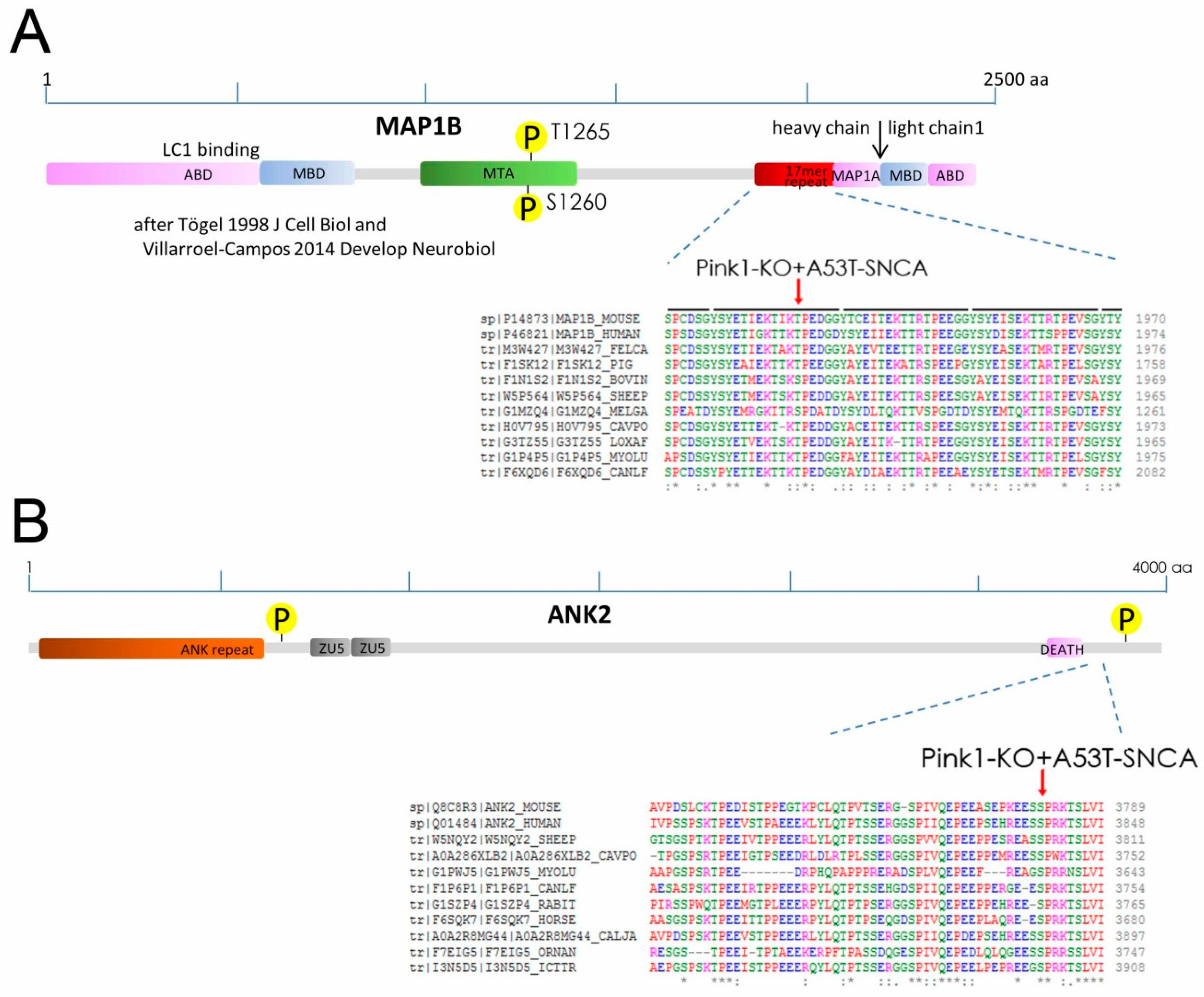
2.2. Phospho-Residues of MAP1B and ANK2 Are Conserved and Fit with PINK1-Target Criteria
2.3. Pathway Enrichments Highlight Microtubule Functions, Synaptic Signaling, and Kinase Domains
2.4. Dysregulated Phosphorylation of pT1928-MAP1B and of Autophagy Factors in DM Mouse Brain
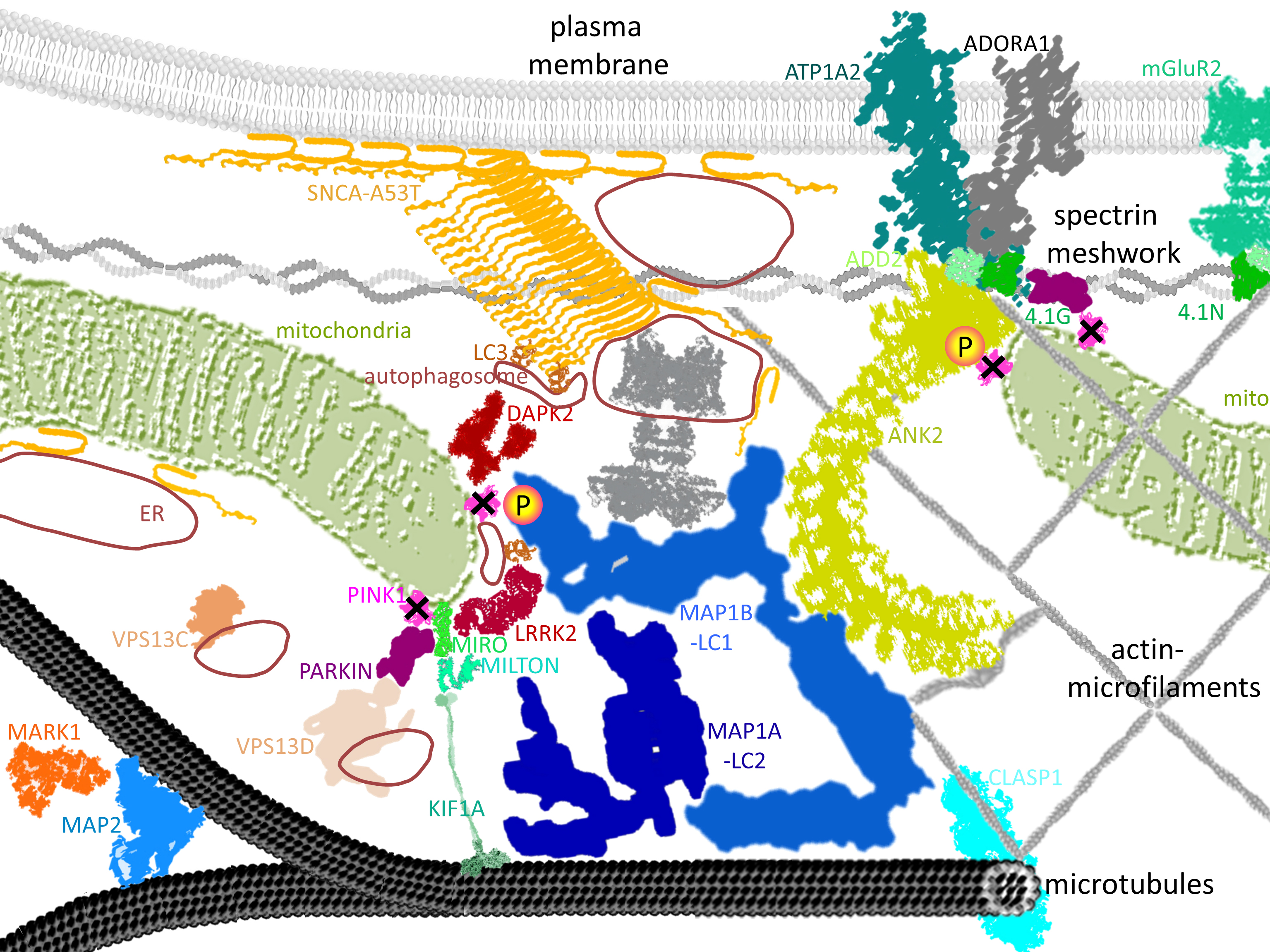
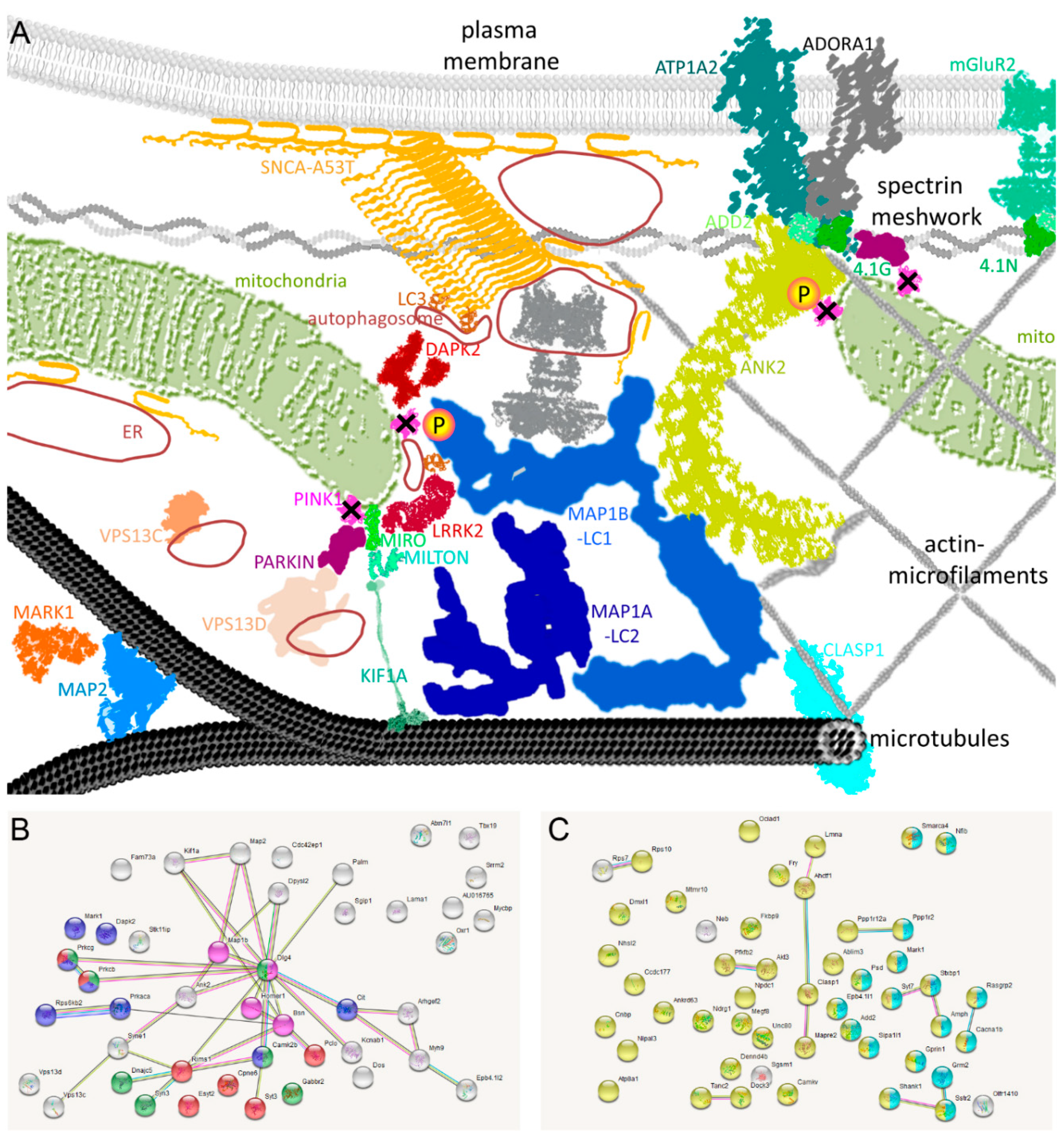
2.5. Known PINK1/SNCA Functional Effects Mirror the Roles of Dephosphorylated Factors in DM Brain
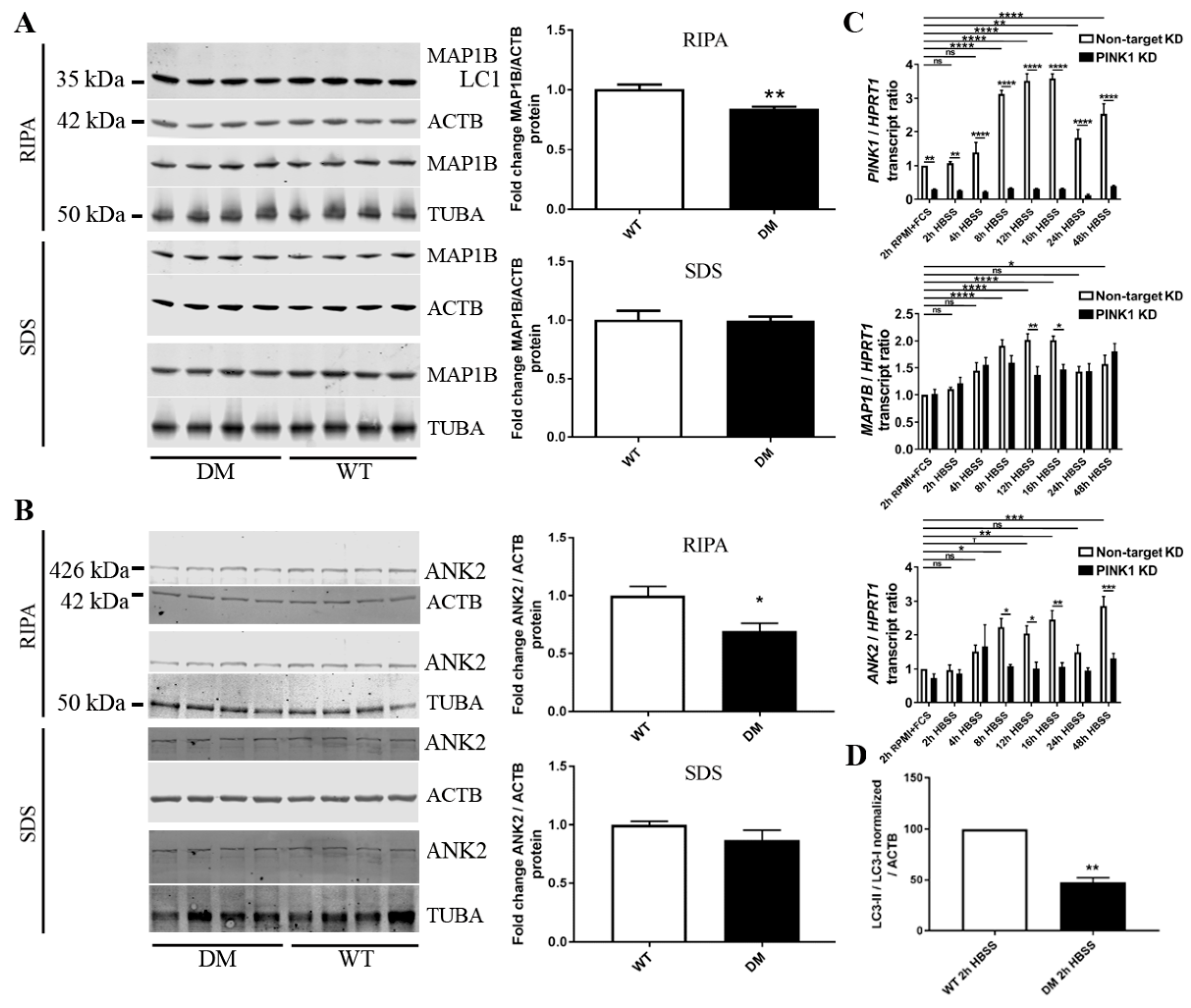
2.6. Validation Tests Confirm PINK1 to Modulate MAP1B/ANK2, Indicate Decreased Autophagy in DM Brain
3. Materials and Methods
3.1. Breeding and Ageing of DM Mice, Homozygous for Pink1−/− and A53T-SNCA Overexpression
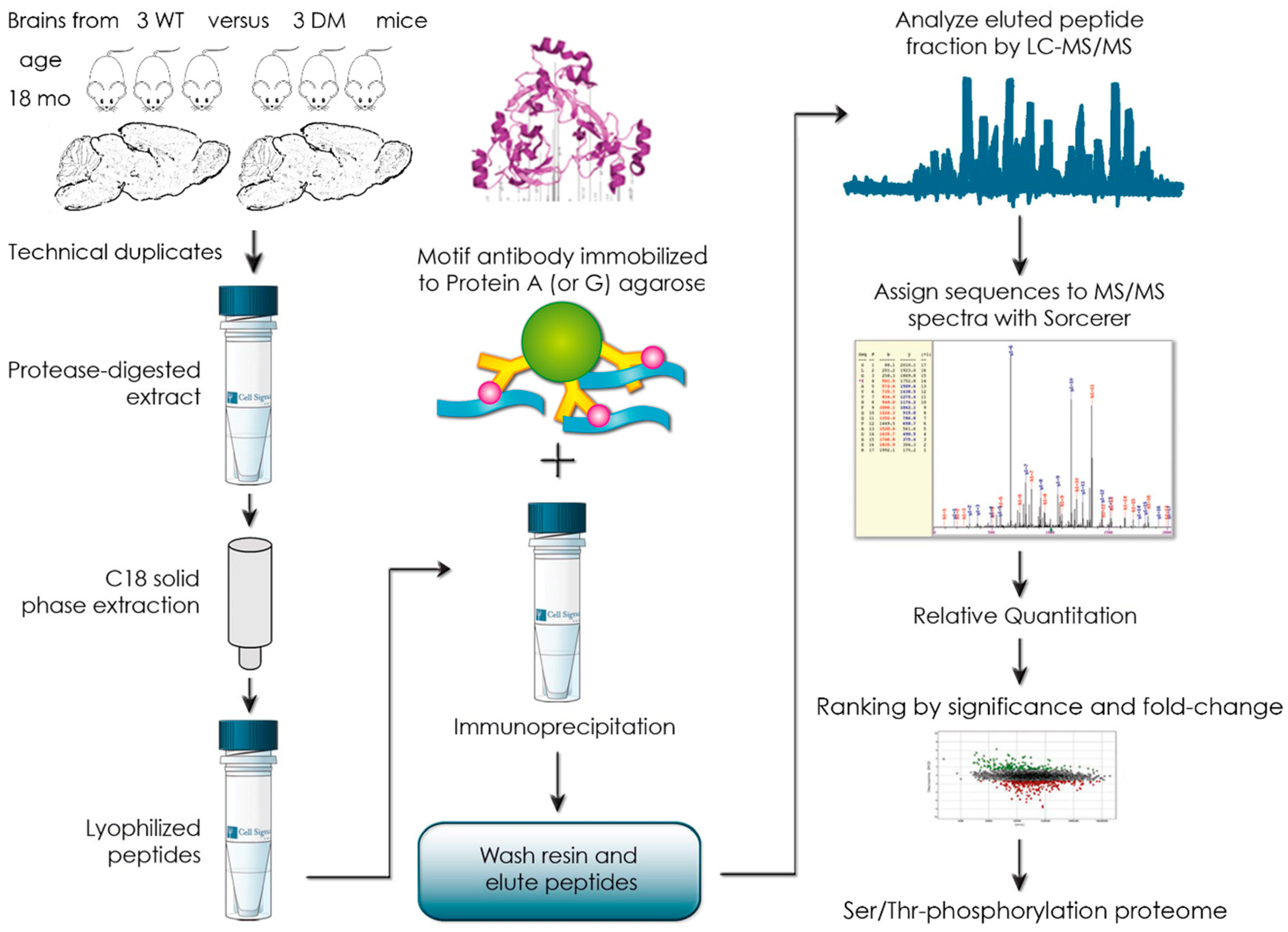
3.2. Global Phospho-Ser/Thr Motif Survey by Label-Free Mass-Spectrometry
3.3. Bioinformatic Pathway Enrichment Analyses
3.4. Validation Experiments via Expression Analysis on Protein and mRNA Level
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Corti, O.; Lesage, S.; Brice, A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef] [PubMed]
- Cabin, D.E.; Gispert-Sanchez, S.; Murphy, D.; Auburger, G.; Myers, R.R.; Nussbaum, R.L. Exacerbated synucleinopathy in mice expressing A53T SNCA on a Snca null background. Neurobiol. Aging 2005, 26, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Tuin, I.; Voss, U.; Kessler, K.; Krakow, K.; Hilker, R.; Morales, B.; Steinmetz, H.; Auburger, G. Sleep quality in a family with hereditary parkinsonism (PARK6). Sleep Med. 2008, 9, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Lahut, S.; Gispert, S.; Omur, O.; Depboylu, C.; Seidel, K.; Dominguez-Bautista, J.A.; Brehm, N.; Tireli, H.; Hackmann, K.; Pirkevi, C.; et al. Blood RNA biomarkers in prodromal PARK4 and rapid eye movement sleep behavior disorder show role of complexin 1 loss for risk of Parkinson’s disease. Dis. Model. Mech. 2017, 10, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Exner, N.; Treske, B.; Paquet, D.; Holmstrom, K.; Schiesling, C.; Gispert, S.; Carballo-Carbajal, I.; Berg, D.; Hoepken, H.H.; Gasser, T.; et al. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J. Neurosci. 2007, 27, 12413–12418. [Google Scholar] [CrossRef]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef]
- Durcan, T.M.; Fon, E.A. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef]
- Ordureau, A.; Paulo, J.A.; Zhang, W.; Ahfeldt, T.; Zhang, J.; Cohn, E.F.; Hou, Z.; Heo, J.M.; Rubin, L.L.; Sidhu, S.S.; et al. Dynamics of PARKIN-Dependent Mitochondrial Ubiquitylation in Induced Neurons and Model Systems Revealed by Digital Snapshot Proteomics. Mol. Cell 2018, 70, 211–227 e218. [Google Scholar] [CrossRef] [PubMed]
- Key, J.; Mueller, A.K.; Gispert, S.; Matschke, L.; Wittig, I.; Corti, O.; Munch, C.; Decher, N.; Auburger, G. Ubiquitylome profiling of Parkin-null brain reveals dysregulation of calcium homeostasis factors ATP1A2, Hippocalcin and GNA11, reflected by altered firing of noradrenergic neurons. Neurobiol. Dis. 2019, 127, 114–130. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, S.; Wu, Z.; Klinkenberg, M.; Sun, Y.; Auburger, G.; Guo, S.; Lu, B. PINK1 and Parkin control localized translation of respiratory chain component mRNAs on mitochondria outer membrane. Cell Metab. 2015, 21, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Klinkenberg, M.; Auburger, G.; Bereiter-Hahn, J.; Jendrach, M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J. Cell Sci. 2010, 123, 917–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinkenberg, M.; Thurow, N.; Gispert, S.; Ricciardi, F.; Eich, F.; Prehn, J.H.; Auburger, G.; Kogel, D. Enhanced vulnerability of PARK6 patient skin fibroblasts to apoptosis induced by proteasomal stress. Neuroscience 2010, 166, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Parganlija, D.; Klinkenberg, M.; Dominguez-Bautista, J.; Hetzel, M.; Gispert, S.; Chimi, M.A.; Drose, S.; Mai, S.; Brandt, U.; Auburger, G.; et al. Loss of PINK1 impairs stress-induced autophagy and cell survival. PLoS ONE 2014, 9, e95288. [Google Scholar] [CrossRef] [PubMed]
- Torres-Odio, S.; Key, J.; Hoepken, H.H.; Canet-Pons, J.; Valek, L.; Roller, B.; Walter, M.; Morales-Gordo, B.; Meierhofer, D.; Harter, P.N.; et al. Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J. Neuroinflamm. 2017, 14, 154. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Ricciardi, F.; Kurz, A.; Azizov, M.; Hoepken, H.H.; Becker, D.; Voos, W.; Leuner, K.; Muller, W.E.; Kudin, A.P.; et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS ONE 2009, 4, e5777. [Google Scholar] [CrossRef] [PubMed]
- Dehorter, N.; Lozovaya, N.; Mdzomba, B.J.; Michel, F.J.; Lopez, C.; Tsintsadze, V.; Tsintsadze, T.; Klinkenberg, M.; Gispert, S.; Auburger, G.; et al. Subthalamic lesion or levodopa treatment rescues giant GABAergic currents of PINK1-deficient striatum. J. Neurosci. 2012, 32, 18047–18053. [Google Scholar] [CrossRef] [PubMed]
- Carron, R.; Filipchuk, A.; Nardou, R.; Singh, A.; Michel, F.J.; Humphries, M.D.; Hammond, C. Early hypersynchrony in juvenile PINK1(-)/(-) motor cortex is rescued by antidromic stimulation. Front. Syst Neurosci. 2014, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Pearlstein, E.; Michel, F.J.; Save, L.; Ferrari, D.C.; Hammond, C. Abnormal Development of Glutamatergic Synapses Afferent to Dopaminergic Neurons of the Pink1(-/-) Mouse Model of Parkinson’s Disease. Front. Cell Neurosci. 2016, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinkenberg, M.; Gispert, S.; Dominguez-Bautista, J.A.; Braun, I.; Auburger, G.; Jendrach, M. Restriction of trophic factors and nutrients induces PARKIN expression. Neurogenetics 2012, 13, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Jendrach, M.; Gispert, S.; Ricciardi, F.; Klinkenberg, M.; Schemm, R.; Auburger, G. The mitochondrial kinase PINK1, stress response and Parkinson’s disease. J. Bioenerg. Biomembr. 2009, 41, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrimsdottir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rub, C.; Liu, Y.; Magrane, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. alpha-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci 2014, 34, 249–259. [Google Scholar] [CrossRef]
- Kamp, F.; Exner, N.; Lutz, A.K.; Wender, N.; Hegermann, J.; Brunner, B.; Nuscher, B.; Bartels, T.; Giese, A.; Beyer, K.; et al. Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010, 29, 3571–3589. [Google Scholar] [CrossRef] [PubMed]
- Menges, S.; Minakaki, G.; Schaefer, P.M.; Meixner, H.; Prots, I.; Schlotzer-Schrehardt, U.; Friedland, K.; Winner, B.; Outeiro, T.F.; Winklhofer, K.F.; et al. Alpha-synuclein prevents the formation of spherical mitochondria and apoptosis under oxidative stress. Sci. Rep. 2017, 7, 42942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoepken, H.H.; Gispert, S.; Azizov, M.; Klinkenberg, M.; Ricciardi, F.; Kurz, A.; Morales-Gordo, B.; Bonin, M.; Riess, O.; Gasser, T.; et al. Parkinson patient fibroblasts show increased alpha-synuclein expression. Exp. Neurol. 2008, 212, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Hoepken, H.H.; Gispert, S.; Morales, B.; Wingerter, O.; Del Turco, D.; Mulsch, A.; Nussbaum, R.L.; Muller, K.; Drose, S.; Brandt, U.; et al. Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol. Dis. 2007, 25, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Auburger, G.; Klinkenberg, M.; Drost, J.; Marcus, K.; Morales-Gordo, B.; Kunz, W.S.; Brandt, U.; Broccoli, V.; Reichmann, H.; Gispert, S.; et al. Primary skin fibroblasts as a model of Parkinson’s disease. Mol. Neurobiol 2012, 46, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Del Turco, D.; Garrett, L.; Chen, A.; Bernard, D.J.; Hamm-Clement, J.; Korf, H.W.; Deller, T.; Braak, H.; Auburger, G.; et al. Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence of aggregate formation. Mol. Cell Neurosci 2003, 24, 419–429. [Google Scholar] [CrossRef]
- Kurz, A.; Double, K.L.; Lastres-Becker, I.; Tozzi, A.; Tantucci, M.; Bockhart, V.; Bonin, M.; Garcia-Arencibia, M.; Nuber, S.; Schlaudraff, F.; et al. A53T-alpha-synuclein overexpression impairs dopamine signaling and striatal synaptic plasticity in old mice. PLoS ONE 2010, 5, e11464. [Google Scholar] [CrossRef]
- Tozzi, A.; Costa, C.; Siliquini, S.; Tantucci, M.; Picconi, B.; Kurz, A.; Gispert, S.; Auburger, G.; Calabresi, P. Mechanisms underlying altered striatal synaptic plasticity in old A53T-alpha synuclein overexpressing mice. Neurobiol Aging 2012, 33, 1792–1799. [Google Scholar] [CrossRef]
- Platt, N.J.; Gispert, S.; Auburger, G.; Cragg, S.J. Striatal dopamine transmission is subtly modified in human A53Talpha-synuclein overexpressing mice. PLoS ONE 2012, 7, e36397. [Google Scholar] [CrossRef]
- Subramaniam, M.; Althof, D.; Gispert, S.; Schwenk, J.; Auburger, G.; Kulik, A.; Fakler, B.; Roeper, J. Mutant alpha-synuclein enhances firing frequencies in dopamine substantia nigra neurons by oxidative impairment of A-type potassium channels. J. Neurosci. 2014, 34, 13586–13599. [Google Scholar] [CrossRef]
- Brehm, N.; Bez, F.; Carlsson, T.; Kern, B.; Gispert, S.; Auburger, G.; Cenci, M.A. A Genetic Mouse Model of Parkinson’s Disease Shows Involuntary Movements and Increased Postsynaptic Sensitivity to Apomorphine. Mol. Neurobiol 2015, 52, 1152–1164. [Google Scholar] [CrossRef] [PubMed]
- Kurz, A.; May, C.; Schmidt, O.; Muller, T.; Stephan, C.; Meyer, H.E.; Gispert, S.; Auburger, G.; Marcus, K. A53T-alpha-synuclein-overexpression in the mouse nigrostriatal pathway leads to early increase of 14-3-3 epsilon and late increase of GFAP. J. Neural Transm (Vienna) 2012, 119, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Brehm, N.; Rau, K.; Kurz, A.; Gispert, S.; Auburger, G. Age-Related Changes of 14-3-3 Isoforms in Midbrain of A53T-SNCA Overexpressing Mice. J. Parkinsons Dis. 2015, 5, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Kurz, A.; Brehm, N.; Rau, K.; Walter, M.; Riess, O.; Auburger, G. Complexin-1 and Foxp1 Expression Changes Are Novel Brain Effects of Alpha-Synuclein Pathology. Mol. Neurobiol 2015, 52, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.; Zimmermann, Z.; Gispert, S.; Auburger, G.; Korf, H.W.; von Gall, C. Impaired Photic Entrainment of Spontaneous Locomotor Activity in Mice Overexpressing Human Mutant alpha-Synuclein. Int. J. Mol. Sci. 2018, 19, 1651. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Brehm, N.; Weil, J.; Seidel, K.; Rub, U.; Kern, B.; Walter, M.; Roeper, J.; Auburger, G. Potentiation of neurotoxicity in double-mutant mice with Pink1 ablation and A53T-SNCA overexpression. Hum. Mol. Genet. 2015, 24, 1061–1076. [Google Scholar] [CrossRef] [PubMed]
- Auburger, G.; Gispert, S.; Jendrach, M. Mitochondrial acetylation and genetic models of Parkinson’s disease. Prog. Mol. Biol. Transl. Sci. 2014, 127, 155–182. [Google Scholar]
- Auburger, G.; Gispert, S.; Brehm, N. Methyl-Arginine Profile of Brain from Aged PINK1-KO+A53T-SNCA Mice Suggests Altered Mitochondrial Biogenesis. Parkinsons Dis. 2016, 2016, 4686185. [Google Scholar] [CrossRef]
- Riederer, B.M. Microtubule-associated protein 1B, a growth-associated and phosphorylated scaffold protein. Brain Res. Bull. 2007, 71, 541–558. [Google Scholar] [CrossRef]
- Li, J.; Wilkinson, B.; Clementel, V.A.; Hou, J.; O’Dell, T.J.; Coba, M.P. Long-term potentiation modulates synaptic phosphorylation networks and reshapes the structure of the postsynaptic interactome. Sci. Signal. 2016, 9, rs8. [Google Scholar] [CrossRef]
- Herskowitz, J.H.; Seyfried, N.T.; Duong, D.M.; Xia, Q.; Rees, H.D.; Gearing, M.; Peng, J.; Lah, J.J.; Levey, A.I. Phosphoproteomic analysis reveals site-specific changes in GFAP and NDRG2 phosphorylation in frontotemporal lobar degeneration. J. Proteome Res. 2010, 9, 6368–6379. [Google Scholar] [CrossRef] [PubMed]
- Bulat, V.; Rast, M.; Pielage, J. Presynaptic CK2 promotes synapse organization and stability by targeting Ankyrin2. J. Cell Biol. 2014, 204, 77–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephan, R.; Goellner, B.; Moreno, E.; Frank, C.A.; Hugenschmidt, T.; Genoud, C.; Aberle, H.; Pielage, J. Hierarchical microtubule organization controls axon caliber and transport and determines synaptic structure and stability. Dev. Cell 2015, 33, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Migh, E.; Gotz, T.; Foldi, I.; Szikora, S.; Gombos, R.; Darula, Z.; Medzihradszky, K.F.; Maleth, J.; Hegyi, P.; Sigrist, S.; et al. Microtubule organization in presynaptic boutons relies on the formin DAAM. Development 2018, 145, dev158519. [Google Scholar] [CrossRef] [PubMed]
- Nelson, W.J.; Hammerton, R.W. A membrane-cytoskeletal complex containing Na+,K+-ATPase, ankyrin, and fodrin in Madin-Darby canine kidney (MDCK) cells: Implications for the biogenesis of epithelial cell polarity. J. Cell Biol. 1989, 108, 893–902. [Google Scholar] [CrossRef]
- Mohler, P.J.; Schott, J.J.; Gramolini, A.O.; Dilly, K.W.; Guatimosim, S.; duBell, W.H.; Song, L.S.; Haurogne, K.; Kyndt, F.; Ali, M.E.; et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 2003, 421, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Baines, A.J.; Lu, H.C.; Bennett, P.M. The Protein 4.1 family: Hub proteins in animals for organizing membrane proteins. Biochim. Biophys. Acta 2014, 1838, 605–619. [Google Scholar] [CrossRef] [Green Version]
- Baines, A.J. The spectrin-ankyrin-4.1-adducin membrane skeleton: Adapting eukaryotic cells to the demands of animal life. Protoplasma 2010, 244, 99–131. [Google Scholar] [CrossRef]
- Weiner, A.T.; Seebold, D.Y.; Michael, N.L.; Guignet, M.; Feng, C.; Follick, B.; Yusko, B.A.; Wasilko, N.P.; Torres-Gutierrez, P.; Rolls, M.M. Identification of Proteins Required for Precise Positioning of Apc2 in Dendrites. G3 2018, 8, 1841–1853. [Google Scholar] [CrossRef] [Green Version]
- Shlevkov, E.; Kramer, T.; Schapansky, J.; LaVoie, M.J.; Schwarz, T.L. Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc. Natl. Acad. Sci. USA 2016, 113, E6097–E6106. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Domenech, G.; Covill-Cooke, C.; Ivankovic, D.; Halff, E.F.; Sheehan, D.F.; Norkett, R.; Birsa, N.; Kittler, J.T. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 2018, 37, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Mateos, E.M.; Gonzalez-Billault, C.; Dawson, H.N.; Vitek, M.P.; Avila, J. Role of MAP1B in axonal retrograde transport of mitochondria. Biochem. J. 2006, 397, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Siraj, S.; Liu, L.; Chen, Q. MARCH5-FUNDC1 axis fine-tunes hypoxia-induced mitophagy. Autophagy 2017, 13, 1244–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, P.H.; Islam, K.; Kenney, J.; Nielsen, M.S.; Power, J.; Gai, W.P. Microtubule-associated protein 1B is a component of cortical Lewy bodies and binds alpha-synuclein filaments. J. Biol. Chem. 2000, 275, 21500–21507. [Google Scholar] [CrossRef] [PubMed]
- Gai, W.P.; Blumbergs, P.C.; Blessing, W.W. Microtubule-associated protein 5 is a component of Lewy bodies and Lewy neurites in the brainstem and forebrain regions affected in Parkinson’s disease. Acta Neuropathol. 1996, 91, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Liu, H.P.; Lin, W.Y.; Guo, H.; Lu, B. LRRK2 kinase regulates synaptic morphology through distinct substrates at the presynaptic and postsynaptic compartments of the Drosophila neuromuscular junction. J. Neurosci. 2010, 30, 16959–16969. [Google Scholar] [CrossRef]
- Islam, M.S.; Nolte, H.; Jacob, W.; Ziegler, A.B.; Putz, S.; Grosjean, Y.; Szczepanowska, K.; Trifunovic, A.; Braun, T.; Heumann, H.; et al. Human R1441C LRRK2 regulates the synaptic vesicle proteome and phosphoproteome in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet. 2016, 25, 5365–5382. [Google Scholar] [PubMed]
- Chan, S.L.; Chua, L.L.; Angeles, D.C.; Tan, E.K. MAP1B rescues LRRK2 mutant-mediated cytotoxicity. Mol. Brain 2014, 7, 29. [Google Scholar] [CrossRef]
- Mohler, P.J.; Gramolini, A.O.; Bennett, V. The ankyrin-B C-terminal domain determines activity of ankyrin-B/G chimeras in rescue of abnormal inositol 1,4,5-trisphosphate and ryanodine receptor distribution in ankyrin-B (-/-) neonatal cardiomyocytes. J. Biol. Chem. 2002, 277, 10599–10607. [Google Scholar] [CrossRef]
- Woodroof, H.I.; Pogson, J.H.; Begley, M.; Cantley, L.C.; Deak, M.; Campbell, D.G.; van Aalten, D.M.; Whitworth, A.J.; Alessi, D.R.; Muqit, M.M. Discovery of catalytically active orthologues of the Parkinson’s disease kinase PINK1: Analysis of substrate specificity and impact of mutations. Open Biol. 2011, 1, 110012. [Google Scholar] [CrossRef]
- Bodaleo, F.J.; Montenegro-Venegas, C.; Henriquez, D.R.; Court, F.A.; Gonzalez-Billault, C. Microtubule-associated protein 1B (MAP1B)-deficient neurons show structural presynaptic deficiencies in vitro and altered presynaptic physiology. Sci. Rep. 2016, 6, 30069. [Google Scholar] [CrossRef] [PubMed]
- Stehbens, S.J.; Paszek, M.; Pemble, H.; Ettinger, A.; Gierke, S.; Wittmann, T. CLASPs link focal-adhesion-associated microtubule capture to localized exocytosis and adhesion site turnover. Nat. Cell Biol. 2014, 16, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Galjart, N. CLIPs and CLASPs and cellular dynamics. Nat. Rev. Mol. Cell Biol. 2005, 6, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Harrison, B.; Kraus, M.; Burch, L.; Stevens, C.; Craig, A.; Gordon-Weeks, P.; Hupp, T.R. DAPK-1 binding to a linear peptide motif in MAP1B stimulates autophagy and membrane blebbing. J. Biol. Chem. 2008, 283, 9999–10014. [Google Scholar] [CrossRef] [PubMed]
- Capoccia, B.J.; Jin, R.U.; Kong, Y.Y.; Peek, R.M., Jr.; Fassan, M.; Rugge, M.; Mills, J.C. The ubiquitin ligase Mindbomb 1 coordinates gastrointestinal secretory cell maturation. J. Clin. Invest. 2013, 123, 1475–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, S.S.; Hammarback, J.A. Molecular characterization of light chain 3. A microtubule binding subunit of MAP1A and MAP1B. J. Biol. Chem. 1994, 269, 11492–11497. [Google Scholar]
- Ashrafi, G.; Schlehe, J.S.; LaVoie, M.J.; Schwarz, T.L. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 2014, 206, 655–670. [Google Scholar] [CrossRef]
- Lee, Y.K.; Lee, J.A. Role of the mammalian ATG8/LC3 family in autophagy: Differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016, 49, 424–430. [Google Scholar] [CrossRef]
- Arasaki, K.; Nagashima, H.; Kurosawa, Y.; Kimura, H.; Nishida, N.; Dohmae, N.; Yamamoto, A.; Yanagi, S.; Wakana, Y.; Inoue, H.; et al. MAP1B-LC1 prevents autophagosome formation by linking syntaxin 17 to microtubules. EMBO Rep. 2018, 19, e45584. [Google Scholar] [CrossRef]
- Gandini, M.A.; Sandoval, A.; Zamponi, G.W.; Felix, R. The MAP1B-LC1/UBE2L3 complex catalyzes degradation of cell surface CaV2.2 channels. Channels (Austin) 2014, 8, 452–457. [Google Scholar] [CrossRef]
- Grimaldo, L.; Sandoval, A.; Garza-Lopez, E.; Felix, R. Involvement of Parkin in the ubiquitin proteasome system-mediated degradation of N-type voltage-gated Ca2+ channels. PLoS ONE 2017, 12, e0185289. [Google Scholar] [CrossRef] [PubMed]
- Palenzuela, R.; Gutierrez, Y.; Draffin, J.E.; Lario, A.; Benoist, M.; Esteban, J.A. MAP1B Light Chain Modulates Synaptic Transmission via AMPA Receptor Intracellular Trapping. J. Neurosci. 2017, 37, 9945–9963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anding, A.L.; Wang, C.; Chang, T.K.; Sliter, D.A.; Powers, C.M.; Hofmann, K.; Youle, R.J.; Baehrecke, E.H. Vps13D Encodes a Ubiquitin-Binding Protein that Is Required for the Regulation of Mitochondrial Size and Clearance. Curr. Biol. 2018, 28, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Seong, E.; Insolera, R.; Dulovic, M.; Kamsteeg, E.J.; Trinh, J.; Bruggemann, N.; Sandford, E.; Li, S.; Ozel, A.B.; Li, J.Z.; et al. Mutations in VPS13D lead to a new recessive ataxia with spasticity and mitochondrial defects. Ann. Neurol. 2018, 83, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S.; et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, N.; Leonzino, M.; Hancock-Cerutti, W.; Horenkamp, F.A.; Li, P.; Lees, J.A.; Wheeler, H.; Reinisch, K.M.; De Camilli, P. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol. 2018, 217, 3625–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marongiu, R.; Spencer, B.; Crews, L.; Adame, A.; Patrick, C.; Trejo, M.; Dallapiccola, B.; Valente, E.M.; Masliah, E. Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson’s disease by disturbing calcium flux. J. Neurochem. 2009, 108, 1561–1574. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.M.; Falomir-Lockhart, L.J.; Botelho, M.G.; Lin, K.H.; Wales, P.; Koch, J.C.; Gerhardt, E.; Taschenberger, H.; Outeiro, T.F.; Lingor, P.; et al. Elevated alpha-synuclein caused by SNCA gene triplication impairs neuronal differentiation and maturation in Parkinson’s patient-derived induced pluripotent stem cells. Cell Death Dis. 2015, 6, e1994. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Lee, K.; Im, H. Alpha-Synuclein modulates neurite outgrowth by interacting with SPTBN1. Biochem. Biophys. Res. Commun 2012, 424, 497–502. [Google Scholar] [CrossRef]
- Kim, S.; Lim, J.; Bang, Y.; Moon, J.; Kwon, M.S.; Hong, J.T.; Jeon, J.; Seo, H.; Choi, H.J. Alpha-Synuclein Suppresses Retinoic Acid-Induced Neuronal Differentiation by Targeting the Glycogen Synthase Kinase-3beta/beta-Catenin Signaling Pathway. Mol. Neurobiol 2018, 55, 1607–1619. [Google Scholar] [CrossRef]
- Ronzitti, G.; Bucci, G.; Emanuele, M.; Leo, D.; Sotnikova, T.D.; Mus, L.V.; Soubrane, C.H.; Dallas, M.L.; Thalhammer, A.; Cingolani, L.A.; et al. Exogenous alpha-synuclein decreases raft partitioning of Cav2.2 channels inducing dopamine release. J. Neurosci. 2014, 34, 10603–10615. [Google Scholar] [CrossRef] [PubMed]
- Baranov, S.V.; Baranova, O.V.; Yablonska, S.; Suofu, Y.; Vazquez, A.L.; Kozai, T.D.Y.; Cui, X.T.; Ferrando, L.M.; Larkin, T.M.; Tyurina, Y.Y.; et al. Mitochondria modulate programmed neuritic retraction. Proc. Natl. Acad. Sci. USA 2019, 116, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Dagda, R.K.; Pien, I.; Wang, R.; Zhu, J.; Wang, K.Z.; Callio, J.; Banerjee, T.D.; Dagda, R.Y.; Chu, C.T. Beyond the mitochondrion: Cytosolic PINK1 remodels dendrites through protein kinase A. J. Neurochem 2014, 128, 864–877. [Google Scholar] [CrossRef] [PubMed]
- Samann, J.; Hegermann, J.; von Gromoff, E.; Eimer, S.; Baumeister, R.; Schmidt, E. Caenorhabditits elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J. Biol. Chem. 2009, 284, 16482–16491. [Google Scholar] [CrossRef] [PubMed]
- Azkona, G.; Lopez de Maturana, R.; Del Rio, P.; Sousa, A.; Vazquez, N.; Zubiarrain, A.; Jimenez-Blasco, D.; Bolanos, J.P.; Morales, B.; Auburger, G.; et al. LRRK2 Expression Is Deregulated in Fibroblasts and Neurons from Parkinson Patients with Mutations in PINK1. Mol. Neurobiol 2018, 55, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Safaei, R.; Fischer, I. Cloning of a cDNA encoding MAP1B in rat brain: Regulation of mRNA levels during development. J. Neurochem 1989, 52, 1871–1879. [Google Scholar] [CrossRef]
- Zhu, M.; Li, X.; Tian, X.; Wu, C. Mask loss-of-function rescues mitochondrial impairment and muscle degeneration of Drosophila pink1 and parkin mutants. Hum. Mol. Genet. 2015, 24, 3272–3285. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhu, R.; Chen, K.; Zheng, H.; Zhao, H.; Yuan, C.; Zhang, H.; Wang, C.; Zhang, M. Potent and specific Atg8-targeting autophagy inhibitory peptides from giant ankyrins. Nat. Chem Biol 2018, 14, 778–787. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.J.; Ding, Y.; Kohtz, D.S.; Mizushima, N.; Cristea, I.M.; Rout, M.P.; Chait, B.T.; Zhong, Y.; Heintz, N.; Yue, Z. Induction of autophagy in axonal dystrophy and degeneration. J. Neurosci. 2006, 26, 8057–8068. [Google Scholar] [CrossRef]
- Rush, J.; Moritz, A.; Lee, K.A.; Guo, A.; Goss, V.L.; Spek, E.J.; Zhang, H.; Zha, X.M.; Polakiewicz, R.D.; Comb, M.J. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 2005, 23, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Gu, H.; Zhou, J.; Mulhern, D.; Wang, Y.; Lee, K.A.; Yang, V.; Aguiar, M.; Kornhauser, J.; Jia, X.; et al. Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell Proteomics 2014, 13, 372–387. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normalized Fold Change | Max % CV | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DM all: Control | DM 1: Control | DM 2: Control | DM 3: Control | Max Intensity | Biological | Gene Name | Protein Name | Site | Peptide | Count in Details |
| −314.3 | −255.3 | −438.7 | −279.5 | 6,559,198 | 35.4 | Mtap1b | MAP1B | §1928 | TIKT*PEDGGYTCEITEK | 16 |
| −3.3 | −543.3 | −715.4 | −1.1 | 858,179 | 155.1 | Ank2 | ANK2 iso2 + iso3 | §3781; §3809; §872 | GSPIVQEPEEASEPKEESS*PRK | 29 |
| −3.0 | -1.0 | −47.8 | −31.0 | 947,741 | 143.6 | Vps13d | VPS13D | §2429 | NAS*SESAVVPK | 8 |
| −2.3 | −2.9 | −2.9 | −1.6 | 26,223 | 54.5 | Mtap2 | MAP2 iso6 | §1801; §471 | RLSNVSSS*GSINLLESPQLATLAEDVTAALAK | 3 |
| −2.3 | −2.9 | −2.9 | −1.6 | 26,223 | 54.5 | Mtap2 | MAP2 iso6 | §1803; §473 | RLSNVSSSGS*INLLESPQLATLAEDVTAALAK | 3 |
| −2.1 | −2.1 | −3.1 | −1.4 | 436,205 | 32.6 | Epb4.1l2 | EPB41L2 iso3 | §86 | QRS*YNLVVAK | 3 |
| −1.9 | −4.5 | −1.5 | −1.4 | 543,218 | 47.1 | Dapk2 | DAPK2 | §299 | RES*VVNLENFKK | 11 |
| −1.8 | −2.2 | −1.4 | −2.0 | 27,656 | 33.9 | Mark1 | MARK1 | §394, §403 | SRPSS*DLNNSTLQS*PAHLK | 19 |
| −1.7 | −2.1 | −1.7 | −1.4 | 405,747 | 33.8 | Vps13c | VPS13C | 2481 | QESS*LFTLTFVPYGYTEVASVPVAR | 5 |
| −1.6 | −1.2 | −5.2 | −1.1 | 400,265 | 53.3 | Mtap1a | MAP1A | §1794 | VPSAPGQESPVPDT*KSTPPTR | 2 |
| −1.6 | −1.2 | −5.2 | −1.1 | 400,265 | 53.3 | Mtap1a | MAP1A | 1796 | VPSAPGQESPVPDTKS*TPPTR | 2 |
| −1.6 | −1.2 | −5.2 | −1.1 | 400,265 | 53.3 | Mtap1a | MAP1A | §1797 | VPSAPGQESPVPDTKST*PPTR | 31 |
| −1.6 | −2.1 | −1.4 | −1.4 | 2,098,225 | 24.4 | Kif1a; Kif1a | KIF1A iso3 | §1537; §1540 | SRPAS*PEPELLPELDSK | 22 |
| −1.4 | −1.1 | −2.6 | -1.1 | 454,218 | 35.3 | Mtap1a | MAP1A | §1789, 1796 | VPSAPGQES*PVPDTKS*TPPTR | 3 |
| −1.4 | −1.1 | −2.6 | −1.1 | 454,218 | 35.3 | Mtap1a | MAP1A | §1789, §1797 | VPSAPGQES*PVPDTKST*PPTR | 43 |
| 1.5 | 2.0 | 1.5 | 1.1 | 674,946 | 30.3 | Mapre2 | RP1 | §218, §222 | SSPASKPGSTPS*RPSS*AK | 11 |
| 1.6 | 2.3 | 1.5 | 1.1 | 563,552 | 32.4 | Mapre2 | RP1 | §215, §222 | SSPASKPGS*TPSRPSS*AK | 2 |
| 1.7 | 1.8 | 2.1 | 1.3 | 12,258,948 | 25.6 | Add2 | ADD2 | §528, §532, §535 | S*RS*PS*TES*QLMSK | 26 |
| 1.7 | 2.2 | 1.9 | 1.2 | 519,692 | 28.2 | Mark1 | MARK1 | §504 | RNT*YVCER | 8 |
| 1.7 | 2.0 | 1.9 | 1.2 | 39,365 | 30.1 | Clasp1 | CLASP1 iso3 | §646, §649 | RQS*SGS*TTNVASTPSDSR | 5 |
| 2.2 | 1.2 | 4.4 | 1.0 | 1,375,910 | 82.0 | Epb4.1l1 | EPB41L1 iso2 + iso4 | §546; §545 | RLPSSPASPS*PK | 29 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Auburger, G.; Gispert, S.; Torres-Odio, S.; Jendrach, M.; Brehm, N.; Canet-Pons, J.; Key, J.; Sen, N.-E. SerThr-PhosphoProteome of Brain from Aged PINK1-KO+A53T-SNCA Mice Reveals pT1928-MAP1B and pS3781-ANK2 Deficits, as Hub between Autophagy and Synapse Changes. Int. J. Mol. Sci. 2019, 20, 3284. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133284
Auburger G, Gispert S, Torres-Odio S, Jendrach M, Brehm N, Canet-Pons J, Key J, Sen N-E. SerThr-PhosphoProteome of Brain from Aged PINK1-KO+A53T-SNCA Mice Reveals pT1928-MAP1B and pS3781-ANK2 Deficits, as Hub between Autophagy and Synapse Changes. International Journal of Molecular Sciences. 2019; 20(13):3284. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133284
Chicago/Turabian StyleAuburger, Georg, Suzana Gispert, Sylvia Torres-Odio, Marina Jendrach, Nadine Brehm, Júlia Canet-Pons, Jana Key, and Nesli-Ece Sen. 2019. "SerThr-PhosphoProteome of Brain from Aged PINK1-KO+A53T-SNCA Mice Reveals pT1928-MAP1B and pS3781-ANK2 Deficits, as Hub between Autophagy and Synapse Changes" International Journal of Molecular Sciences 20, no. 13: 3284. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133284