Loss of Mevalonate/Cholesterol Homeostasis in the Brain: A Focus on Autism Spectrum Disorder and Rett Syndrome


Abstract
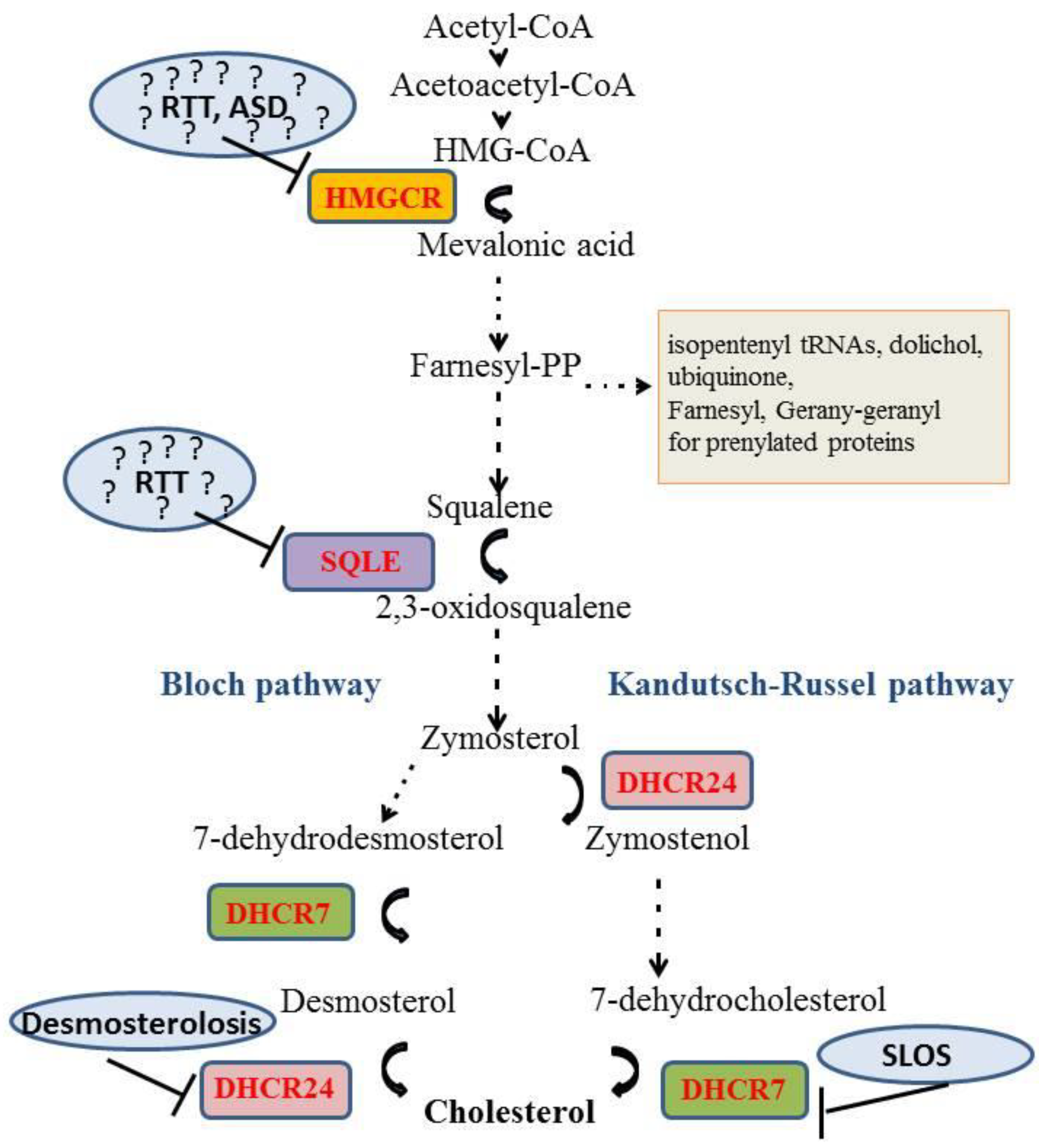
:1. Introduction
Cholesterol Metabolism in the Brain
2. Diseases with Direct Links to Cholesterol Homeostasis
3. Neurodegenerative Diseases with Suspected Links to Cholesterol Metabolism
4. Autism Spectrum Disorder
5. Rett Syndrome
6. Conclusions and Outlook
Funding
Conflicts of Interest
Abbreviations
| 7-DHC | 7-dehydrocholesterol |
| 24-S-OH | 24-S hydroxycholestero |
| AD | Alzheimer’s disease |
| ASD | Autism Spectrum Disorder |
| ABCA1 | ATP binding cassette 1 protein |
| CNS | Central Nervous system |
| CDKL5 | Cyclin-dependent kinase-like 5 |
| CYP46A1 | Cholesterol 24-hydroxylase |
| DHCR7 | 17-Dehydrocholesterol reductase |
| ER | Endoplasmic reticulum |
| HDL | High Density Lipoprotein |
| HD | Huntington’s disease |
| HMG-CoA | 3-hydroxy-3-methylglutaryl Coenzyme A |
| HMGCR | 3-hydroxy-3-methylglutaryl Coenzyme A reductase |
| HTT | Huntingtin |
| LDL | Low density lipoprotein |
| LDLR | Low density lipoprotein receptor |
| LRP1 | LDLR related protein |
| MeCP2 | Methyl CpG binding protein 2 |
| MVA | Mevalonate |
| NPC | Niemann-Pick type C |
| PCSK9 | Proprotein Convertase Subtilisin/Kexin type 9 |
| RTT | Rett syndrome |
| SLOS | Smith–Lemli–Opitz syndrome |
| SQLE | Squalene epoxidase |
| SR-B1 | Scavenger Receptor class B type 1 |
| SREBP2 | Sterol Regulatory Element Binding Protein 2 |
| VPA | Valproic acid |
References
- Dietschy, J.M.; Turley, S.D. Thematic review series: Brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 2004, 45, 1375–1397. [Google Scholar] [PubMed] [Green Version]
- Pfrieger, F.W. Outsourcing in the brain: Do neurons depend on cholesterol delivery by astrocytes? Bioessays 2003, 25, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Segatto, M.; Leboffe, L.; Trapani, L.; Pallottini, V. Cholesterol homeostasis failure in the brain: Implications for synaptic dysfunction and cognitive decline. Curr. Med. Chem. 2014, 21, 2788–2802. [Google Scholar] [CrossRef] [PubMed]
- Mondal, M.; Mesmin, B.; Mukherjee, S.; Maxfield, F.R. Sterols are mainly in the cytoplasmic leaflet of the plasma membrane and the endocytic recycling compartment in CHO cells. Mol. Biol. Cell 2009, 20, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Wang, H. Lipid rafts: A signaling platform linking cholesterol metabolism to synaptic deficits in autism spectrum disorders. Front. Behav. Neurosci. 2014, 8, 104. [Google Scholar] [CrossRef]
- Suzuki, T. Lipid rafts at postsynaptic sites: Distribution, function and linkage to postsynaptic density. Neurosci. Res. 2002, 44, 1–9. [Google Scholar] [CrossRef]
- Gil, C.; Cubi, R.; Blasi, J.; Aguilera, J. Synaptic proteins associate with a sub-set of lipid rafts when isolated from nerve endings at physiological temperature. Biochem. Biophys. Res. Commun. 2006, 348, 1334–1342. [Google Scholar] [CrossRef]
- Allen, J.A.; Halverson-Tamboli, R.A.; Rasenick, M.M. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 2007, 8, 128–140. [Google Scholar] [CrossRef]
- Wasser, C.R.; Kavalali, E.T. Leaky synapses: Regulation of spontaneous neurotransmission in central synapses. Neuroscience 2009, 158, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailman, T.; Hariharan, M.; Karten, B. Inhibition of neuronal cholesterol biosynthesis with lovastatin leads to impaired synaptic vesicle release even in the presence of lipoproteins or geranylgeraniol. J. Neurochem. 2011, 119, 1002–1015. [Google Scholar] [CrossRef]
- Sooksawate, T.; Simmonds, M.A. Effects of membrane cholesterol on the sensitivity of the GABA(A) receptor to GABA in acutely dissociated rat hippocampal neurones. Neuropharmacology 2001, 40, 178–184. [Google Scholar] [CrossRef]
- Mauch, D.H.; Nagler, K.; Schumacher, S.; Goritz, C.; Muller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef] [PubMed]
- Pfrieger, F.W.; Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid. Res. 2011, 50, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Fracassi, A.; Marangoni, M.; Rosso, P.; Pallottini, V.; Fioramonti, M.; Siteni, S.; Segatto, M. Statins and the Brain: More than Lipid Lowering Agents? Curr. Neuropharmacol. 2019, 17, 59–83. [Google Scholar] [CrossRef] [PubMed]
- Trapani, L.; Pallottini, V. Hypercholesterolemia and 3-hydroxy 3-methylglutaryl Coenzyme A reductase regulation during ageing. Sci. World J. 2009, 9, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Sever, N.; Yang, T.; Brown, M.S.; Goldstein, J.L.; DeBose-Boyd, R.A. Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol. Cell 2003, 11, 25–33. [Google Scholar] [CrossRef]
- Ikonen, E. Mechanisms for cellular cholesterol transport: Defects and human disease. Physiol. Rev. 2006, 86, 1237–1261. [Google Scholar] [CrossRef]
- Cartocci, V.; Servadio, M.; Trezza, V.; Pallottini, V. Can Cholesterol Metabolism Modulation Affect Brain Function and Behavior? J. Cell Physiol. 2017, 232, 281–286. [Google Scholar] [CrossRef]
- Leoni, V.; Caccia, C. 24S-hydroxycholesterol in plasma: A marker of cholesterol turnover in neurodegenerative diseases. Biochimie 2013, 95, 595–612. [Google Scholar] [CrossRef]
- Boussicault, L.; Alves, S.; Lamaziere, A.; Planques, A.; Heck, N.; Moumne, L.; Despres, G.; Bolte, S.; Hu, A.; Pages, C.; et al. CYP46A1, the rate-limiting enzyme for cholesterol degradation, is neuroprotective in Huntington’s disease. Brain 2016, 139, 953–970. [Google Scholar] [CrossRef]
- Iuliano, L.; Crick, P.J.; Zerbinati, C.; Tritapepe, L.; Abdel-Khalik, J.; Poirot, M.; Wang, Y.; Griffiths, W.J. Cholesterol metabolites exported from human brain. Steroids 2015, 99, 189–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segatto, M.; Trapani, L.; Lecis, C.; Pallottini, V. Regulation of cholesterol biosynthetic pathway in different regions of the rat central nervous system. Acta. Physiol. (Oxf) 2012, 206, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Segatto, M.; Trapani, L.; Marino, M.; Pallottini, V. Age- and sex-related differences in extra-hepatic low-density lipoprotein receptor. J. Cell Physiol. 2011, 226, 2610–2616. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.G.; Pfrieger, F.; Dotti, C.G. Cholesterol in brain disease: Sometimes determinant and frequently implicated. EMBO. Rep. 2014, 15, 1036–1052. [Google Scholar] [CrossRef] [PubMed]
- Cunniff, C.; Kratz, L.E.; Moser, A.; Natowicz, M.R.; Kelley, R.I. Clinical and biochemical spectrum of patients with RSH/Smith-Lemli-Opitz syndrome and abnormal cholesterol metabolism. Am. J. Med. Genet. 1997, 68, 263–269. [Google Scholar] [CrossRef]
- Porter, J.A.; Ekker, S.C.; Park, W.J.; von Kessler, D.P.; Young, K.E.; Chen, C.H.; Ma, Y.; Woods, A.S.; Cotter, R.J.; Koonin, E.V.; et al. Hedgehog patterning activity: Role of a lipophilic modification mediated by the carboxy-terminal autoprocessing domain. Cell 1996, 86, 21–34. [Google Scholar] [CrossRef]
- Cooper, M.K.; Porter, J.A.; Young, K.E.; Beachy, P.A. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 1998, 280, 1603–1607. [Google Scholar] [CrossRef]
- Koide, T.; Hayata, T.; Cho, K.W. Negative regulation of Hedgehog signaling by the cholesterogenic enzyme 7-dehydrocholesterol reductase. Development 2006, 133, 2395–2405. [Google Scholar] [CrossRef] [Green Version]
- DeBarber, A.E.; Eroglu, Y.; Merkens, L.S.; Pappu, A.S.; Steiner, R.D. Smith-Lemli-Opitz syndrome. Expert. Rev. Mol. Med. 2011, 13, e24. [Google Scholar] [CrossRef]
- Wassif, C.A.; Yu, J.; Cui, J.; Porter, F.D.; Javitt, N.B. 27-Hydroxylation of 7- and 8-dehydrocholesterol in Smith-Lemli-Opitz syndrome: A novel metabolic pathway. Steroids 2003, 68, 497–502. [Google Scholar] [CrossRef]
- Elias, E.R.; Irons, M.B.; Hurley, A.D.; Tint, G.S.; Salen, G. Clinical effects of cholesterol supplementation in six patients with the Smith-Lemli-Opitz syndrome (SLOS). Am. J. Med. Genet. 1997, 68, 305–310. [Google Scholar] [CrossRef]
- Nwokoro, N.A.; Mulvihill, J.J. Cholesterol and bile acid replacement therapy in children and adults with Smith-Lemli-Opitz (SLO/RSH) syndrome. Am. J. Med. Genet. 1997, 68, 315–321. [Google Scholar] [CrossRef]
- Ryan, A.K.; Bartlett, K.; Clayton, P.; Eaton, S.; Mills, L.; Donnai, D.; Winter, R.M.; Burn, J. Smith-Lemli-Opitz syndrome: A variable clinical and biochemical phenotype. J. Med. Genet. 1998, 35, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.P.; Olah, A.V.; Kozak, L.; Balogh, E.; Nagy, A.; Blahakova, I.; Olah, E. A patient with Smith-Lemli-Opitz syndrome: Novel mutation of the DHCR7 gene and effects of therapy with simvastatin and cholesterol supplement. Eur. J. Pediatr. 2010, 169, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Fliesler, S.J.; Xu, L. Oxysterols and Retinal Degeneration in a Rat Model of Smith-Lemli-Opitz Syndrome: Implications for an Improved Therapeutic Intervention. Molecules 2018, 23. [Google Scholar] [CrossRef]
- Vanier, M.T.; Millat, G. Niemann-Pick disease type C. Clin. Genet. 2003, 64, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Sevin, M.; Lesca, G.; Baumann, N.; Millat, G.; Lyon-Caen, O.; Vanier, M.T.; Sedel, F. The adult form of Niemann-Pick disease type C. Brain 2007, 130, 120–133. [Google Scholar] [CrossRef]
- Walterfang, M.; Fietz, M.; Abel, L.; Bowman, E.; Mocellin, R.; Velakoulis, D. Gender dimorphism in siblings with schizophrenia-like psychosis due to Niemann-Pick disease type C. J. Inherit. Metab. Dis. 2009, 32 (Suppl. 1), S221–S226. [Google Scholar] [CrossRef]
- Probert, F.; Ruiz-Rodado, V.; Zhang, X.; te Vruchte, D.; Claridge, T.D.; Edgar, M.; Tocchio, A.Z.; Lachmann, R.H.; Platt, F.M.; Grootveld, M. Urinary excretion and metabolism of miglustat and valproate in patients with Niemann-Pick type C1 disease: One- and two-dimensional solution-state (1)H NMR studies. J. Pharm. Biomed. Anal. 2016, 117, 276–288. [Google Scholar] [CrossRef]
- Vite, C.H.; Bagel, J.H.; Swain, G.P.; Prociuk, M.; Sikora, T.U.; Stein, V.M.; O’Donnell, P.; Ruane, T.; Ward, S.; Crooks, A.; et al. Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease. Sci. Transl. Med. 2015, 7, 276ra26. [Google Scholar] [CrossRef]
- Demais, V.; Barthelemy, A.; Perraut, M.; Ungerer, N.; Keime, C.; Reibel, S.; Pfrieger, F.W. Reversal of Pathologic Lipid Accumulation in NPC1-Deficient Neurons by Drug-Promoted Release of LAMP1-Coated Lamellar Inclusions. J. Neurosci. 2016, 36, 8012–8025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerenturk, E.J.; Sharpe, L.J.; Ikonen, E.; Brown, A.J. Desmosterol and DHCR24: Unexpected new directions for a terminal step in cholesterol synthesis. Prog. Lipid. Res. 2013, 52, 666–680. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wilson, W.A.; Moore, S.D.; Mace, B.E.; Maeda, N.; Schmechel, D.E.; Sullivan, P.M. Human apoE4-targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiol. Dis. 2005, 18, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.; DeKosky, S.T.; Kamboh, M.I. Genetic variation in the cholesterol 24-hydroxylase (CYP46) gene and the risk of Alzheimer’s disease. Neurosci. Lett. 2002, 328, 9–12. [Google Scholar] [CrossRef]
- Kolsch, H.; Lutjohann, D.; Ludwig, M.; Schulte, A.; Ptok, U.; Jessen, F.; von Bergmann, K.; Rao, M.L.; Maier, W.; Heun, R. Polymorphism in the cholesterol 24S-hydroxylase gene is associated with Alzheimer’s disease. Mol. Psychiatry 2002, 7, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Wollmer, M.A.; Streffer, J.R.; Lutjohann, D.; Tsolaki, M.; Iakovidou, V.; Hegi, T.; Pasch, T.; Jung, H.H.; Bergmann, K.; Nitsch, R.M.; et al. ABCA1 modulates CSF cholesterol levels and influences the age at onset of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 421–426. [Google Scholar] [CrossRef]
- Burton, J.K.; Papworth, R.; Haig, C.; McCowan, C.; Ford, I.; Stott, D.J.; Quinn, T.J. Statin Use is Not Associated with Future Long-Term Care Admission: Extended Follow-Up of Two Randomised Controlled Trials. Drugs Aging 2018, 35, 657–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakour, N.; Bianconi, V.; Pirro, M.; Barreto, G.E.; Hadizadeh, F.; Sahebkar, A. In silico evidence of direct interaction between statins and beta-amyloid. J. Cell Biochem. 2019, 120, 4710–4715. [Google Scholar] [CrossRef]
- Trettel, F.; Rigamonti, D.; Hilditch-Maguire, P.; Wheeler, V.C.; Sharp, A.H.; Persichetti, F.; Cattaneo, E.; MacDonald, M.E. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum. Mol. Genet. 2000, 9, 2799–2809. [Google Scholar] [CrossRef] [PubMed]
- Sipione, S.; Rigamonti, D.; Valenza, M.; Zuccato, C.; Conti, L.; Pritchard, J.; Kooperberg, C.; Olson, J.M.; Cattaneo, E. Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum. Mol. Genet. 2002, 11, 1953–1965. [Google Scholar] [CrossRef]
- Valenza, M.; Marullo, M.; Di Paolo, E.; Cesana, E.; Zuccato, C.; Biella, G.; Cattaneo, E. Disruption of astrocyte-neuron cholesterol cross talk affects neuronal function in Huntington’s disease. Cell Death Differ. 2015, 22, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, M.; Di Paolo, E.; Leoni, V.; Caccia, C.; Ferrari Bardile, C.; Mohammed, H.; Di Donato, S.; Kwak, S.; Marchionini, D.; Turner, S.; et al. Early and brain region-specific decrease of de novo cholesterol biosynthesis in Huntington’s disease: A cross-validation study in Q175 knock-in mice. Neurobiol. Dis. 2017, 98, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Valenza, M.; Chen, J.Y.; Di Paolo, E.; Ruozi, B.; Belletti, D.; Ferrari Bardile, C.; Leoni, V.; Caccia, C.; Brilli, E.; Di Donato, S.; et al. Cholesterol-loaded nanoparticles ameliorate synaptic and cognitive function in Huntington’s disease mice. EMBO. Mol. Med. 2015, 7, 1547–1564. [Google Scholar] [CrossRef] [PubMed]
- Servadio, M.; Vanderschuren, L.J.; Trezza, V. Modeling autism-relevant behavioral phenotypes in rats and mice: Do ‘autistic’ rodents exist? Behav. Pharmacol. 2015, 26, 522–540. [Google Scholar] [CrossRef] [PubMed]
- Cartocci, V.; Catallo, M.; Tempestilli, M.; Segatto, M.; Pfrieger, F.W.; Bronzuoli, M.R.; Scuderi, C.; Servadio, M.; Trezza, V.; Pallottini, V. Altered Brain Cholesterol/Isoprenoid Metabolism in a Rat Model of Autism Spectrum Disorders. Neuroscience 2018, 372, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Cartocci, V.; Tonini, C.; Di Pippo, T.; Vuono, F.; Schiavi, S.; Marino, M.; Trezza, V.; Pallottini, V. Prenatal exposure to valproate induces sex-, age-, and tissue-dependent alterations of cholesterol metabolism: Potential implications on autism. J. Cell Physiol. 2019, 234, 4362–4374. [Google Scholar] [CrossRef] [PubMed]
- Melancia, F.; Schiavi, S.; Servadio, M.; Cartocci, V.; Campolongo, P.; Palmery, M.; Pallottini, V.; Trezza, V. Sex-specific autistic endophenotypes induced by prenatal exposure to valproic acid involve anandamide signalling. Br. J. Pharmacol. 2018, 175, 3699–3712. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Regier, D.A.; Kuhl, E.A.; Kupfer, D.J. The DSM-5: Classification and criteria changes. World Psychiatry 2013, 12, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Torrico, B.; Shaw, A.D.; Mosca, R.; Vivo-Luque, N.; Hervas, A.; Fernandez-Castillo, N.; Aloy, P.; Bayes, M.; Fullerton, J.M.; Cormand, B.; et al. Truncating variant burden in high-functioning autism and pleiotropic effects of LRP1 across psychiatric phenotypes. J. Psychiatry Neurosci. 2019, 44, 1–10. [Google Scholar]
- Ionita-Laza, I.; Makarov, V.; Buxbaum, J.D. Scan-statistic approach identifies clusters of rare disease variants in LRP2, a gene linked and associated with autism spectrum disorders, in three datasets. Am. J. Hum. Genet. 2012, 90, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Rett, A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien. Med. Wochenschr. 1966, 116, 723–726. [Google Scholar] [PubMed]
- Hagberg, B.; Aicardi, J.; Dias, K.; Ramos, O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: Report of 35 cases. Ann. Neurol. 1983, 14, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Patankar, J.V. Cholesterol metabolism is a potential therapeutic target for Rett syndrome. Clin. Genet. 2014, 85, 229–230. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.P.; Jugloff, D.G.; Zhang, G.; Logan, R.; Brown, S.; Eubanks, J.H. The expression of methyl CpG binding factor MeCP2 correlates with cellular differentiation in the developing rat brain and in cultured cells. J. Neurobiol. 2003, 55, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Kyle, S.M.; Vashi, N.; Justice, M.J. Rett syndrome: A neurological disorder with metabolic components. Open Biol. 2018, 8. [Google Scholar] [CrossRef]
- Jellinger, K.; Seitelberger, F. Neuropathology of Rett syndrome. Am. J. Med. Genet. Suppl. 1986, 1, 259–288. [Google Scholar] [CrossRef]
- Jellinger, K.; Armstrong, D.; Zoghbi, H.Y.; Percy, A.K. Neuropathology of Rett syndrome. Acta. Neuropathol. 1988, 76, 142–158. [Google Scholar] [CrossRef]
- Armstrong, D.D. The neuropathology of the Rett syndrome. Brain Dev. 1992, 14, S89–S98. [Google Scholar]
- Zoghbi, H.Y.; Milstien, S.; Butler, I.J.; Smith, E.O.; Kaufman, S.; Glaze, D.G.; Percy, A.K. Cerebrospinal fluid biogenic amines and biopterin in Rett syndrome. Ann. Neurol. 1989, 25, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Acampa, M.; Guideri, F.; Hayek, J.; Blardi, P.; De Lalla, A.; Zappella, M.; Auteri, A. Sympathetic overactivity and plasma leptin levels in Rett syndrome. Neurosci. Lett. 2008, 432, 69–72. [Google Scholar] [CrossRef]
- Blardi, P.; De Lalla, A.; D’Ambrogio, T.; Vonella, G.; Ceccatelli, L.; Auteri, A.; Hayek, J. Long-term plasma levels of leptin and adiponectin in Rett syndrome. Clin. Endocrinol. (Oxf) 2009, 70, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Matsuishi, T.; Urabe, F.; Percy, A.K.; Komori, H.; Yamashita, Y.; Schultz, R.S.; Ohtani, Y.; Kuriya, N.; Kato, H. Abnormal carbohydrate metabolism in cerebrospinal fluid in Rett syndrome. J. Child. Neurol. 1994, 9, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Segatto, M.; Trapani, L.; Di Tunno, I.; Sticozzi, C.; Valacchi, G.; Hayek, J.; Pallottini, V. Cholesterol metabolism is altered in Rett syndrome: A study on plasma and primary cultured fibroblasts derived from patients. PLoS ONE 2014, 9, e104834. [Google Scholar] [CrossRef] [PubMed]
- Taneja, P.; Ogier, M.; Brooks-Harris, G.; Schmid, D.A.; Katz, D.M.; Nelson, S.B. Pathophysiology of locus ceruleus neurons in a mouse model of Rett syndrome. J. Neurosci. 2009, 29, 12187–12195. [Google Scholar] [CrossRef] [PubMed]
- Buchovecky, C.M.; Turley, S.D.; Brown, H.M.; Kyle, S.M.; McDonald, J.G.; Liu, B.; Pieper, A.A.; Huang, W.; Katz, D.M.; Russell, D.W.; et al. A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nat. Genet. 2013, 45, 1013–1020. [Google Scholar] [CrossRef] [Green Version]
- Lopez, A.M.; Chuang, J.C.; Posey, K.S.; Turley, S.D. Suppression of brain cholesterol synthesis in male Mecp2-deficient mice is age dependent and not accompanied by a concurrent change in the rate of fatty acid synthesis. Brain Res. 2017, 1654, 77–84. [Google Scholar] [CrossRef]
- Lutjohann, D.; Lopez, A.M.; Chuang, J.C.; Kerksiek, A.; Turley, S.D. Identification of Correlative Shifts in Indices of Brain Cholesterol Metabolism in the C57BL6/Mecp2(tm1.1Bird) Mouse, a Model for Rett Syndrome. Lipids 2018, 53, 363–373. [Google Scholar] [CrossRef]
- Pacheco, N.L.; Heaven, M.R.; Holt, L.M.; Crossman, D.K.; Boggio, K.J.; Shaffer, S.A.; Flint, D.L.; Olsen, M.L. RNA sequencing and proteomics approaches reveal novel deficits in the cortex of Mecp2-deficient mice, a model for Rett syndrome. Mol. Autism. 2017, 8, 56. [Google Scholar] [CrossRef]
- Villani, C.; Sacchetti, G.; Bagnati, R.; Passoni, A.; Fusco, F.; Carli, M.; Invernizzi, R.W. Lovastatin fails to improve motor performance and survival in methyl-CpG-binding protein2-null mice. Elife 2016, 5, e22409. [Google Scholar] [CrossRef] [PubMed]
- Golomb, B.A.; Evans, M.A. Statin adverse effects: A review of the literature and evidence for a mitochondrial mechanism. Am. J. Cardiovasc. Drugs 2008, 8, 373–418. [Google Scholar] [CrossRef] [PubMed]
- Jose, M.A.; Anandkumar, S.; Narmadha, M.P.; Sandeep, M. A comparative effect of atorvastatin with other statins in patients of hyperlipidemia. Indian J. Pharmacol. 2012, 44, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Reilly, D.; Cham, S.; Golomb, B.A. First-degree relatives with behavioural adverse effects on statins. BMJ. Case Rep. 2011, 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.D.; Seguin, M.; Therrien, N.; Riopel, G.; Chawky, N.; Lesage, A.D.; Turecki, G. Familial aggregation of suicidal behavior: A family study of male suicide completers from the general population. Am. J. Psychiatry 2005, 162, 1017–1019. [Google Scholar] [CrossRef] [PubMed]
- Ruljancic, N.; Mihanovic, M.; Cepelak, I. Thrombocyte serotonin and serum cholesterol concentration in suicidal and non-suicidal depressed patients. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- De Berardis, D.; Serroni, N.; Campanella, D.; Olivieri, L.; Marini, S.; Moschetta, F.S.; Martinotti, G.; Di Giannantonio, M. Safety and efficacy of combined clozapine-azathioprine treatment in a case of resistant schizophrenia associated with Behcet’s disease: A 2-year follow-up. Gen. Hosp. Psychiatry 2012, 35, 213.e9–213.e11. [Google Scholar] [CrossRef]
- Tedders, S.H.; Fokong, K.D.; McKenzie, L.E.; Wesley, C.; Yu, L.; Zhang, J. Low cholesterol is associated with depression among US household population. J. Affect. Disord. 2011, 135, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Papakostas, G.I.; Perlis, R.H.; Scalia, M.J.; Petersen, T.J.; Fava, M. A meta-analysis of early sustained response rates between antidepressants and placebo for the treatment of major depressive disorder. J. Clin. Psychopharmacol. 2006, 26, 56–60. [Google Scholar] [CrossRef]
- Wysokinski, A.; Strzelecki, D.; Kloszewska, I. Levels of triglycerides, cholesterol, LDL, HDL and glucose in patients with schizophrenia, unipolar depression and bipolar disorder. Diabetes Metab. Syndr. 2015, 9, 168–176. [Google Scholar] [CrossRef]
- Nadeau, G.; Larue, G.H. Blood Cholesterol in Schizophrenia. Can. Med. Assoc. J. 1952, 66, 320–323. [Google Scholar] [PubMed]
- Goff, D.C.; Sullivan, L.M.; McEvoy, J.P.; Meyer, J.M.; Nasrallah, H.A.; Daumit, G.L.; Lamberti, S.; D’Agostino, R.B.; Stroup, T.S.; Davis, S.; et al. A comparison of ten-year cardiac risk estimates in schizophrenia patients from the CATIE study and matched controls. Schizophr. Res. 2005, 80, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Jow, G.M.; Yang, T.T.; Chen, C.L. Leptin and cholesterol levels are low in major depressive disorder, but high in schizophrenia. J. Affect. Disord. 2006, 90, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Zachor, D.A.; Curatolo, P. Recommendations for early diagnosis and intervention in autism spectrum disorders: An Italian-Israeli consensus conference. Eur. J. Paediatr. Neurol. 2014, 18, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Jeste, S.S.; Geschwind, D.H. Disentangling the heterogeneity of autism spectrum disorder through genetic findings. Nat. Rev. Neurol. 2014, 10, 74–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaste, P.; Leboyer, M. Autism risk factors: Genes, environment, and gene-environment interactions. Dialogues Clin. Neurosci. 2012, 14, 281–292. [Google Scholar] [PubMed]
- DiCicco-Bloom, E.; Lord, C.; Zwaigenbaum, L.; Courchesne, E.; Dager, S.R.; Schmitz, C.; Schultz, R.T.; Crawley, J.; Young, L.J. The developmental neurobiology of autism spectrum disorder. J. Neurosci. 2006, 26, 6897–6906. [Google Scholar] [CrossRef]
- Loomes, R.; Hull, L.; Mandy, W.P.L. What Is the Male-to-Female Ratio in Autism Spectrum Disorder? A Systematic Review and Meta-Analysis. J. Am. Acad. Child. Adolesc. Psychiatry 2017, 56, 466–474. [Google Scholar] [CrossRef]
- De Marinis, E.; Martini, C.; Trentalance, A.; Pallottini, V. Sex differences in hepatic regulation of cholesterol homeostasis. J. Endocrinol. 2008, 198, 635–643. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Trotter, J.; Zhang, J.; Peters, M.M.; Cheng, H.; Bao, J.; Han, X.; Weeber, E.J.; Bu, G. Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J. Neurosci. 2010, 30, 17068–17078. [Google Scholar] [CrossRef]
- Kaufmann, W.E.; Moser, H.W. Dendritic anomalies in disorders associated with mental retardation. Cereb. Cortex. 2000, 10, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Sticozzi, C.; Belmonte, G.; Pecorelli, A.; Cervellati, F.; Leoncini, S.; Signorini, C.; Ciccoli, L.; De Felice, C.; Hayek, J.; Valacchi, G. Scavenger receptor B1 post-translational modifications in Rett syndrome. FEBS. Lett. 2013, 587, 2199–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapani, L.; Melli, L.; Segatto, M.; Trezza, V.; Campolongo, P.; Jozwiak, A.; Swiezewska, E.; Pucillo, L.P.; Moreno, S.; Fanelli, F.; et al. Effects of myosin heavy chain (MHC) plasticity induced by HMGCoA-reductase inhibition on skeletal muscle functions. FASEB J. 2011, 25, 4037–4047. [Google Scholar] [CrossRef] [PubMed]
- Cartocci, V.; Segatto, M.; Di Tunno, I.; Leone, S.; Pfrieger, F.W.; Pallottini, V. Modulation of the Isoprenoid/Cholesterol Biosynthetic Pathway During Neuronal Differentiation In Vitro. J. Cell Biochem. 2016, 117, 2036–2044. [Google Scholar] [CrossRef] [PubMed]
- Segatto, M.; Di Giovanni, A.; Marino, M.; Pallottini, V. Analysis of the protein network of cholesterol homeostasis in different brain regions: An age and sex dependent perspective. J. Cell Physiol. 2013, 228, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Segatto, M.; Manduca, A.; Lecis, C.; Rosso, P.; Jozwiak, A.; Swiezewska, E.; Moreno, S.; Trezza, V.; Pallottini, V. Simvastatin treatment highlights a new role for the isoprenoid/cholesterol biosynthetic pathway in the modulation of emotional reactivity and cognitive performance in rats. Neuropsychopharmacology 2014, 39, 841–854. [Google Scholar] [CrossRef] [PubMed]
- Trapani, L.; Segatto, M.; Pallottini, V. New compounds able to control hepatic cholesterol metabolism: Is it possible to avoid statin treatment in aged people? World J. Hepatol. 2013, 5, 676–684. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Disease | Direct/Indirect Involvement of MVA Pathway | Experimental Model | References |
|---|---|---|---|
| SLOS | Direct DHCR7 | Human | [24,25,26,27,28,29,30,31,32,33,34] |
| Rodents | [35] | ||
| NPC | Direct NPC1 or/and NPC2 | Human | [36,37,38,39] |
| Rodents | [40,41] | ||
| Desmosterolosis | Direct DHCR24 | Human | [42] |
| AD | Indirect | Rodents | [43] |
| Human | [44,45,46,47] | ||
| Silico | [48] | ||
| HD | Indirect | Striatal mouse cell line | [49,50] |
| Rodents | [20,51,52,53] | ||
| ASD | Indirect | Rodents | [54,55,56,57] |
| Human | [58,59,60,61] | ||
| RTT | Indirect | Human | [62,63,64,65,66,67,68,69,70,71,72,73,74] |
| Primary human fibroblasts | [67,75] | ||
| Rodents | [76,77,78,79,80,81] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segatto, M.; Tonini, C.; Pfrieger, F.W.; Trezza, V.; Pallottini, V. Loss of Mevalonate/Cholesterol Homeostasis in the Brain: A Focus on Autism Spectrum Disorder and Rett Syndrome. Int. J. Mol. Sci. 2019, 20, 3317. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133317
Segatto M, Tonini C, Pfrieger FW, Trezza V, Pallottini V. Loss of Mevalonate/Cholesterol Homeostasis in the Brain: A Focus on Autism Spectrum Disorder and Rett Syndrome. International Journal of Molecular Sciences. 2019; 20(13):3317. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133317
Chicago/Turabian StyleSegatto, Marco, Claudia Tonini, Frank W. Pfrieger, Viviana Trezza, and Valentina Pallottini. 2019. "Loss of Mevalonate/Cholesterol Homeostasis in the Brain: A Focus on Autism Spectrum Disorder and Rett Syndrome" International Journal of Molecular Sciences 20, no. 13: 3317. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133317