Glucagon Receptor Signaling and Glucagon Resistance

 , ,
, ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
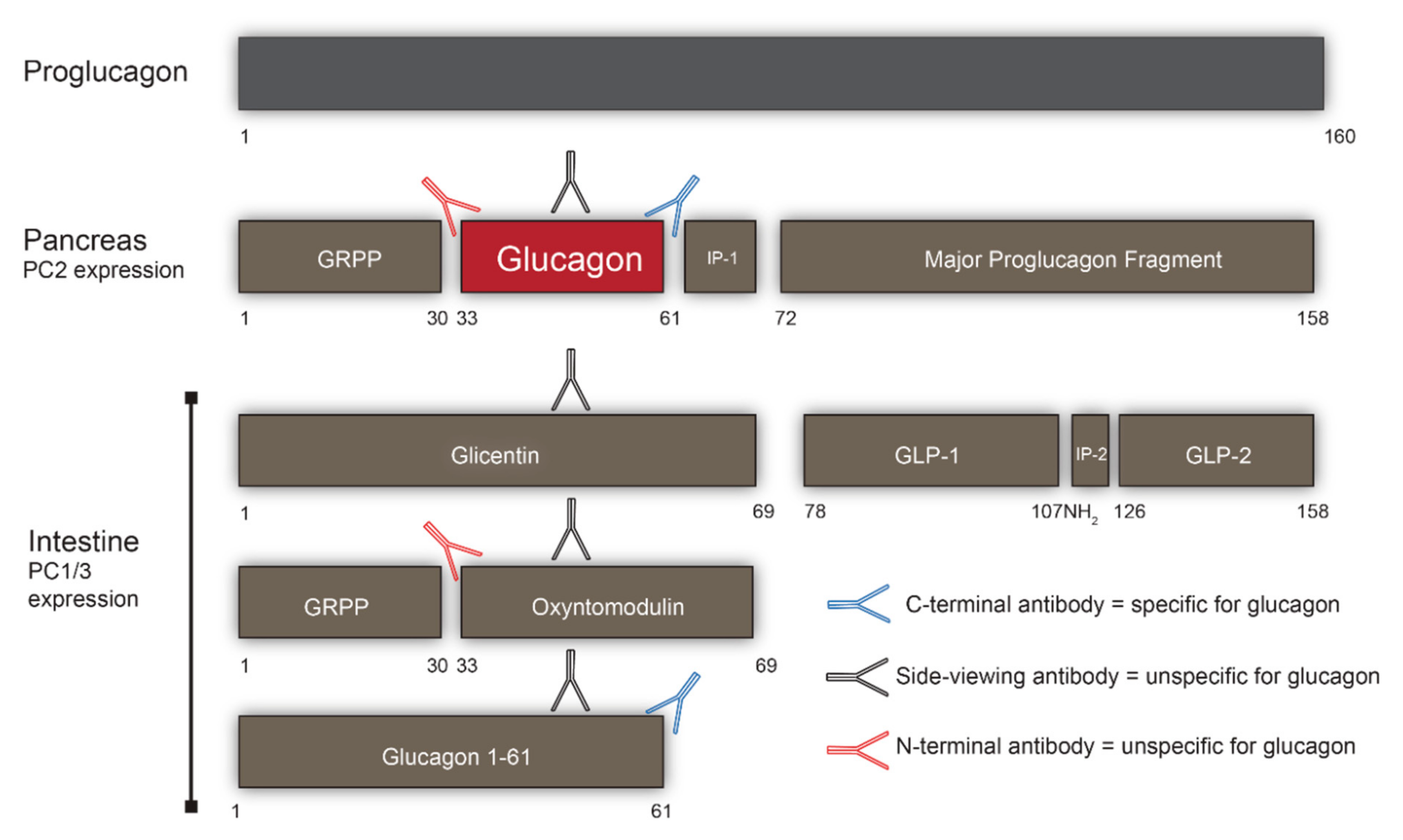
2. Processing of Proglucagon
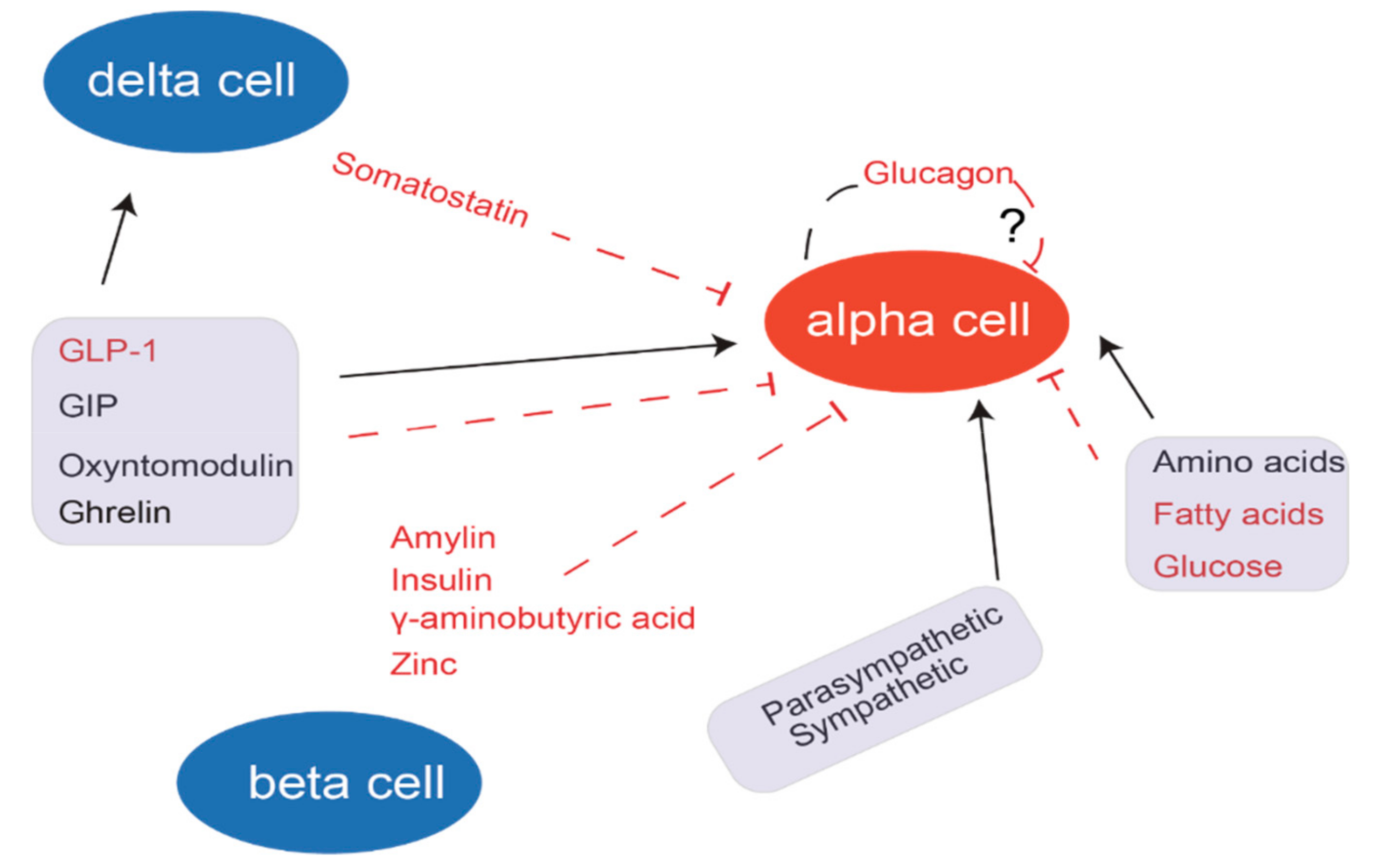
3. Secretion of Glucagon
4. Accurate Measurement of Glucagon
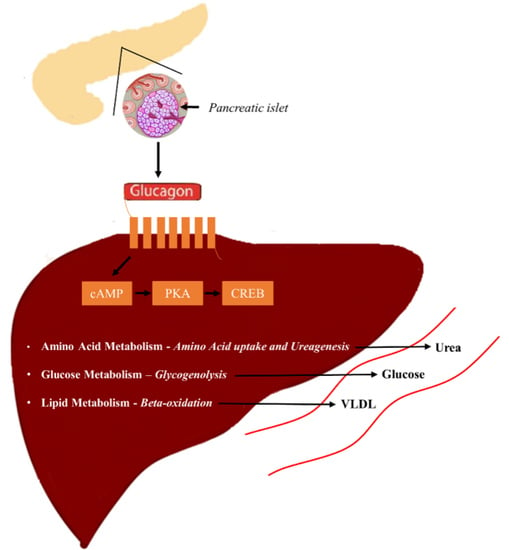
5. Glucagon Receptor Signaling
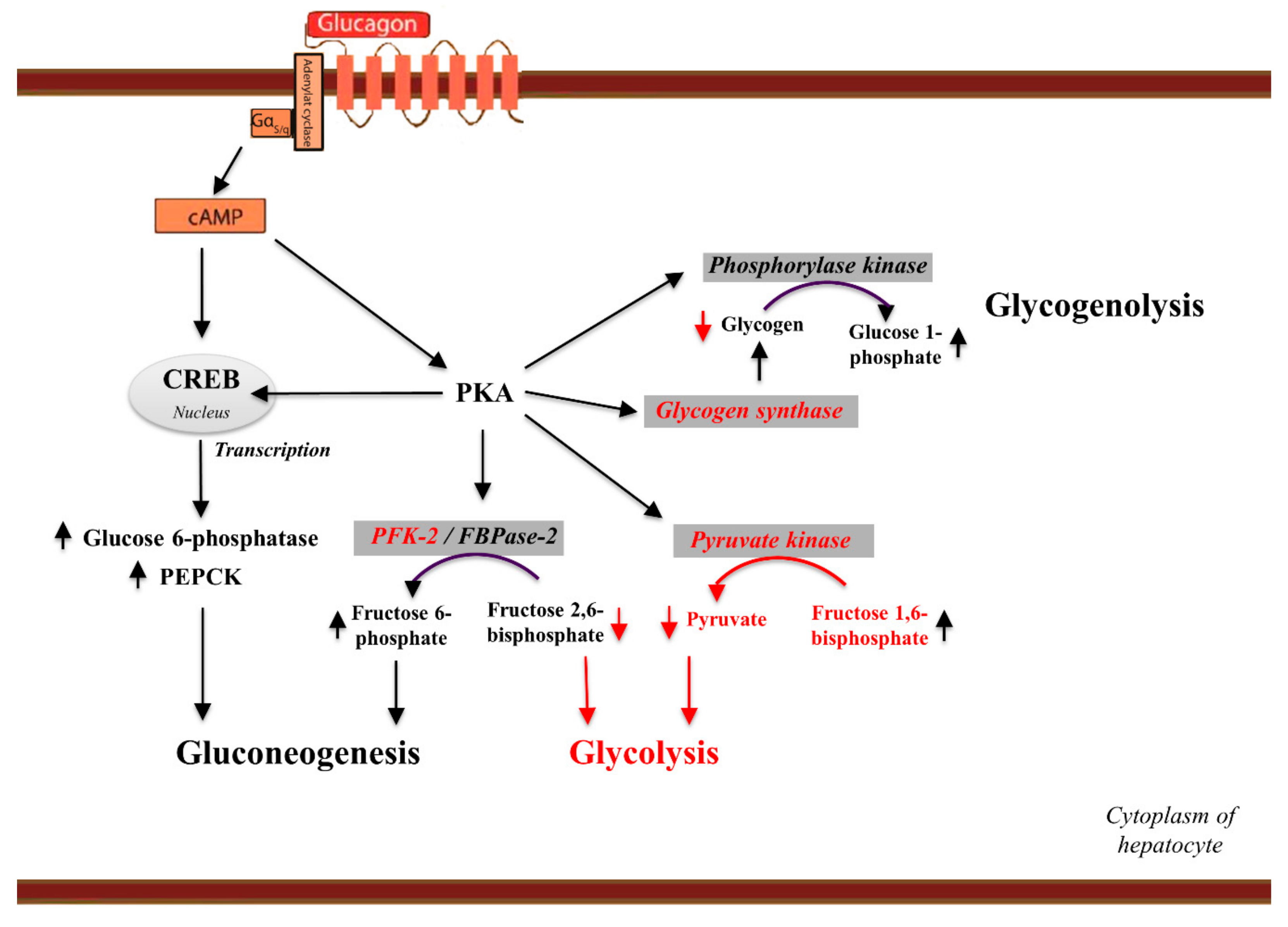
5.1. Glucagon and Glucose Homeostasis
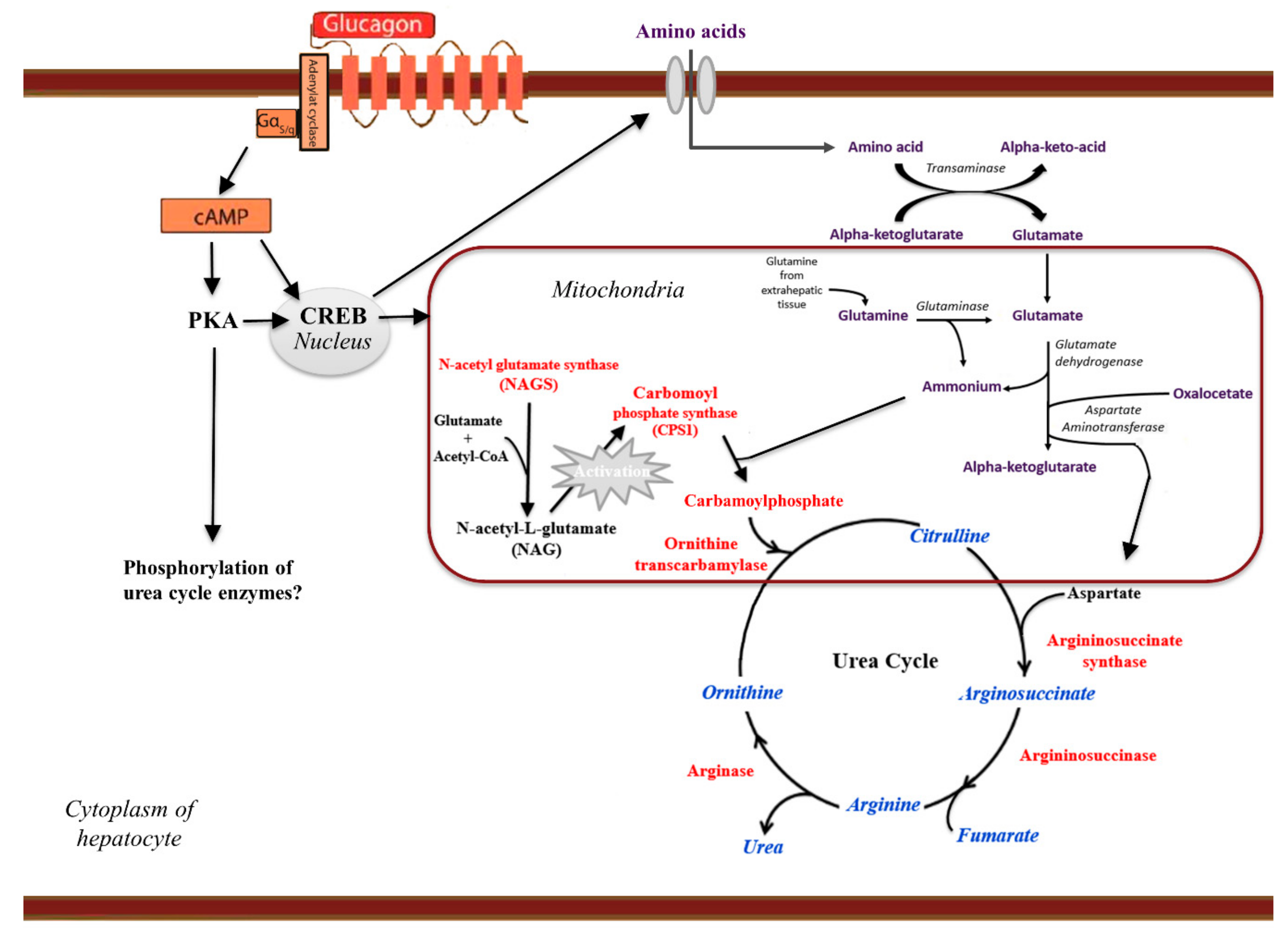
5.2. Glucagon and Amino Acid Metabolism
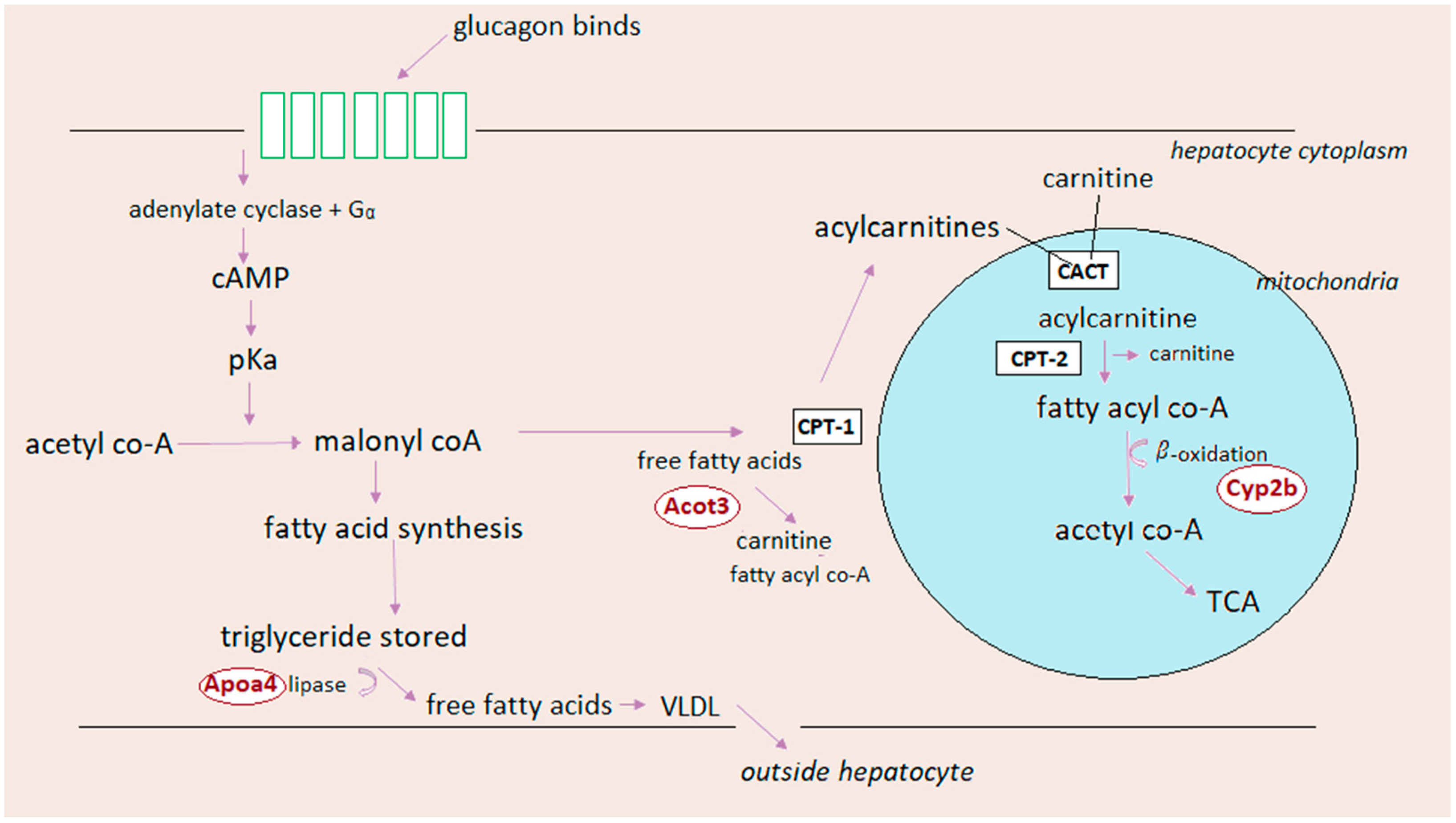
5.3. Glucagon and Lipid Metabolism
6. Glucagon Resistance and Potential Biomarkers
7. Outlook and Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CPS-1 | carbamyl phosphate synthetase-1 |
| CPT-1 | carnitine acyl transferase-1 |
| CREB | cAMP response element-binding |
| FBPase2 | Fructose 2,6-bisphosphatase |
| FFA | Free fatty acid |
| Gcgr−/− | Glucagon receptor knockout |
| GLP-1 | Glucagon-like peptide-1 |
| GLP-2 | Glucagon-like peptide-2 |
| GIP | Glucose-dependent insulinotropic polypeptide |
| GRA | Glucagon receptor antagonist |
| HCD | High carbohydrate diet |
| HSL | Hormone sensitive lipase |
| IRA | Insulin receptor antagonist |
| LCD | Low carbohydrate diet |
| LDL | Low density lipoprotein |
| NAFLD | Non alcoholic fatty liver disease |
| NAG | N-acetyl glutamate |
| NAGS | N-acetyl glutamate synthetase |
| PEPCK | Phosphoenolpyruvatecarboxykinase |
| PFK-2 | Phospho-fructokinase 2 |
| PKA | Protein kinase A |
| TG | Triglyceride |
| VLDL | Very-low density lipoprotein |
References
- Scott, R.V.; Bloom, S.R. Problem or solution: The strange story of glucagon. Peptides 2018, 100, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Kimball, C.P.; Murlin, J.R. Aqueous Extracts of Pancreas: III. Some Precipitation Reactions of Insulin. J. Biol. Chem. 1923, 58, 337–346. [Google Scholar]
- Sutherland, E.W.; Cori, C.F.; Haynes, R.; Olsen, N.S. Purification of the hyperglycemic-glycogenolytic factor from insulin and from gastric mucosa. J. Biol. Chem. 1949, 180, 825–837. [Google Scholar] [PubMed]
- Muller, T.D.; Finan, B.; Clemmensen, C.; DiMarchi, R.D.; Tschop, M.H. The New Biology and Pharmacology of Glucagon. Physiol. Rev. 2017, 97, 721–766. [Google Scholar] [CrossRef] [PubMed]
- Vilstrup, H.; Hansen, B.A.; Almdal, T.P. Glucagon increases hepatic efficacy for urea synthesis. J. Hepatol. 1990, 10, 46–50. [Google Scholar] [CrossRef]
- Gromada, J.; Franklin, I.; Wollheim, C.B. α-Cells of the Endocrine Pancreas: 35 Years of Research but the Enigma Remains. Endocr. Rev. 2007, 28, 84–116. [Google Scholar] [CrossRef] [PubMed]
- Wewer Albrechtsen, N.J.; Pedersen, J.; Galsgaard, K.D.; Winther-Sorensen, M.; Suppli, M.P.; Janah, L.; Gromada, J.; Vilstrup, H.; Knop, F.K.; Holst, J.J. The liver-alpha cell axis and type 2 diabetes. Endocr. Rev. 2019. [Google Scholar] [CrossRef]
- Pearson, M.J.; Unger, R.H.; Holland, W.L. Clinical Trials, Triumphs, and Tribulations of Glucagon Receptor Antagonists. Diabetes Care 2016, 39, 1075–1077. [Google Scholar] [CrossRef] [Green Version]
- Capozzi, M.E.; DiMarchi, R.D.; Tschop, M.H.; Finan, B.; Campbell, J.E. Targeting the Incretin/Glucagon System With Triagonists to Treat Diabetes. Endocr. Rev. 2018, 39, 719–738. [Google Scholar] [CrossRef]
- Ambery, P.; Parker, V.E.; Stumvoll, M.; Posch, M.G.; Heise, T.; Plum-Moerschel, L.; Tsai, L.-F.; Robertson, D.; Jain, M.; Petrone, M.; et al. MEDI0382, a GLP-1 and glucagon receptor dual agonist, in obese or overweight patients with type 2 diabetes: A randomised, controlled, double-blind, ascending dose and phase 2a study. Lancet 2018, 391, 2607–2618. [Google Scholar] [CrossRef]
- Kazda, C.M.; Ding, Y.; Kelly, R.P.; Garhyan, P.; Shi, C.; Lim, C.N.; Fu, H.; Watson, D.E.; Lewin, A.J.; Landschulz, W.H.; et al. Evaluation of Efficacy and Safety of the Glucagon Receptor Antagonist LY2409021 in Patients with Type 2 Diabetes: 12- and 24-Week Phase 2 Studies. Diabetes Care 2016, 39, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Raskin, P.; Unger, R.H. Hyperglucagonemia and Its Suppression. N. Engl. J. Med. 1978, 299, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Hare, K.J.; Vilsboll, T.; Holst, J.J.; Knop, F.K. Inappropriate glucagon response after oral compared with isoglycemic intravenous glucose administration in patients with type 1 diabetes. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E832–E837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faerch, K.; Vistisen, D.; Pacini, G.; Torekov, S.S.; Johansen, N.B.; Witte, D.R.; Jonsson, A.; Pedersen, O.; Hansen, T.; Lauritzen, T.; et al. Insulin Resistance is Accompanied by Increased Fasting Glucagon and Delayed Glucagon Suppression in Individuals with Normal and Impaired Glucose Regulation. Diabetes 2016, 65, 3473–3481. [Google Scholar] [CrossRef] [PubMed]
- Cegla, J.; Jones, B.J.; Gardiner, J.V.; Hodson, D.J.; Marjot, T.; McGlone, E.R.; Tan, T.M.; Bloom, S.R. RAMP2 Influences Glucagon Receptor Pharmacology via Trafficking and Signaling. Endocrinology 2017, 158, 2680–2693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman, C.B.; Zhang, X.M.; Liu, R.; Regev, A.; Shankar, S.; Garhyan, P.; Pillai, S.G.; Kazda, C.; Chalasani, N.; Hardy, T.A. Treatment with LY2409021, a Glucagon Receptor Antagonist, Increases Liver Fat in Patients with Type 2 Diabetes. Diabetes Obes. Metab. 2017, 19, 1521–1528. [Google Scholar] [CrossRef]
- Bell, G.I.; Santerre, R.F.; Mullenbach, G.T. Hamster preproglucagon contains the sequence of glucagon and two related peptides. Nature 1983, 302, 716–718. [Google Scholar] [CrossRef]
- Wewer Albrechtsen, N.J. Glucagon receptor signaling in metabolic diseases. Peptides 2018, 100, 42–47. [Google Scholar] [CrossRef]
- Steiner, D.F.; Wewer Albrechtsen, N.J.; Rehfeld, J.F.; Holst, J.J. Glucagon Processing. In Reference Module in Neuroscience and Biobehavioral Psychology; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Wewer Albrechtsen, N.J.; Kuhre, R.E.; Torang, S.; Holst, J.J. The intestinal distribution pattern of appetite-and glucose regulatory peptides in mice, rats and pigs. BMC Res. Notes 2016, 9, 60. [Google Scholar] [CrossRef]
- Roberts, G.P.; Larraufie, P.; Richards, P.; Kay, R.G.; Galvin, S.G.; Miedzybrodzka, E.L.; Leiter, A.; Li, H.J.; Glass, L.L.; Ma, M.K.L.; et al. Comparison of Human and Murine Enteroendocrine Cells by Transcriptomic and Peptidomic Profiling. Diabetes 2019, 68, 1062–1072. [Google Scholar] [CrossRef] [Green Version]
- Drucker, D.J.; Habener, J.F.; Holst, J.J. Discovery, characterization, and clinical development of the glucagon-like peptides. J. Clin. Investig. 2017, 127, 4217–4227. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Yusta, B. Physiology and Pharmacology of the Enteroendocrine Hormone Glucagon-Like Peptide-2. Annu. Rev. Physiol. 2014, 76, 561–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estall, J.L.; Drucker, D.J. Glucagon and glucagon-like peptide receptors as drug targets. Curr. Pharm. Des. 2006, 12, 1731–1750. [Google Scholar] [CrossRef] [PubMed]
- Chaudhri, O.B.; Field, B.C.; Bloom, S.R. Gastrointestinal satiety signals. Int. J. Obes. (Lond.) 2008, 32, S28–S31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holst, J.J. The Physiology of Glucagon-like Peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef] [PubMed]
- Mineo, I.; Matsumura, T.; Shingu, R.; Namba, M.; Kuwajima, M.; Matsuzawa, Y. The role of prohormone convertases PC1 (PC3) and PC2 in the cell-specific processing of proglucagon. Biochem. Biophys. Res. Commun. 1995, 207, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, M.E.; Eilertson, C.D.; Klein, K.; Zhou, Y.; Lindberg, I.; McDonald, J.K.; Mackin, R.B.; Noe, B.D. Processing of mouse proglucagon by recombinant prohormone convertase 1 and immunopurified prohormone convertase 2 in vitro. J. Biol. Chem. 1995, 270, 10136–10146. [Google Scholar] [CrossRef]
- Dhanvantari, S.; Brubaker, P.L. Proglucagon processing in an islet cell line: Effects of PC1 overexpression and PC2 depletion. Endocrinology 1998, 139, 1630–1637. [Google Scholar] [CrossRef]
- Ugleholdt, R.; Zhu, X.; Deacon, C.F.; Orskov, C.; Steiner, D.F.; Holst, J.J. Impaired intestinal proglucagon processing in mice lacking prohormone convertase 1. Endocrinology 2004, 145, 1349–1355. [Google Scholar] [CrossRef]
- Webb, G.C.; Dey, A.; Wang, J.; Stein, J.; Milewski, M.; Steiner, D.F. Altered proglucagon processing in an alpha-cell line derived from prohormone convertase 2 null mouse islets. J. Biol. Chem. 2004, 279, 31068–31075. [Google Scholar] [CrossRef]
- Wideman, R.D.; Covey, S.D.; Webb, G.C.; Drucker, D.J.; Kieffer, T.J. A switch from prohormone convertase (PC)-2 to PC1/3 expression in transplanted alpha-cells is accompanied by differential processing of proglucagon and improved glucose homeostasis in mice. Diabetes 2007, 56, 2744–2752. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B. Brain glucose sensing and neural regulation of insulin and glucagon secretion. Diabetesobes. Metab. 2011, 13, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Cherrington, A.; Vranic, M. Role of glucagon and insulin in control of glucose turnover. Metabolism 1971, 20, 625–628. [Google Scholar] [CrossRef]
- Unger, R.H.; Ohneda, A.; Aguilar-Parada, E.; Eisentraut, A.M. The role of aminogenic glucagon secretion in blood glucose homeostasis. J. Clin. Investig. 1969, 48, 810–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerich, J.E.; Langlois, M.; Schneider, V.; Karam, J.H.; Noacco, C. Effects of alternations of plasma free fatty acid levels on pancreatic glucagon secretion in man. J. Clin. Investig. 1974, 53, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Bloom, S.R.; Daniel, P.M.; Johnston, D.I.; Ogawa, O.; Pratt, O.E. Release of glucagon, induced by stress. Q. J. Exp. Physiol. Cogn. Med. Sci. 1973, 58, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Rorsman, P. K(ATP) channels and islet hormone secretion: New insights and controversies. Nat. Rev. Endocrinol. 2013, 9, 660–669. [Google Scholar] [CrossRef]
- Gaisano, H.; MacDonald, P.; Vranic, M. Glucagon secretion and signaling in the development of diabetes. Front. Physiol. 2012, 3, 349. [Google Scholar] [CrossRef] [Green Version]
- Rorsman, P.; Braun, M.; Zhang, Q. Regulation of calcium in pancreatic alpha- and beta-cells in health and disease. Cell Calcium 2012, 51, 300–308. [Google Scholar] [CrossRef]
- Wewer Albrechtsen, N.J.; Kuhre, R.E.; Pedersen, J.; Knop, F.K.; Holst, J.J. The biology of glucagon and the consequences of hyperglucagonemia. Biomark. Med. 2016, 10, 1141–1151. [Google Scholar] [CrossRef] [Green Version]
- Quesada, I.; Tudurí, E.; Ripoll, C.; Nadal, Á. Physiology of the pancreatic α-cell and glucagon secretion: Role in glucose homeostasis and diabetes. J. Endocrinol. 2008, 199, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Drucker, D.J. Islet alpha cells and glucagon critical regulators of energy homeostasis. Nat. Rev. Endocrinol. 2015, 11, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Briant, L.; Salehi, A.; Vergari, E.; Zhang, Q.; Rorsman, P. Glucagon secretion from pancreatic α-cells. Upsala J. Med. Sci. 2016, 121, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.N.; Ramracheya, R.; Zhang, Q.; Johnson, P.R.; Braun, M.; Rorsman, P. Regulation of glucagon secretion by glucose: Paracrine, intrinsic or both? Diabetesobes. Metab. 2011, 13, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ramracheya, R.; Lahmann, C.; Tarasov, A.; Bengtsson, M.; Braha, O.; Braun, M.; Brereton, M.; Collins, S.; Galvanovskis, J.; et al. Role of KATP channels in glucose-regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell Metab. 2013, 18, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Hevener, A.L.; Bergman, R.N.; Donovan, C.M. Novel glucosensor for hypoglycemic detection localized to the portal vein. Diabetes 1997, 46, 1521–1525. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E.; Charles, M.A.; Grodsky, G.M. Characterization of the effects of arginine and glucose on glucagon and insulin release from the perfused rat pancreas. J. Clin. Investig. 1974, 54, 833–841. [Google Scholar] [CrossRef]
- Basco, D.; Zhang, Q.; Salehi, A.; Tarasov, A.; Dolci, W.; Herrera, P.; Spiliotis, I.; Berney, X.; Tarussio, D.; Rorsman, P.; et al. α-cell glucokinase suppresses glucose-regulated glucagon secretion. Nat. Commun. 2018, 9, 546. [Google Scholar] [CrossRef]
- Chakera, A.J.; Hurst, P.S.; Spyer, G.; Ogunnowo-Bada, E.O.; Marsh, W.J.; Riches, C.H.; Yueh, C.-Y.; Markkula, S.P.; Dalley, J.W.; Cox, R.D.; et al. Molecular Reductions in Glucokinase Activity Increase Counter-regulatory Responses to Hypoglycemia in Mice and Humans with Diabetes. Mol. Metab. 2018, 17, 17–27. [Google Scholar] [CrossRef]
- Salehi, A.; Vieira, E.; Gylfe, E. Paradoxical Stimulation of Glucagon Secretion by High Glucose Concentrations. Diabetes 2006, 55, 2318–2323. [Google Scholar] [CrossRef] [Green Version]
- Vergari, E.; Knudsen, J.G.; Ramracheya, R.; Salehi, A.; Zhang, Q.; Adam, J.; Asterholm, I.W.; Benrick, A.; Briant, L.J.B.; Chibalina, M.V.; et al. Insulin inhibits glucagon release by SGLT2-induced stimulation of somatostatin secretion. Nat. Commun. 2019, 10, 139. [Google Scholar] [CrossRef] [PubMed]
- Lai, B.-K.; Chae, H.; Gómez-Ruiz, A.; Cheng, P.; Gallo, P.; Antoine, N.; Beauloye, C.; Jonas, J.-C.; Seghers, V.; Seino, S.; et al. Somatostatin is Only Partly Required for the Glucagonostatic Effect of Glucose but is Necessary for the Glucagonostatic Effect of KATP Channel Blockers. Diabetes 2018, 67, 2239–2253. [Google Scholar] [CrossRef] [PubMed]
- Van der Meulen, T.; Donaldson, C.J.; Cáceres, E.; Hunter, A.E.; Cowing-Zitron, C.; Pound, L.D.; Adams, M.W.; Zembrzycki, A.; Grove, K.L.; Huising, M.O. Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat. Med. 2015, 21, 769. [Google Scholar] [CrossRef] [PubMed]
- Hardy, A.B.; Serino, A.S.; Wijesekara, N.; Chimienti, F.; Wheeler, M.B. Regulation of glucagon secretion by zinc: Lessons from the beta cell-specific Znt8 knockout mouse model. Diabetesobes. Metab. 2011, 13, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, O.; Jacques-Silva, M.C.; Speier, S.; Yang, S.-N.; Köhler, M.; Fachado, A.; Vieira, E.; Zierath, J.R.; Kibbey, R.; Berman, D.M.; et al. Glutamate is a Positive Autocrine Signal for Glucagon Release. Cell Metab. 2008, 7, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Unger, R.H. Effects of gamma-aminobutyric acid on insulin, glucagon, and somatostatin release from isolated perfused dog pancreas. Endocrinology 1983, 113, 111–113. [Google Scholar] [CrossRef]
- Gedulin, B.R.; Jodka, C.M.; Herrmann, K.; Young, A.A. Role of endogenous amylin in glucagon secretion and gastric emptying in rats demonstrated with the selective antagonist, AC187. Regul. Pept. 2006, 137, 121–127. [Google Scholar] [CrossRef]
- Hutchens, T.; Piston, D.W. EphA4 Receptor Forward Signaling Inhibits Glucagon Secretion From α-Cells. Diabetes 2015, 64, 3839–3851. [Google Scholar] [CrossRef]
- Wettergren, A.; Schjoldager, B.; Mortensen, P.E.; Myhre, J.; Christiansen, J.; Holst, J.J. Truncated GLP-1 (proglucagon 78–107-amide) inhibits gastric and pancreatic functions in man. Dig. Dis. Sci. 1993, 38, 665–673. [Google Scholar] [CrossRef]
- Meier, J.J.; Gallwitz, B.; Siepmann, N.; Holst, J.J.; Deacon, C.F.; Schmidt, W.E.; Nauck, M.A. Gastric inhibitory polypeptide (GIP) dose-dependently stimulates glucagon secretion in healthy human subjects at euglycaemia. Diabetologia 2003, 46, 798–801. [Google Scholar] [CrossRef] [Green Version]
- Meier, J.J.; Nauck, M.A.; Pott, A.; Heinze, K.; Goetze, O.; Bulut, K.; Schmidt, W.E.; Gallwitz, B.; Holst, J.J. Glucagon-like peptide 2 stimulates glucagon secretion, enhances lipid absorption, and inhibits gastric acid secretion in humans. Gastroenterology 2006, 130, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Salehi, A.; Dornonville de la Cour, C.; Hakanson, R.; Lundquist, I. Effects of ghrelin on insulin and glucagon secretion: A study of isolated pancreatic islets and intact mice. Regul. Pept. 2004, 118, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H.; Ketterer, H.; Dupré, J.; Eisentraut, A.M. The Effects of Secretin, Pancreozymin, and Gastrin on Insulin and Glucagon Secretion in Anesthetized Dogs. J. Clin. Investig. 1967, 46, 630–645. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Muscelli, E.; Frascerra, S.; Baldi, S.; Mari, A.; Heise, T.; Broedl, U.C.; Woerle, H.-J. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J. Clin. Investig. 2014, 124, 499–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merovci, A.; Solis-Herrera, C.; Daniele, G.; Eldor, R.; Fiorentino, T.V.; Tripathy, D.; Xiong, J.; Perez, Z.; Norton, L.; Abdul-Ghani, M.A.; et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J. Clin. Investig. 2014, 124, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Bonner, C.; Kerr-Conte, J.; Gmyr, V.; Queniat, G.; Moerman, E.; Thevenet, J.; Beaucamps, C.; Delalleau, N.; Popescu, I.; Malaisse, W.J.; et al. Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat. Med. 2015, 21, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Solini, A.; Sebastiani, G.; Nigi, L.; Santini, E.; Rossi, C.; Dotta, F. Dapagliflozin modulates glucagon secretion in an SGLT2-independent manner in murine alpha cells. Diabetes Metab. 2017, 43, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, E.J.; Shin, H.M.; Jung, H.S.; Kim, T.K.; Kim, T.N.; Kwon, M.J.; Lee, S.H.; Rhee, B.D.; Park, J.H. The effect of PPARgamma agonist on SGLT2 and glucagon expressions in alpha cells under hyperglycemia. J. Endocrinol. Investig. 2017, 40, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.G.; Ahlstedt, I.; El Hachmane, M.F.; Göpel, S.O. Dapagliflozin stimulates glucagon secretion at high glucose: Experiments and mathematical simulations of human A-cells. Sci. Rep. 2016, 6, 31214. [Google Scholar] [CrossRef]
- Wang, M.-Y.; Yu, X.; Lee, Y.; McCorkle, S.K.; Chen, S.; Li, J.; Wang, Z.V.; Davidson, J.A.; Scherer, P.E.; Holland, W.L.; et al. Dapagliflozin suppresses glucagon signaling in rodent models of diabetes. Proc. Natl. Acad. Sci. USA 2017, 114, 6611–6616. [Google Scholar] [CrossRef] [Green Version]
- Jurczak, M.J.; Lee, H.-Y.; Birkenfeld, A.L.; Jornayvaz, F.R.; Frederick, D.W.; Pongratz, R.L.; Zhao, X.; Moeckel, G.W.; Samuel, V.T.; Whaley, J.M.; et al. SGLT2 Deletion Improves Glucose Homeostasis and Preserves Pancreatic β-Cell Function. Diabetes 2011, 60, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.T.W.; Chen, L.; Li, S.Y.T.; Mayoux, E.; Leung, P.S. The Effects of Empagliflozin, an SGLT2 Inhibitor, on Pancreatic β-Cell Mass and Glucose Homeostasis in Type 1 Diabetes. PLoS ONE 2016, 11, e0147391. [Google Scholar] [CrossRef] [PubMed]
- Lundkvist, P.; Pereira, M.J.; Kamble, P.G.; Katsogiannos, P.; Langkilde, A.M.; Esterline, R.; Johnsson, E.; Eriksson, J.W. Glucagon Levels during Short-term SGLT2 Inhibition are Largely Regulated by Glucose Changes in Type 2 Diabetes Patients. J. Clin. Endocrinol. Metab. 2018, 104, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Kuhre, R.E.; Ghiasi, S.M.; Adriaenssens, A.E.; Wewer Albrechtsen, N.J.; Andersen, D.B.; Aivazidis, A.; Chen, L.; Mandrup-Poulsen, T.; Orskov, C.; Gribble, F.M.; et al. No direct effect of SGLT2 activity on glucagon secretion. Diabetologia 2019, 62, 1011–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bak, M.J.; Albrechtsen, N.W.; Pedersen, J.; Hartmann, B.; Christensen, M.; Vilsbøll, T.; Knop, F.K.; Deacon, C.F.; Dragsted, L.O.; Holst, J.J. Specificity and sensitivity of commercially available assays for glucagon and oxyntomodulin measurement in humans. Eur. J. Endocrinol. 2014, 170, 529–538. [Google Scholar] [CrossRef] [Green Version]
- Albrechtsen, N.J.W. Measurement of Gastrointestinal Hormones. Dan. Med. J. 2017, 64, B5425. [Google Scholar]
- Roberts, G.P.; Kay, R.G.; Howard, J.; Hardwick, R.H.; Reimann, F.; Gribble, F.M. Gastrectomy with Roux-en-Y reconstruction as a lean model of bariatric surgery. Surg. Obes. Relat. Dis. Off. J. Am. Soc. Bariatr. Surg. 2018, 14, 562–568. [Google Scholar] [CrossRef] [Green Version]
- Wewer Albrechtsen, N.J.; Kuhre, R.E.; Hornburg, D.; Jensen, C.Z.; Hornum, M.; Dirksen, C.; Svane, M.; Gasbjerg, L.S.; Jorgensen, N.B.; Gabe, M.N.; et al. Circulating Glucagon 1-61 Regulates Blood Glucose by Increasing Insulin Secretion and Hepatic Glucose Production. Cell Rep. 2017, 21, 1452–1460. [Google Scholar] [CrossRef] [Green Version]
- Baldissera, F.G.; Holst, J.J. Glicentin 1-61 probably represents a major fraction of glucagon-related peptides in plasma of anaesthetized uraemic pigs. Diabetologia 1986, 29, 462–467. [Google Scholar] [CrossRef]
- Wewer Albrechtsen, N.; Hartmann, B.; Veedfald, S.; Windeløv, J.; Plamboeck, A.; Bojsen-Møller, K.; Idorn, T.; Feldt-Rasmussen, B.; Knop, F.; Vilsbøll, T.; et al. Hyperglucagonaemia analysed by glucagon sandwich ELISA: Nonspecific interference or truly elevated levels? Diabetologia 2014, 57, 1919–1926. [Google Scholar] [CrossRef]
- Holst, J.J.; Holland, W.; Gromada, J.; Lee, Y.; Unger, R.H.; Yan, H.; Sloop, K.W.; Kieffer, T.J.; Damond, N.; Herrera, P.L. Insulin and Glucagon: Partners for Life. Endocrinology 2017, 158, 696–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramnanan, C.J.; Edgerton, D.S.; Kraft, G.; Cherrington, A.D. Physiologic action of glucagon on liver glucose metabolism. Diabetesobes. Metab. 2011, 13, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Elrick, H.; Witten, T.A.; Arai, Y. Glucagon treatment of insulin reactions. N. Engl. J. Med. 1958, 258, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Reiband, H.K.; Schmidt, S.; Ranjan, A.; Holst, J.J.; Madsbad, S.; Norgaard, K. Dual-hormone treatment with insulin and glucagon in patients with type 1 diabetes. Diabetes Metab. Res. Rev. 2015, 31, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Zhang, B.B. Glucagon and regulation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E671–E678. [Google Scholar] [CrossRef] [Green Version]
- Habegger, K.M.; Heppner, K.M.; Geary, N.; Bartness, T.J.; DiMarchi, R.; Tschop, M.H. The metabolic actions of glucagon revisited. Nat. Rev. Endocrinol. 2010, 6, 689–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cryer, P.E. Minireview: Glucagon in the Pathogenesis of Hypoglycemia and Hyperglycemia in Diabetes. Endocrinology 2012, 153, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marliss, E.B.; Aoki, T.T.; Unger, R.H.; Soeldner, J.S.; Cahill, G.F., Jr. Glucagon levels and metabolic effects in fasting man. J. Clin. Investig. 1970, 49, 2256–2270. [Google Scholar] [CrossRef] [Green Version]
- Brand, C.L.; Jorgensen, P.N.; Knigge, U.; Warberg, J.; Svendsen, I.; Kristensen, J.S.; Holst, J.J. Role of glucagon in maintenance of euglycemia in fed and fasted rats. Am. J. Physiol. 1995, 269, E469–E477. [Google Scholar] [CrossRef]
- Brand, C.L.; Rolin, B.; Jorgensen, P.N.; Svendsen, I.; Kristensen, J.S.; Holst, J.J. Immunoneutralization of endogenous glucagon with monoclonal glucagon antibody normalizes hyperglycaemia in moderately streptozotocin-diabetic rats. Diabetologia 1994, 37, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Longuet, C.; Sinclair, E.M.; Maida, A.; Baggio, L.L.; Maziarz, M.; Charron, M.J.; Drucker, D.J. The glucagon receptor is required for the adaptive metabolic response to fasting. Cell Metab. 2008, 8, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Schmid, R.; Schusdziarra, V.; Schulte-Frohlinde, E.; Maier, V.; Classen, M. Role of amino acids in stimulation of postprandial insulin, glucagon, and pancreatic polypeptide in humans. Pancreas 1989, 4, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H. Glucagon physiology and pathophysiology. N. Engl. J. Med. 1971, 285, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Belanger, P.; Couturier, K.; Latour, M.G.; Lavoie, J.M. Effects of supranormal liver glycogen content on hyperglucagonemia-induced liver glycogen breakdown. Eur. J. Appl. Physiol. 2000, 83, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, O.H.; Dichmann, D.S.; Abrahamsen, N.; Grunnet, N.; Nishimura, E. Identification of a novel human glucagon receptor promoter: Regulation by cAMP and PGC-1alpha. Gene 2007, 393, 127–136. [Google Scholar] [CrossRef]
- Ranjan, A.; Schmidt, S.; Damm-Frydenberg, C.; Steineck, I.; Clausen, T.R.; Holst, J.J.; Madsbad, S.; Norgaard, K. Low-Carbohydrate Diet Impairs the Effect of Glucagon in the Treatment of Insulin-Induced Mild Hypoglycemia: A Randomized Crossover Study. Diabetes Care 2017, 40, 132–135. [Google Scholar] [CrossRef]
- Galsgaard, K.D.; Winther-Sorensen, M.; Pedersen, J.; Kjeldsen, S.A.S.; Rosenkilde, M.M.; Wewer Albrechtsen, N.J.; Holst, J.J. Glucose and amino acid metabolism in mice depend mutually on glucagon and insulin receptor signaling. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E660–E673. [Google Scholar] [CrossRef]
- Ranjan, A.; Schmidt, S.; Madsbad, S.; Holst, J.J.; Norgaard, K. Effects of subcutaneous, low-dose glucagon on insulin-induced mild hypoglycaemia in patients with insulin pump treated type 1 diabetes. Diabetesobes. Metab. 2016, 18, 410–418. [Google Scholar] [CrossRef]
- El Youssef, J.; Castle, J.R.; Bakhtiani, P.A.; Haidar, A.; Branigan, D.L.; Breen, M.; Ward, W.K. Quantification of the glycemic response to microdoses of subcutaneous glucagon at varying insulin levels. Diabetes Care 2014, 37, 3054–3060. [Google Scholar] [CrossRef] [PubMed]
- Blauw, H.; Wendl, I.; DeVries, J.H.; Heise, T.; Jax, T. Pharmacokinetics and pharmacodynamics of various glucagon dosages at different blood glucose levels. Diabetesobes. Metab. 2016, 18, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Haymond, M.W.; Schreiner, B. Mini-Dose Glucagon Rescue for Hypoglycemia in Children with Type 1 Diabetes. Diabetes Care 2001, 24, 643. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, L.; Mallad, A.; Dalla Man, C.; Basu, R.; Cobelli, C.; Carter, R.E.; Kudva, Y.C.; Basu, A. Glucagon sensitivity and clearance in type 1 diabetes: Insights from in vivo and in silico experiments. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E474–E486. [Google Scholar] [CrossRef] [PubMed]
- Charbonneau, A.; Couturier, K.; Gauthier, M.S.; Lavoie, J.M. Evidence of Hepatic Glucagon Resistance Associated with Hepatic Steatosis: Reversal Effect of Training. Int. J. Sports Med. 2005, 26, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Charbonneau, A.; Unson, C.G.; Lavoie, J.M. High-fat diet-induced hepatic steatosis reduces glucagon receptor content in rat hepatocytes: Potential interaction with acute exercise. J. Physiol. 2007, 579, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Suppli, M.P.; Bagger, J.I.; Lund, A.B.; Albrechtsen, N.J.W.; Holst, J.J.; Vilsbøll, T.; Knop, F.K. Glucagon Resistance at the Level of Amino Acid Turnover and Ureagenesis in Obese Subjects with Hepatic Steatosis. Diabetes 2018, 67. [Google Scholar] [CrossRef]
- Gopel, S.O.; Kanno, T.; Barg, S.; Weng, X.G.; Gromada, J.; Rorsman, P. Regulation of glucagon release in mouse -cells by KATP channels and inactivation of TTX-sensitive Na+ channels. J. Physiol. 2000, 528, 509–520. [Google Scholar] [CrossRef]
- Ishihara, H.; Maechler, P.; Gjinovci, A.; Herrera, P.L.; Wollheim, C.B. Islet beta-cell secretion determines glucagon release from neighbouring alpha-cells. Nat. Cell Biol. 2003, 5, 330–335. [Google Scholar] [CrossRef]
- Franklin, I.; Gromada, J.; Gjinovci, A.; Theander, S.; Wollheim, C.B. Beta-cell secretory products activate alpha-cell ATP-dependent potassium channels to inhibit glucagon release. Diabetes 2005, 54, 1808–1815. [Google Scholar] [CrossRef]
- Olsen, H.L.; Theander, S.; Bokvist, K.; Buschard, K.; Wollheim, C.B.; Gromada, J. Glucose stimulates glucagon release in single rat alpha-cells by mechanisms that mirror the stimulus-secretion coupling in beta-cells. Endocrinology 2005, 146, 4861–4870. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Murayama, Y.; Okuda, Y.; Yamashita, K. Postprandial glucose, insulin and glucagon responses to meals with different nutrient compositions in non-insulin-dependent diabetes mellitus. Endocrinol. Jpn. 1987, 34, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A.; Faloona, G.R.; Aguilar-Parada, E.; Unger, R.H. Abnormal alpha-cell function in diabetes. Response to carbohydrate and protein ingestion. N. Engl. J. Med. 1970, 283, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Bagger, J.I.; Knop, F.K.; Lund, A.; Holst, J.J.; Vilsboll, T. Glucagon responses to increasing oral loads of glucose and corresponding isoglycaemic intravenous glucose infusions in patients with type 2 diabetes and healthy individuals. Diabetologia 2014, 57, 1720–1725. [Google Scholar] [CrossRef] [PubMed]
- Ang, T.; Bruce, C.R.; Kowalski, G.M. Postprandial Aminogenic Insulin and Glucagon Secretion Can Stimulate Glucose Flux in Humans. Diabetes 2019, 68, 939–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagger, J.I.; Knop, F.K.; Holst, J.J.; Vilsboll, T. Glucagon antagonism as a potential therapeutic target in type 2 diabetes. Diabetes Obes. Metab. 2011, 13, 965–971. [Google Scholar] [CrossRef]
- Sloop, K.W.; Michael, M.D.; Moyers, J.S. Glucagon as a target for the treatment of Type 2 diabetes. Expert Opin. Ther. Targets 2005, 9, 593–600. [Google Scholar] [CrossRef]
- Scheen, A.J.; Paquot, N.; Lefebvre, P.J. Investigational glucagon receptor antagonists in Phase I and II clinical trials for diabetes. Expert Opin. Investig. Drugs 2017, 26, 1373–1389. [Google Scholar] [CrossRef]
- Gelling, R.W.; Du, X.Q.; Dichmann, D.S.; Romer, J.; Huang, H.; Cui, L.; Obici, S.; Tang, B.; Holst, J.J.; Fledelius, C.; et al. Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc. Natl. Acad. Sci. USA 2003, 100, 1438–1443. [Google Scholar] [CrossRef]
- Kim, J.; Okamoto, H.; Huang, Z.; Anguiano, G.; Chen, S.; Liu, Q.; Cavino, K.; Xin, Y.; Na, E.; Hamid, R.; et al. Amino Acid Transporter Slc38a5 Controls Glucagon Receptor Inhibition-Induced Pancreatic alpha Cell Hyperplasia in Mice. Cell Metab. 2017, 25, 1348–1361. [Google Scholar] [CrossRef]
- Sorensen, H.; Winzell, M.S.; Brand, C.L.; Fosgerau, K.; Gelling, R.W.; Nishimura, E.; Ahren, B. Glucagon receptor knockout mice display increased insulin sensitivity and impaired beta-cell function. Diabetes 2006, 55, 3463–3469. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H.; Cherrington, A.D. Glucagonocentric restructuring of diabetes: A pathophysiologic and therapeutic makeover. J. Clin. Investig. 2012, 122, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H.; Aguilar-Parada, E.; Müller, W.A.; Eisentraut, A.M. Studies of pancreatic alpha cell function in normal and diabetic subjects. J. Clin. Investig. 1970, 49, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sammons, M.F.; Lee, E.C. Recent progress in the development of small-molecule glucagon receptor antagonists. Bioorg. Med. Chem. Lett. 2015, 25, 4057–4064. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Holleman, C.L.; Nason, S.; Arble, D.M.; Ottaway, N.; Chabenne, J.; Loyd, C.; Kim, J.A.; Sandoval, D.; Drucker, D.J.; et al. Hepatic Glucagon Receptor Signaling Enhances Insulin-Stimulated Glucose Disposal in Rodents. Diabetes 2018, 67, 2157–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, T.M.; Field, B.C.; McCullough, K.A.; Troke, R.C.; Chambers, E.S.; Salem, V.; Gonzalez Maffe, J.; Baynes, K.C.; De Silva, A.; Viardot, A.; et al. Coadministration of glucagon-like peptide-1 during glucagon infusion in humans results in increased energy expenditure and amelioration of hyperglycemia. Diabetes 2013, 62, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Hamberg, O.; Vilstrup, H. Regulation of urea synthesis by glucose and glucagon in normal man. Clin. Nutr. 1994, 13, 183–191. [Google Scholar] [CrossRef]
- Snodgrass, P.J.; Lin, R.C.; Muller, W.A.; Aoki, T.T. Induction of urea cycle enzymes of rat liver by glucagon. J. Biol. Chem. 1978, 253, 2748–2753. [Google Scholar]
- Vilstrup, H. On urea synthesis-regulation in vivo. Dan. Med. Bull. 1989, 36, 415–429. [Google Scholar]
- Heibel, S.K.; Lopez, G.Y.; Panglao, M.; Sodha, S.; Marino-Ramirez, L.; Tuchman, M.; Caldovic, L. Transcriptional regulation of N-acetylglutamate synthase. PLoS ONE 2012, 7, e29527. [Google Scholar] [CrossRef]
- Kilberg, M.S.; Barber, E.F.; Handlogten, M.E. Characteristics and hormonal regulation of amino acid transport system A in isolated rat hepatocytes. Curr. Top. Cell. Regul. 1985, 25, 133–163. [Google Scholar] [PubMed]
- Nelson, D.L.; Lehninger, A.L.; Cox, M.M. Lehninger Principles of Biochemistry; W.H. Freeman: New York, NY, USA, 2008. [Google Scholar]
- Morris, S.M., Jr. Regulation of enzymes of the urea cycle and arginine metabolism. Annu. Rev. Nutr. 2002, 22, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Galsgaard, K.D.; Winther-Sorensen, M.; Orskov, C.; Kissow, H.; Poulsen, S.S.; Vilstrup, H.; Prehn, C.; Adamski, J.; Jepsen, S.L.; Hartmann, B.; et al. Disruption of glucagon receptor signaling causes hyperaminoacidemia exposing a possible liver-alpha-cell axis. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E93–E103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, K.F.; Hansen, B.A.; Vilstrup, H. Time dependent stimulating effect of glucagon on the capacity of urea-N synthesis in rats. Horm. Metab. Res. 1987, 19, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Almdal, T.P.; Holst, J.J.; Heindorff, H.; Vilstrup, H. Glucagon immunoneutralization in diabetic rats normalizes urea synthesis and decreases nitrogen wasting. Diabetes 1992, 41, 12–16. [Google Scholar] [CrossRef]
- Hamberg, O.; Vilstrup, H. Effects of insulin and glucose on urea synthesis in normal man, independent of pancreatic hormone secretion. J. Hepatol. 1994, 21, 381–387. [Google Scholar] [CrossRef]
- Kraft, G.; Coate, K.C.; Winnick, J.J.; Dardevet, D.; Donahue, E.P.; Cherrington, A.D.; Williams, P.E.; Moore, M.C. Glucagon’s effect on liver protein metabolism in vivo. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E263–E272. [Google Scholar] [CrossRef]
- Winther-Sørensen, M.; Galsgaard, K.D.; Kuhre, R.E.; Pedersen, J.; Wewer Albrechtsen, N.J.; Holst, J.J. Amino Acid Metabolism is Regulated by Glucagon Receptor Signaling in Mice. In Proceedings of the American Diabetes Association—78th Sceintific Session, Orlando, FL, USA, 22–26 June 2018. [Google Scholar]
- Dean, E.D.; Li, M.; Prasad, N.; Wisniewski, S.N.; Von Deylen, A.; Spaeth, J.; Maddison, L.; Botros, A.; Sedgeman, L.R.; Bozadjieva, N.; et al. Interrupted Glucagon Signaling Reveals Hepatic alpha Cell Axis and Role for L-Glutamine in alpha Cell Proliferation. Cell Metab. 2017, 25, 1362–1373. [Google Scholar] [CrossRef]
- Solloway, M.J.; Madjidi, A.; Gu, C.; Eastham-Anderson, J.; Clarke, H.J.; Kljavin, N.; Zavala-Solorio, J.; Kates, L.; Friedman, B.; Brauer, M.; et al. Glucagon Couples Hepatic Amino Acid Catabolism to mTOR-Dependent Regulation of alpha-Cell Mass. Cell Rep. 2015, 12, 495–510. [Google Scholar] [CrossRef]
- Charlton, M.R.; Adey, D.B.; Nair, K.S. Evidence for a catabolic role of glucagon during an amino acid load. J. Clin. Investig. 1996, 98, 90–99. [Google Scholar] [CrossRef]
- Xiong, Y.; Guan, K.L. Mechanistic insights into the regulation of metabolic enzymes by acetylation. J. Cell Biol. 2012, 198, 155–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Cam, A. Insulin and glucagon regulation of protein phosphorylation in isolated hepatocytes. Persistence, reversibility, and concentration dependence of hormonal effect. Evidence for common phosphorylation sites for both hormones on the Mr = 46,000 protein. J. Biol. Chem. 1982, 257, 8376–8389. [Google Scholar] [PubMed]
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschop, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol. Rev. 2012, 92, 1479–1514. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, R.; Kleemann, E. Hormonal regulation of amino acid transport system N in primary cultures of rat hepatocytes. Eur. J. Biochem. 1987, 166, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J.; Christensen, M.; Lund, A.; de Heer, J.; Svendsen, B.; Kielgast, U.; Knop, F.K. Regulation of glucagon secretion by incretins. Diabetes Obes. Metab. 2011, 13, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Bonadonna, R.C.; Saccomani, M.P.; Cobelli, C.; DeFronzo, R.A. Effect of insulin on system A amino acid transport in human skeletal muscle. J. Clin. Investig. 1993, 91, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Longuet, C.; Robledo, A.M.; Dean, E.D.; Dai, C.; Ali, S.; McGuinness, I.; de Chavez, V.; Vuguin, P.M.; Charron, M.J.; Powers, A.C.; et al. Liver-specific disruption of the murine glucagon receptor produces alpha-cell hyperplasia: Evidence for a circulating alpha-cell growth factor. Diabetes 2013, 62, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Dominguez Gutierrez, G.; Xin, Y.; Cavino, K.; Sung, B.; Sipos, B.; Kloeppel, G.; Gromada, J.; Okamoto, H. Increased SLC38A4 Amino Acid Transporter Expression in Human Pancreatic alpha-Cells After Glucagon Receptor Inhibition. Endocrinology 2019, 160, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Willcox, A.; Richardson, S.J.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. Evidence of increased islet cell proliferation in patients with recent-onset type 1 diabetes. Diabetologia 2010, 53, 2020–2028. [Google Scholar] [CrossRef] [Green Version]
- Baron, A.D.; Schaeffer, L.; Shragg, P.; Kolterman, O.G. Role of hyperglucagonemia in maintenance of increased rates of hepatic glucose output in type II diabetics. Diabetes 1987, 36, 274–283. [Google Scholar] [CrossRef]
- Reaven, G.M.; Chen, Y.D.; Golay, A.; Swislocki, A.L.; Jaspan, J.B. Documentation of hyperglucagonemia throughout the day in nonobese and obese patients with noninsulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 1987, 64, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Wewer Albrechtsen, N.J.; Junker, A.E.; Christensen, M.; Haedersdal, S.; Wibrand, F.; Lund, A.M.; Galsgaard, K.D.; Holst, J.J.; Knop, F.K.; Vilsboll, T. Hyperglucagonemia correlates with plasma levels of non-branched-chain amino acids in patients with liver disease independent of type 2 diabetes. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G91–G96. [Google Scholar] [CrossRef] [PubMed]
- Wewer Albrechtsen, N.J.; Faerch, K.; Jensen, T.M.; Witte, D.R.; Pedersen, J.; Mahendran, Y.; Jonsson, A.E.; Galsgaard, K.D.; Winther-Sorensen, M.; Torekov, S.S.; et al. Evidence of a liver-alpha cell axis in humans: Hepatic insulin resistance attenuates relationship between fasting plasma glucagon and glucagonotropic amino acids. Diabetologia 2018, 61, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Gruppuso, P.A.; Susa, J.B.; Domenech, M.; Cha, C.J.; Schwartz, R. Chronic hyperglucagonemia in rats: Effects on insulin, substrates, and hepatic enzymes of intermediary metabolism. Metabolism 1983, 32, 911–918. [Google Scholar] [CrossRef]
- Almdal, T.P.; Vilstrup, H. Exogenous hyperglucagonaemia in insulin controlled diabetic rats increases urea excretion and nitrogen loss from organs. Diabetologia 1988, 31, 836–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chastain, M.A. The glucagonoma syndrome: A review of its features and discussion of new perspectives. Am. J. Med. Sci. 2001, 321, 306–320. [Google Scholar] [CrossRef] [PubMed]
- Mallinson, C.N.; Bloom, S.R.; Warin, A.P.; Salmon, P.R.; Cox, B. A glucagonoma syndrome. Lancet (Lond. Engl.) 1974, 2, 1–5. [Google Scholar] [CrossRef]
- Marchesini, G.; Bianchi, G.P.; Vilstrup, H.; Checchia, G.A.; Patrono, D.; Zoli, M. Plasma clearances of branched-chain amino acids in control subjects and in patients with cirrhosis. J. Hepatol. 1987, 4, 108–117. [Google Scholar] [CrossRef]
- Boden, G.; Rezvani, I.; Owen, O.E. Effects of glucagon on plasma amino acids. J. Clin. Investig. 1984, 73, 785–793. [Google Scholar] [CrossRef]
- Almdal, T.P.; Heindorff, H.; Bardram, L.; Vilstrup, H. Increased amino acid clearance and urea synthesis in a patient with glucagonoma. Gut 1990, 31, 946–948. [Google Scholar] [CrossRef]
- Kay, R.G.; Challis, B.G.; Casey, R.T.; Roberts, G.P.; Meek, C.L.; Reimann, F.; Gribble, F.M. Peptidomic analysis of endogenous plasma peptides from patients with pancreatic neuroendocrine tumours. Rapid Commun. Mass Spectrom. 2018, 32, 1414–1424. [Google Scholar] [CrossRef] [PubMed]
- Norton, J.A.; Kahn, C.R.; Schiebinger, R.; Gorschboth, C.; Brennan, M.F. Amino acid deficiency and the skin rash associated with glucagonoma. Ann. Intern. Med. 1979, 91, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Alexander, E.K.; Robinson, M.; Staniec, M.; Dluhy, R.G. Peripheral amino acid and fatty acid infusion for the treatment of necrolytic migratory erythema in the glucagonoma syndrome. Clin. Endocrinol. 2002, 57, 827–831. [Google Scholar] [CrossRef]
- Larger, E.; Wewer Albrechtsen, N.J.; Hansen, L.H.; Gelling, R.W.; Capeau, J.; Deacon, C.F.; Madsen, O.D.; Yakushiji, F.; De Meyts, P.; Holst, J.J.; et al. Pancreatic alpha-cell hyperplasia and hyperglucagonemia due to a glucagon receptor splice mutation. Endocrinol. Diabetes Metab. Case Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Dhall, D.; Nissen, N.N.; Chen, C.R.; Yu, R. Homozygous P86S mutation of the human glucagon receptor is associated with hyperglucagonemia, alpha cell hyperplasia, and islet cell tumor. Pancreas 2009, 38, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Master, R.W.; Rezvani, I.; Palmer, J.P.; Lobe, T.E.; Owen, O.E. Glucagon deficiency and hyperaminoacidemia after total pancreatectomy. J. Clin. Investig. 1980, 65, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Hickner, R.C.; Cortright, R.L.; Dohm, G.L.; Houmard, J.A. Lipid oxidation is reduced in obese human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E1039–E1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, F.B.; Constantin-Teodosiu, D.; Greenhaff, P.L. New insights concerning the role of carnitine in the regulation of fuel metabolism in skeletal muscle. J. Physiol. 2007, 581, 431–444. [Google Scholar] [CrossRef]
- DiMarco, J.P.; Hoppel, C. Hepatic mitochondrial function in ketogenic states. Diabetes, starvation, and after growth hormone administration. J. Clin. Investig. 1975, 55, 1237–1244. [Google Scholar] [CrossRef]
- Berglund, E.D.; Lee-Young, R.S.; Lustig, D.G.; Lynes, S.E.; Donahue, E.P.; Camacho, R.C.; Meredith, M.E.; Magnuson, M.A.; Charron, M.J.; Wasserman, D.H. Hepatic energy state is regulated by glucagon receptor signaling in mice. J. Clin. Investig. 2009, 119, 2412–2422. [Google Scholar] [CrossRef] [Green Version]
- Von Meyenn, F.; Porstmann, T.; Gasser, E.; Selevsek, N.; Schmidt, A.; Aebersold, R.; Stoffel, M. Glucagon-induced acetylation of Foxa2 regulates hepatic lipid metabolism. Cell Metab. 2013, 17, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Stoffel, M. Coactivation of Foxa2 through Pgc-1β promotes liver fatty acid oxidation and triglyceride/VLDL secretion. Cell Metab. 2006, 3, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Galsgaard, K.D.; Pedersen, J.; Knop, F.K.; Holst, J.J.; Wewer Albrechtsen, N.J. Glucagon Receptor Signaling and Lipid Metabolism. Front. Physiol. 2019, 10, 413. [Google Scholar] [CrossRef] [PubMed]
- Guettet, C.; Mathe, D.; Riottot, M.; Lutton, C. Effects of chronic glucagon administration on cholesterol and bile acid metabolism. Biochim. Biophys. Acta 1988, 963, 215–223. [Google Scholar] [CrossRef]
- Penhos, J.C.; Wu, C.H.; Daunas, J.; Reitman, M.; Levine, R. Effect of glucagon on the metabolism of lipids and on urea formation by the perfused rat liver. Diabetes 1966, 15, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Heimberg, M.; Weinstein, I.; Kohout, M. The effects of glucagon, dibutyryl cyclic adenosine 3’,5’-monophosphate, and concentration of free fatty acid on hepatic lipid metabolism. J. Biol. Chem. 1969, 244, 5131–5139. [Google Scholar] [PubMed]
- Eaton, R.P. Hypolipemic action of glucagon in experimental endogenous lipemia in the rat. J. Lipid Res. 1973, 14, 312–318. [Google Scholar]
- Xiao, C.; Pavlic, M.; Szeto, L.; Patterson, B.W.; Lewis, G.F. Effects of acute hyperglucagonemia on hepatic and intestinal lipoprotein production and clearance in healthy humans. Diabetes 2011, 60, 383–390. [Google Scholar] [CrossRef]
- Prip-Buus, C.; Pegorier, J.P.; Duee, P.H.; Kohl, C.; Girard, J. Evidence that the sensitivity of carnitine palmitoyltransferase I to inhibition by malonyl-CoA is an important site of regulation of hepatic fatty acid oxidation in the fetal and newborn rabbit. Perinatal development and effects of pancreatic hormones in cultured rabbit hepatocytes. Biochem. J. 1990, 269, 409–415. [Google Scholar] [CrossRef]
- Sloop, K.W.; Cao, J.X.; Siesky, A.M.; Zhang, H.Y.; Bodenmiller, D.M.; Cox, A.L.; Jacobs, S.J.; Moyers, J.S.; Owens, R.A.; Showalter, A.D.; et al. Hepatic and glucagon-like peptide-1-mediated reversal of diabetes by glucagon receptor antisense oligonucleotide inhibitors. J. Clin. Investig. 2004, 113, 1571–1581. [Google Scholar] [CrossRef]
- Liang, Y.; Osborne, M.C.; Monia, B.P.; Bhanot, S.; Gaarde, W.A.; Reed, C.; She, P.; Jetton, T.L.; Demarest, K.T. Reduction in glucagon receptor expression by an antisense oligonucleotide ameliorates diabetic syndrome in db/db mice. Diabetes 2004, 53, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; MacDougall, M.L.; McDowell, M.T.; Xi, L.; Wei, R.; Zavadoski, W.J.; Molloy, M.P.; Baker, J.D.; Kuhn, M.; Cabrera, O.; et al. Polyomic profiling reveals significant hepatic metabolic alterations in glucagon-receptor (GCGR) knockout mice: Implications on anti-glucagon therapies for diabetes. BMC Genom. 2011, 12, 281. [Google Scholar] [CrossRef] [PubMed]
- Conarello, S.L.; Jiang, G.; Mu, J.; Li, Z.; Woods, J.; Zycband, E.; Ronan, J.; Liu, F.; Roy, R.S.; Zhu, L.; et al. Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia 2007, 50, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Pocai, A.; Carrington, P.E.; Adams, J.R.; Wright, M.; Eiermann, G.; Zhu, L.; Du, X.; Petrov, A.; Lassman, M.E.; Jiang, G.; et al. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes 2009, 58, 2258–2266. [Google Scholar] [CrossRef] [PubMed]
- Day, J.W.; Ottaway, N.; Patterson, J.T.; Gelfanov, V.; Smiley, D.; Gidda, J.; Findeisen, H.; Bruemmer, D.; Drucker, D.J.; Chaudhary, N.; et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat. Chem. Biol. 2009, 5, 749–757. [Google Scholar] [CrossRef]
- More, V.R.; Lao, J.; McLaren, D.G.; Cumiskey, A.M.; Murphy, B.A.; Chen, Y.; Previs, S.; Stout, S.; Patel, R.; Satapati, S.; et al. Glucagon like receptor 1/ glucagon dual agonist acutely enhanced hepatic lipid clearance and suppressed de novo lipogenesis in mice. PLoS ONE 2017, 12, e0186586. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.J.; Konkar, A.; Hornigold, D.C.; Trevaskis, J.L.; Jackson, R.; Fritsch Fredin, M.; Jansson-Lofmark, R.; Naylor, J.; Rossi, A.; Bednarek, M.A.; et al. Robust anti-obesity and metabolic effects of a dual GLP-1/glucagon receptor peptide agonist in rodents and non-human primates. Diabetesobes. Metab. 2016, 18, 1176–1190. [Google Scholar] [CrossRef]
- Zhou, J.; Cai, X.; Huang, X.; Dai, Y.; Sun, L.; Zhang, B.; Yang, B.; Lin, H.; Huang, W.; Qian, H. A novel glucagon-like peptide-1/glucagon receptor dual agonist exhibits weight-lowering and diabetes-protective effects. Eur. J. Med. Chem. 2017, 138, 1158–1169. [Google Scholar] [CrossRef]
- Evers, A.; Haack, T.; Lorenz, M.; Bossart, M.; Elvert, R.; Henkel, B.; Stengelin, S.; Kurz, M.; Glien, M.; Dudda, A.; et al. Design of Novel Exendin-Based Dual Glucagon-like Peptide 1 (GLP-1)/Glucagon Receptor Agonists. J. Med. Chem. 2017, 60, 4293–4303. [Google Scholar] [CrossRef]
- Clemmensen, C.; Chabenne, J.; Finan, B.; Sullivan, L.; Fischer, K.; Kuchler, D.; Sehrer, L.; Ograjsek, T.; Hofmann, S.M.; Schriever, S.C.; et al. GLP-1/glucagon coagonism restores leptin responsiveness in obese mice chronically maintained on an obesogenic diet. Diabetes 2014, 63, 1422–1427. [Google Scholar] [CrossRef]
- Stralfors, P.; Bjorgell, P.; Belfrage, P. Hormonal regulation of hormone-sensitive lipase in intact adipocytes: Identification of phosphorylated sites and effects on the phosphorylation by lipolytic hormones and insulin. Proc. Natl. Acad. Sci. USA 1984, 81, 3317–3321. [Google Scholar] [CrossRef]
- Garton, A.J.; Campbell, D.G.; Cohen, P.; Yeaman, S.J. Primary structure of the site on bovine hormone-sensitive lipase phosphorylated by cyclic AMP-dependent protein kinase. FEBS Lett. 1988, 229, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Anthonsen, M.W.; Ronnstrand, L.; Wernstedt, C.; Degerman, E.; Holm, C. Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J. Biol. Chem. 1998, 273, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Egan, J.J.; Wek, S.A.; Garty, N.B.; Blanchette-Mackie, E.J.; Londos, C. Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J. Biol. Chem. 1991, 266, 11341–11346. [Google Scholar] [PubMed]
- Lass, A.; Zimmermann, R.; Haemmerle, G.; Riederer, M.; Schoiswohl, G.; Schweiger, M.; Kienesberger, P.; Strauss, J.G.; Gorkiewicz, G.; Zechner, R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006, 3, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granneman, J.G.; Moore, H.P.; Krishnamoorthy, R.; Rathod, M. Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl). J. Biol. Chem. 2009, 284, 34538–34544. [Google Scholar] [CrossRef] [PubMed]
- Egan, J.J.; Greenberg, A.S.; Chang, M.K.; Wek, S.A.; Moos, M.C., Jr.; Londos, C. Mechanism of hormone-stimulated lipolysis in adipocytes: Translocation of hormone-sensitive lipase to the lipid storage droplet. Proc. Natl. Acad. Sci. USA 1992, 89, 8537–8541. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.J.; Patel, S.; Miyoshi, H.; Greenberg, A.S.; Kraemer, F.B. Functional interaction of hormone-sensitive lipase and perilipin in lipolysis. J. Lipid Res. 2009, 50, 2306–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Hu, L.; Dalen, K.; Dorward, H.; Marcinkiewicz, A.; Russell, D.; Gong, D.; Londos, C.; Yamaguchi, T.; Holm, C.; et al. Activation of hormone-sensitive lipase requires two steps, protein phosphorylation and binding to the PAT-1 domain of lipid droplet coat proteins. J. Biol. Chem. 2009, 284, 32116–32125. [Google Scholar] [CrossRef]
- Schweiger, M.; Eichmann, T.O.; Taschler, U.; Zimmermann, R.; Zechner, R.; Lass, A. Measurement of lipolysis. Methods Enzymol. 2014, 538, 171–193. [Google Scholar] [CrossRef]
- Hansen, L.H.; Abrahamsen, N.; Nishimura, E. Glucagon receptor mRNA distribution in rat tissues. Peptides 1995, 16, 1163–1166. [Google Scholar] [CrossRef]
- Svoboda, M.; Tastenoy, M.; Vertongen, P.; Robberecht, P. Relative quantitative analysis of glucagon receptor mRNA in rat tissues. Mol. Cell. Endocrinol. 1994, 105, 131–137. [Google Scholar] [CrossRef] [Green Version]
- Slavin, B.G.; Ong, J.M.; Kern, P.A. Hormonal regulation of hormone-sensitive lipase activity and mRNA levels in isolated rat adipocytes. J. Lipid Res. 1994, 35, 1535–1541. [Google Scholar] [PubMed]
- Vaughan, M.; Berger, J.E.; Steinberg, D. Hormone-Sensitive Lipase and Monoglyceride Lipase Activities in Adipose Tissue. J. Biol. Chem. 1964, 239, 401–409. [Google Scholar] [PubMed]
- Rodbell, M.; Jones, A.B. Metabolism of isolated fat cells. 3. The similar inhibitory action of phospholipase C (Clostridium perfringens alpha toxin) and of insulin on lipolysis stimulated by lipolytic hormones and theophylline. J. Biol. Chem. 1966, 241, 140–142. [Google Scholar] [PubMed]
- Prigge, W.F.; Grande, F. Effects of glucagon, epinephrine and insulin on in vitro lipolysis of adipose tissue from mammals and birds. Comp. Biochem. Physiol. B Comp. Biochem. 1971, 39, 69–82. [Google Scholar] [CrossRef]
- Vaughan, M.; Steinberg, D. Effect of hormones on lipolysis and esterification of free fatty acids during incubation of adipose tissue in vitro. J. Lipid Res. 1963, 4, 193–199. [Google Scholar]
- Manganiello, V.; Vaughan, M. Selective loss of adipose cell responsiveness to glucagon with growth in the rat. J. Lipid Res. 1972, 13, 12–16. [Google Scholar]
- Livingston, J.N.; Cuatrecasas, P.; Lockwood, D.H. Studies of glucagon resistance in large rat adipocytes: 125I-labeled glucagon binding and lipolytic capacity. J. Lipid Res. 1974, 15, 26–32. [Google Scholar]
- Lefebvre, P.; Luyckx, A.; Bacq, Z.M. Effects of denervation on the metabolism and the response to glucagon of white adipose tissue of rats. Horm. Metab. Res. Metab. 1973, 5, 245–250. [Google Scholar] [CrossRef]
- Honnor, R.C.; Dhillon, G.S.; Londos, C. cAMP-dependent protein kinase and lipolysis in rat adipocytes. I. Cell preparation, manipulation, and predictability in behavior. J. Biol. Chem. 1985, 260, 15122–15129. [Google Scholar] [PubMed]
- Mosinger, B.; Kuhn, E.; Kujalova, V. Action of adipokinetic hormones on human adipose tissue in vitro. J. Lab. Clin. Med. 1965, 66, 380–389. [Google Scholar] [PubMed]
- Vizek, K.; Razova, M.; Melichar, V. Lipolytic effect of TSH, glucagon and hydrocortisone on the adipose tissue of newborns and adults in vitro. Physiol. Bohemoslov. 1979, 28, 325–331. [Google Scholar] [PubMed]
- Gravholt, C.H.; Moller, N.; Jensen, M.D.; Christiansen, J.S.; Schmitz, O. Physiological levels of glucagon do not influence lipolysis in abdominal adipose tissue as assessed by microdialysis. J. Clin. Endocrinol. Metab. 2001, 86, 2085–2089. [Google Scholar] [CrossRef] [PubMed]
- Pozza, G.; Pappalettera, A.; Melogli, O.; Viberti, G.; Ghidoni, A. Lipolytic effect of intra-arterial injection of glucagon in man. Horm. Metab. Res. Metab. 1971, 3, 291–292. [Google Scholar] [CrossRef]
- Pozefsky, T.; Tancredi, R.G.; Moxley, R.T.; Dupre, J.; Tobin, J.D. Metabolism of forearm tissues in man. Studies with glucagon. Diabetes 1976, 25, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.D.; Heiling, V.J.; Miles, J.M. Effects of glucagon on free fatty acid metabolism in humans. J. Clin. Endocrinol. Metab. 1991, 72, 308–315. [Google Scholar] [CrossRef]
- Wu, M.S.; Jeng, C.Y.; Hollenbeck, C.B.; Chen, Y.D.; Jaspan, J.; Reaven, G.M. Does glucagon increase plasma free fatty acid concentration in humans with normal glucose tolerance? J. Clin. Endocrinol. Metab. 1990, 70, 410–416. [Google Scholar] [CrossRef]
- Schade, D.S.; Eaton, R.P. Modulation of fatty acid metabolism by glucagon in man. I. Effects in normal subjects. Diabetes 1975, 24, 502–509. [Google Scholar] [CrossRef]
- Schneider, S.H.; Fineberg, S.E.; Blackburn, G.L. The acute metabolic effects of glucagon and its interactions with insulin in forearm tissue. Diabetologia 1981, 20, 616–621. [Google Scholar] [CrossRef]
- Lefebvre, P.J.; Luyckx, A.S. Glucagon and Catecholamines. In Glucagon II; Lefebvre, P.J., Ed.; Springer: Berlin/Heidelberg, Germany, 1983; pp. 537–543. [Google Scholar]
- Liljenquist, J.E.; Bomboy, J.D.; Lewis, S.B.; Sinclair-Smith, B.C.; Felts, P.W.; Lacy, W.W.; Crofford, O.B.; Liddle, G.W. Effects of glucagon on lipolysis and ketogenesis in normal and diabetic men. J. Clin. Investig. 1974, 53, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Goldfine, I.D.; Cerasi, E.; Luft, R. Glucagon Stimulation of Insulin Release in Man: Inhibition during Hypoglycemia. J. Clin. Endocrinol. Metab. 1972, 35, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Samols, E.; Marri, G.; Marks, V. Promotion of insulin secretion by glucagon. Lancet (Lond. Engl.) 1965, 2, 415–416. [Google Scholar] [CrossRef]
- Gerich, J.E.; Lorenzi, M.; Bier, D.M.; Tsalikian, E.; Schneider, V.; Karam, J.H.; Forsham, P.H. Effects of physiologic levels of glucagon and growth hormone on human carbohydrate and lipid metabolism. Studies involving administration of exogenous hormone during suppression of endogenous hormone secretion with somatostatin. J. Clin. Investig. 1976, 57, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.J.; Luyckx, A.S. Effect of insulin on glucagon enhanced lipolysis in vitro. Diabetologia 1969, 5, 195–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, M.G.; Snead, W.L.; Campbell, P.J. Regulation of free fatty acid metabolism by glucagon. J. Clin. Endocrinol. Metab. 1993, 77, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Lizarraga-Mollinedo, E.; Fernández-Millán, E.; Martín, J.d.T.; Martínez-Honduvilla, C.; Escrivá, F.; Álvarez, C. Early undernutrition induces glucagon resistance and insulin hypersensitivity in the liver of suckling rats. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1070–E1077. [Google Scholar] [CrossRef] [PubMed]
- Charbonneau, A.; Melancon, A.; Lavoie, C.; Lavoie, J.M. Alterations in hepatic glucagon receptor density and in Gsalpha and Gialpha2 protein content with diet-induced hepatic steatosis: Effects of acute exercise. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E8–E14. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, F.; Heeboll, S.; Marrone, G.; Montoliu, C.; Hamilton-Dutoit, S.; Ferrandez, A.; Andreola, F.; Rombouts, K.; Gronbaek, H.; Felipo, V.; et al. Urea cycle dysregulation in non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 905–915. [Google Scholar] [CrossRef] [Green Version]
- Glavind, E.; Aagaard, N.K.; Gronbaek, H.; Moller, H.J.; Orntoft, N.W.; Vilstrup, H.; Thomsen, K.L. Alcoholic Hepatitis Markedly Decreases the Capacity for Urea Synthesis. PLoS ONE 2016, 11, e0158388. [Google Scholar] [CrossRef]
- Cheng, X.; Kim, S.Y.; Okamoto, H.; Xin, Y.; Yancopoulos, G.D.; Murphy, A.J.; Gromada, J. Glucagon contributes to liver zonation. Proc. Natl. Acad. Sci. USA 2018, 115, E4111–E4119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, A.; Zeng, L.; Houslay, M.D. A role for protein kinase C-mediated phosphorylation in eliciting glucagon desensitization in rat hepatocytes. Biochem. J. 1995, 307, 281–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Authier, F.; Desbuquois, B.; De Galle, B. Ligand-mediated internalization of glucagon receptors in intact rat liver. Endocrinology 1992, 131, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Krilov, L.; Nguyen, A.; Miyazaki, T.; Unson, C.G.; Williams, R.; Lee, N.H.; Ceryak, S.; Bouscarel, B. Dual mode of glucagon receptor internalization: Role of PKCalpha, GRKs and beta-arrestins. Exp. Cell Res. 2011, 317, 2981–2994. [Google Scholar] [CrossRef] [PubMed]
- Krilov, L.; Nguyen, A.; Miyazaki, T.; Unson, C.G.; Bouscarel, B. Glucagon receptor recycling: Role of carboxyl terminus, beta-arrestins, and cytoskeleton. Am. J. Physiol. Cell Physiol. 2008, 295, C1230–C1237. [Google Scholar] [CrossRef] [PubMed]
- Elkeles, R.S.; Hambley, J. Glucagon resistance as a cause of hypertriglyceride. Lancet 1976, 308, 18–20. [Google Scholar] [CrossRef]
- Suppli, M.P.; Lund, A.; Bagger, J.I.; Vilsboll, T.; Knop, F.K. Involvement of steatosis-induced glucagon resistance in hyperglucagonaemia. Med. Hypotheses 2016, 86, 100–103. [Google Scholar] [CrossRef]
- Haidar, A.; Legault, L.; Messier, V.; Mitre, T.M.; Leroux, C.; Rabasa-Lhoret, R. Comparison of dual-hormone artificial pancreas, single-hormone artificial pancreas, and conventional insulin pump therapy for glycaemic control in patients with type 1 diabetes: An open-label randomised controlled crossover trial. Lancet Diabetes Endocrinol. 2015, 3, 17–26. [Google Scholar] [CrossRef]
- Finan, B.; Capozzi, M.E.; Campbell, J.E. Repositioning Glucagon Action in the Physiology and Pharmacology of Diabetes. Diabetes 2019, 68. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janah, L.; Kjeldsen, S.; Galsgaard, K.D.; Winther-Sørensen, M.; Stojanovska, E.; Pedersen, J.; Knop, F.K.; Holst, J.J.; Wewer Albrechtsen, N.J. Glucagon Receptor Signaling and Glucagon Resistance. Int. J. Mol. Sci. 2019, 20, 3314. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133314
Janah L, Kjeldsen S, Galsgaard KD, Winther-Sørensen M, Stojanovska E, Pedersen J, Knop FK, Holst JJ, Wewer Albrechtsen NJ. Glucagon Receptor Signaling and Glucagon Resistance. International Journal of Molecular Sciences. 2019; 20(13):3314. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133314
Chicago/Turabian StyleJanah, Lina, Sasha Kjeldsen, Katrine D. Galsgaard, Marie Winther-Sørensen, Elena Stojanovska, Jens Pedersen, Filip K. Knop, Jens J. Holst, and Nicolai J. Wewer Albrechtsen. 2019. "Glucagon Receptor Signaling and Glucagon Resistance" International Journal of Molecular Sciences 20, no. 13: 3314. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133314