Structure-Based Virtual Screening Allows the Identification of Efficient Modulators of E-Cadherin-Mediated Cell–Cell Adhesion

 and
and
Abstract
:
1. Introduction
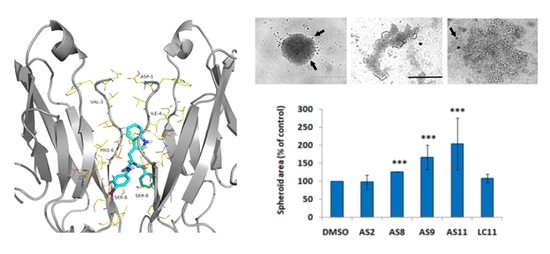
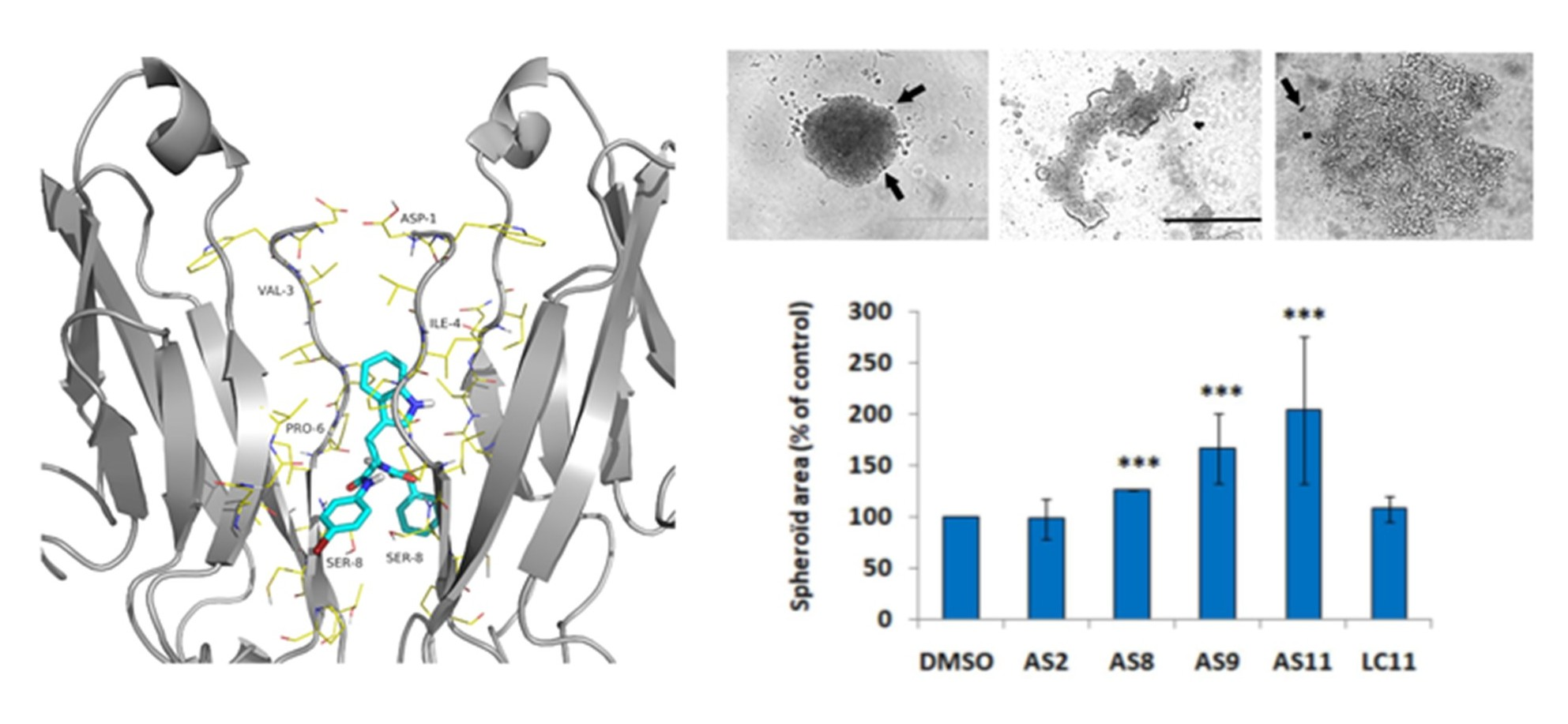
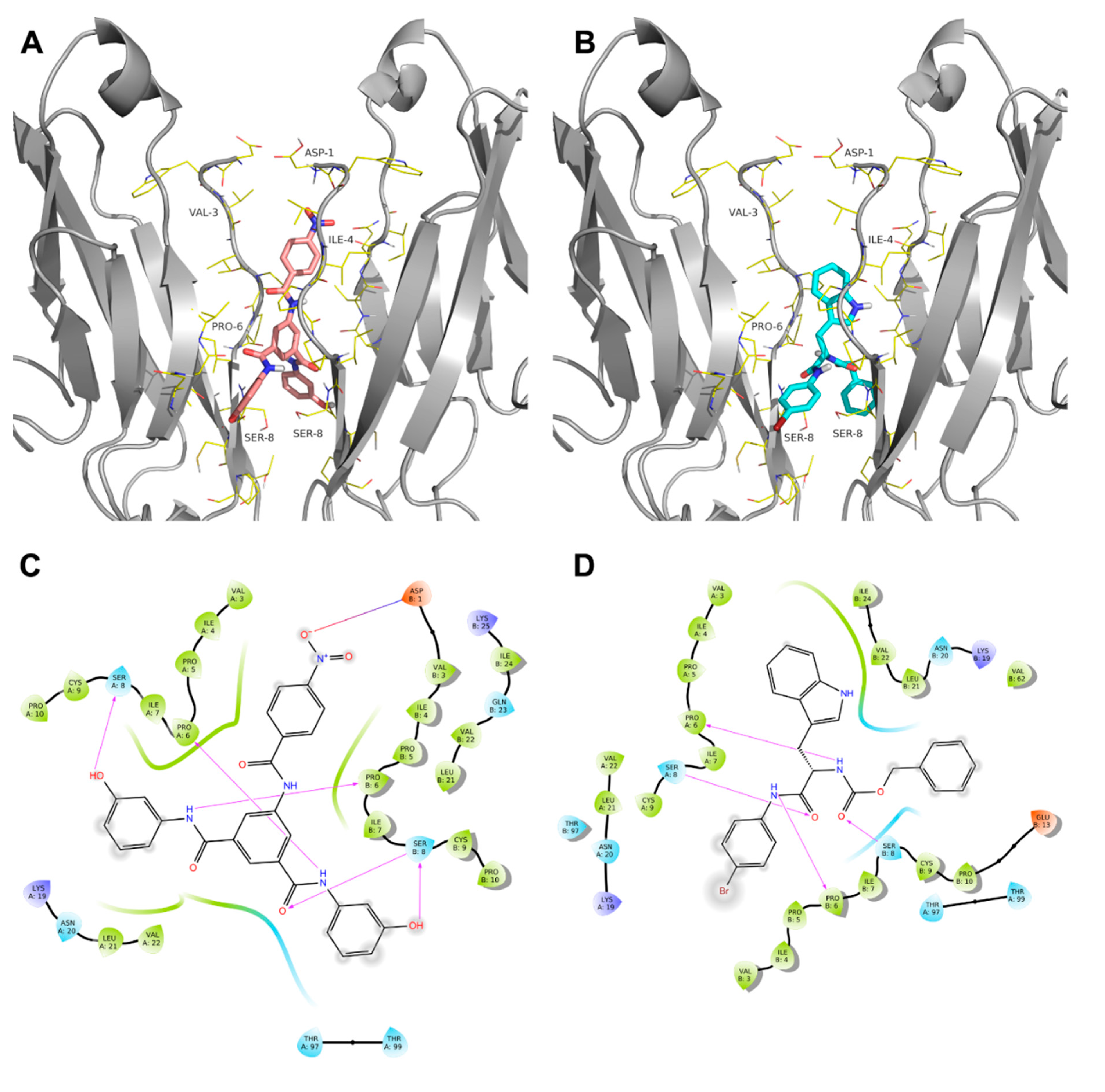
2. Results
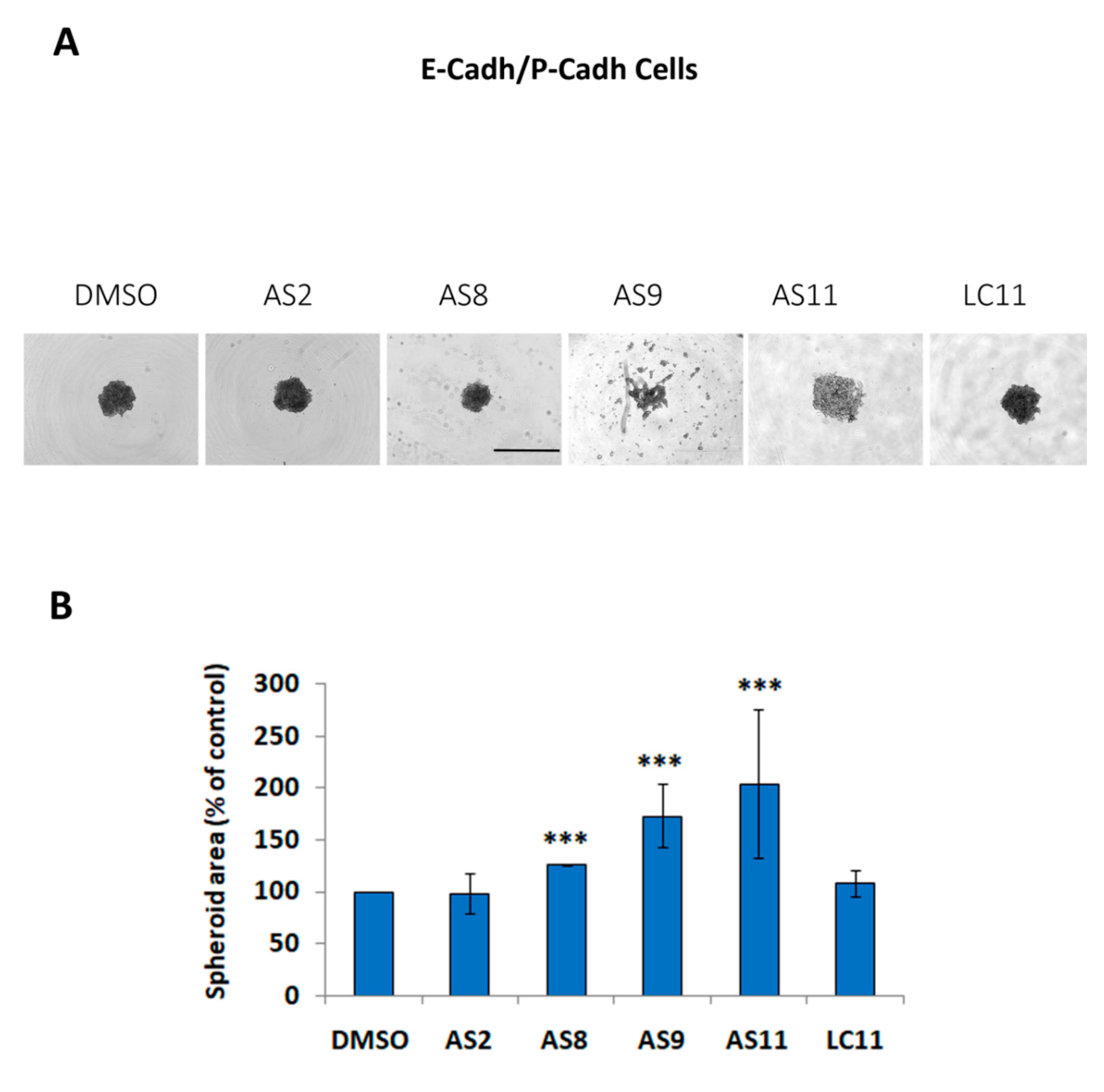
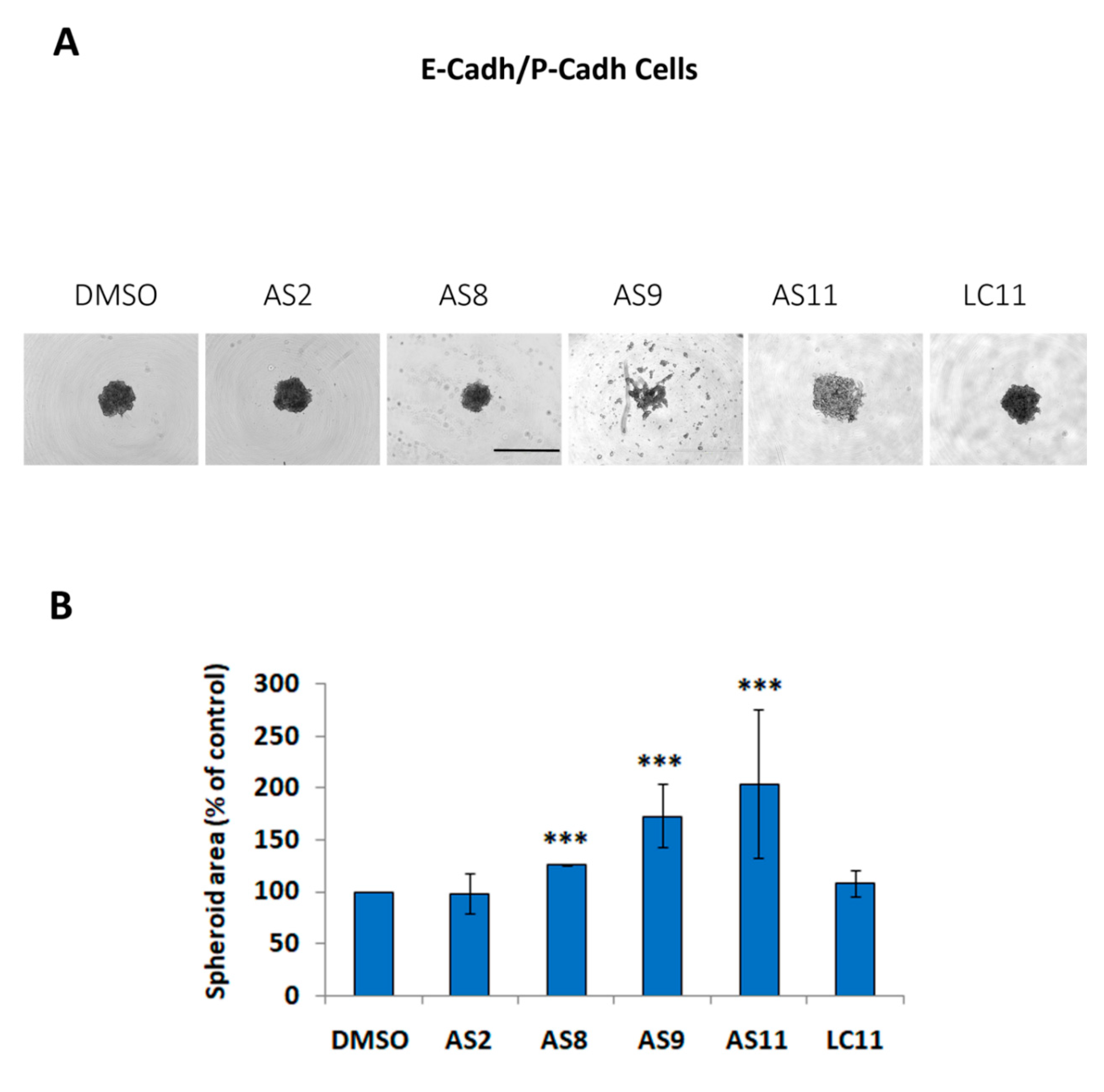
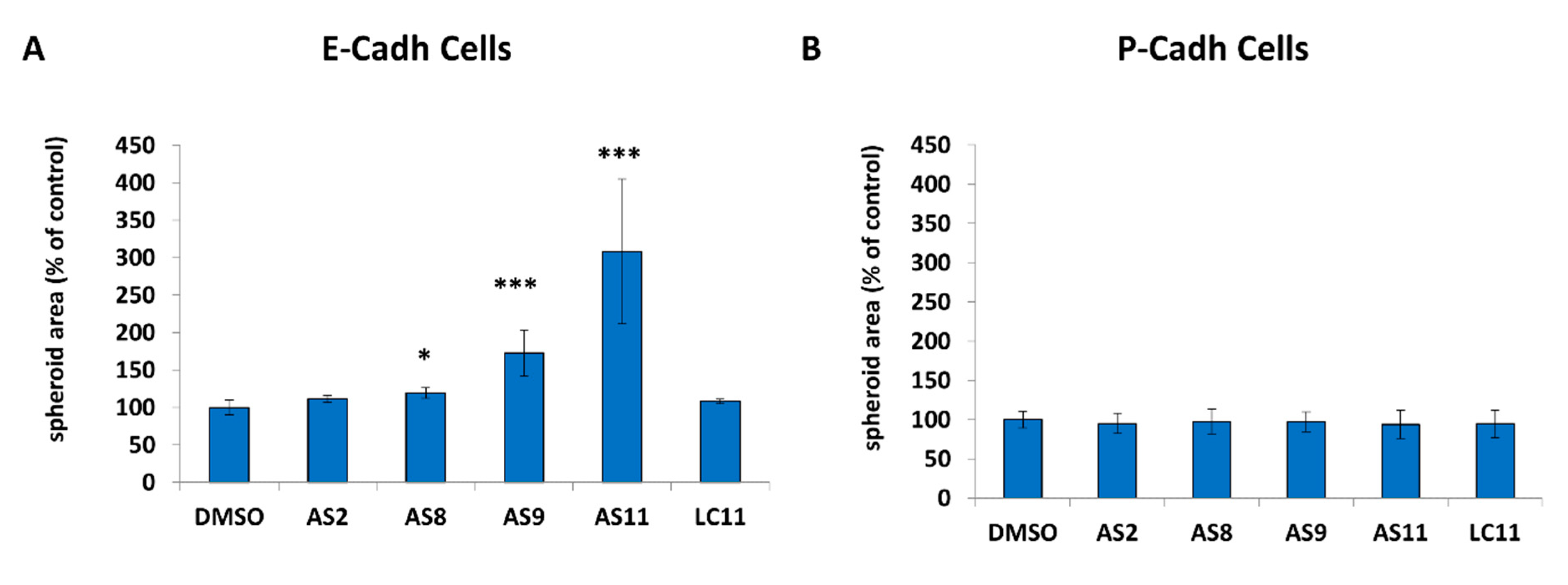
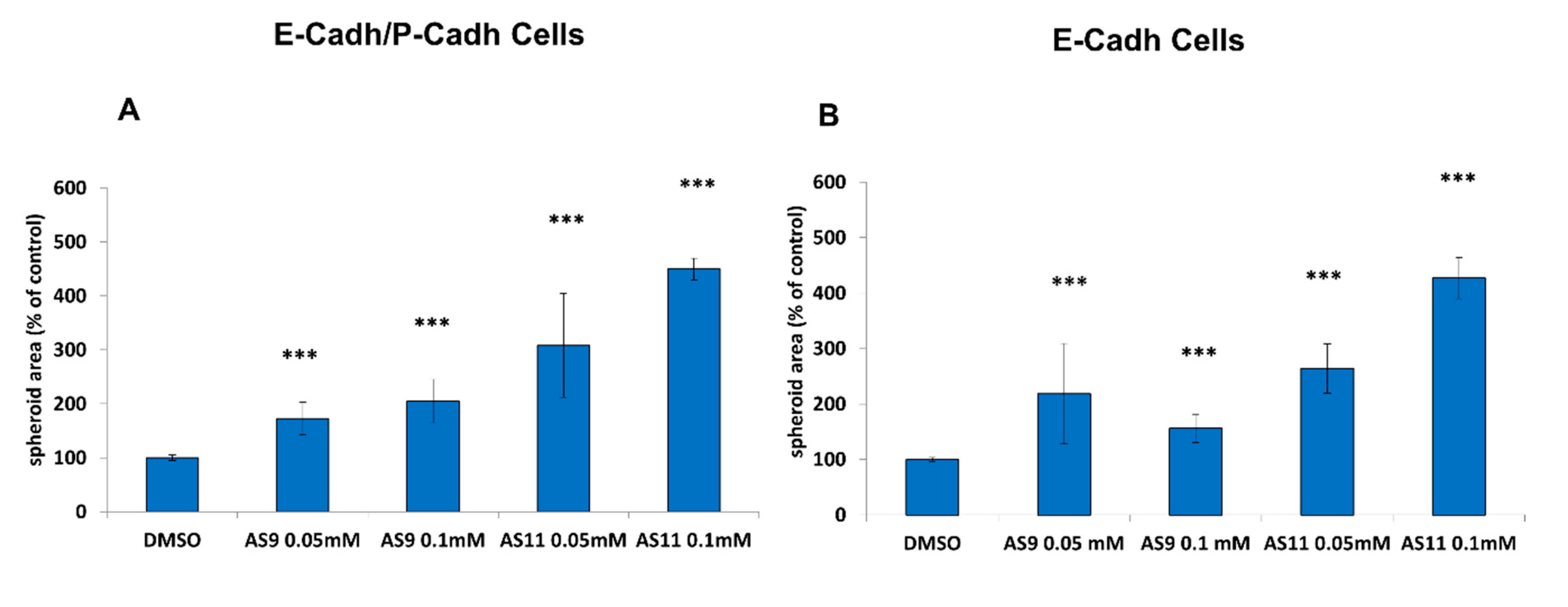
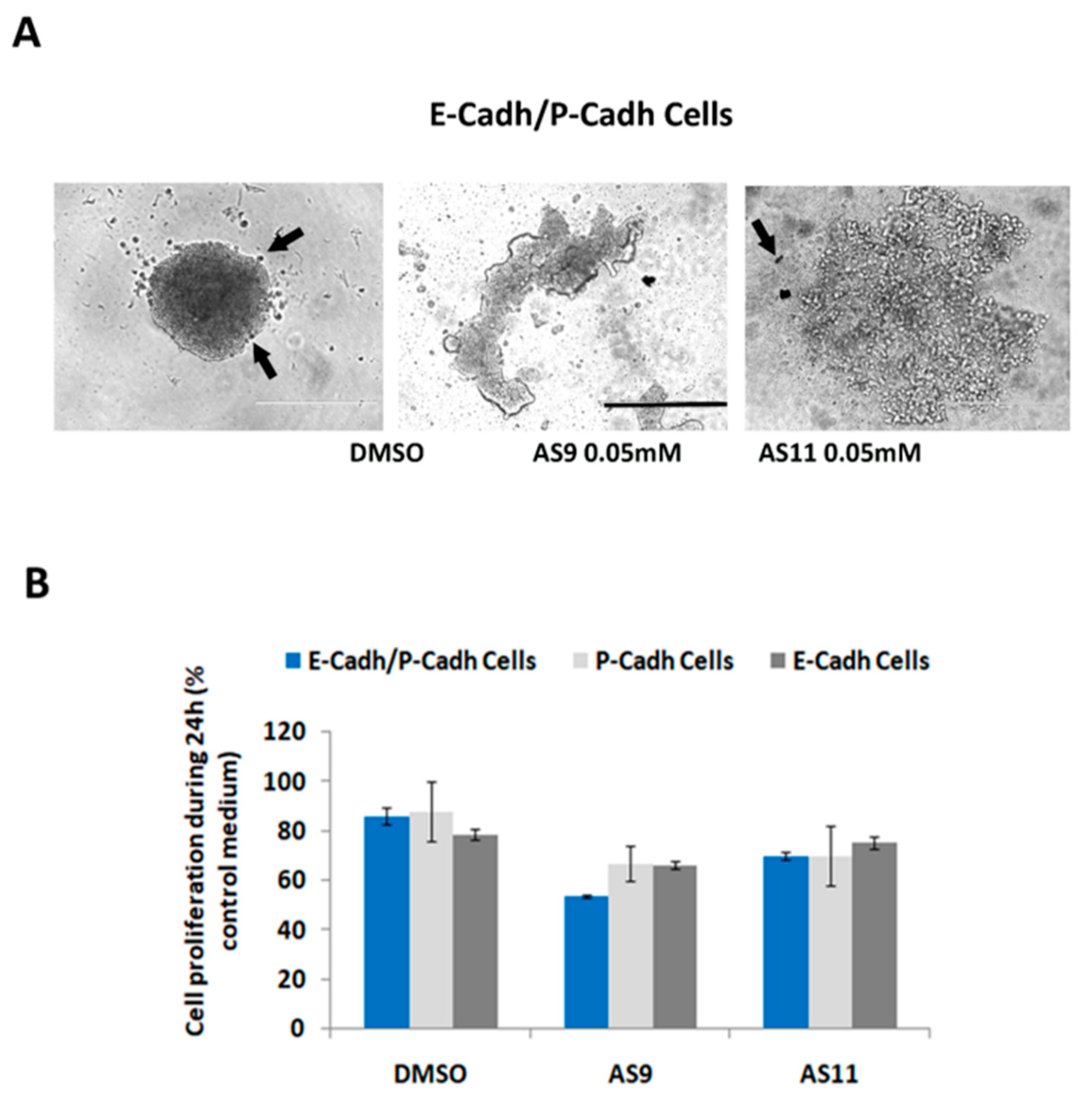
2.1. Cell Adhesion
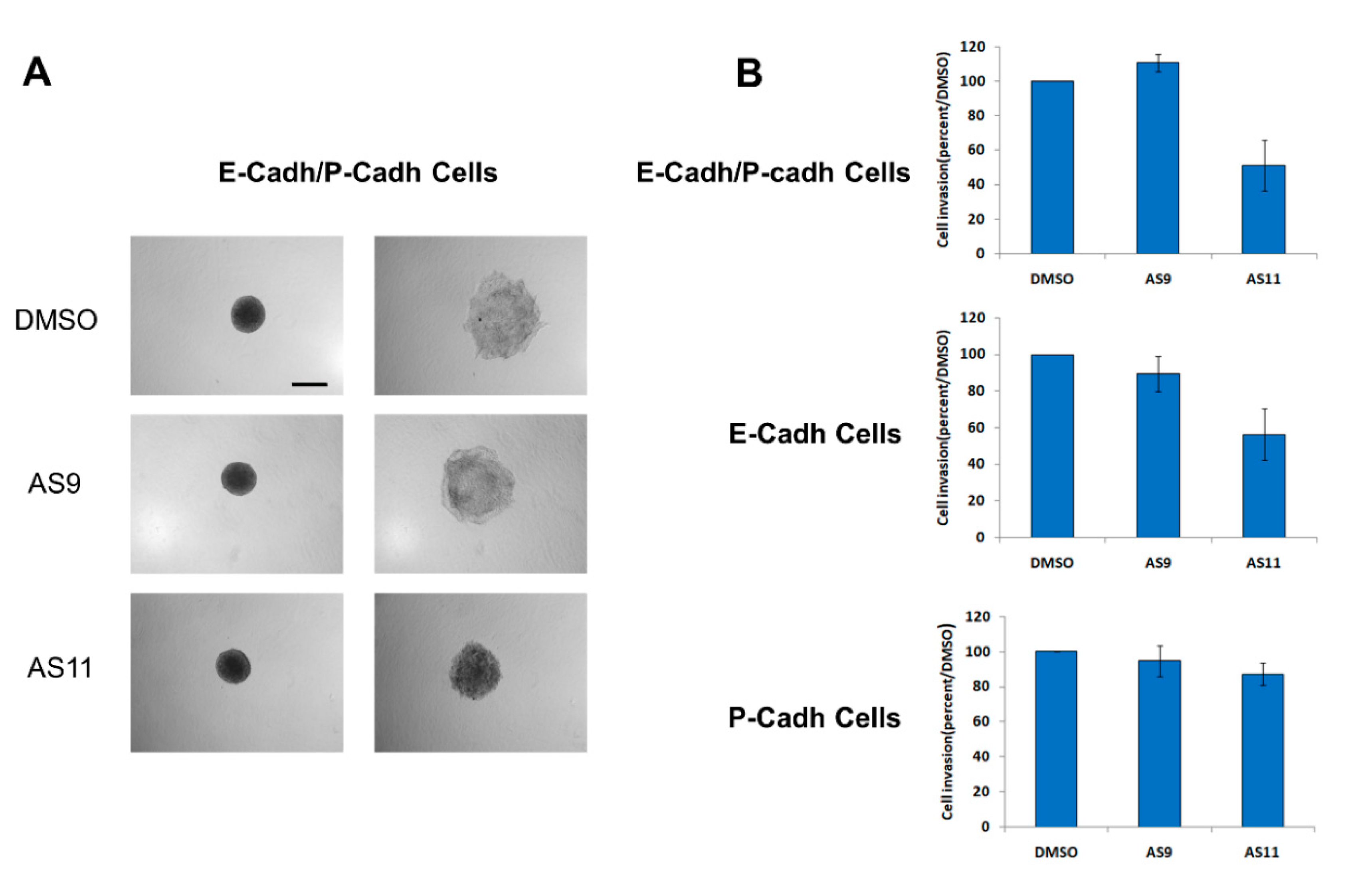
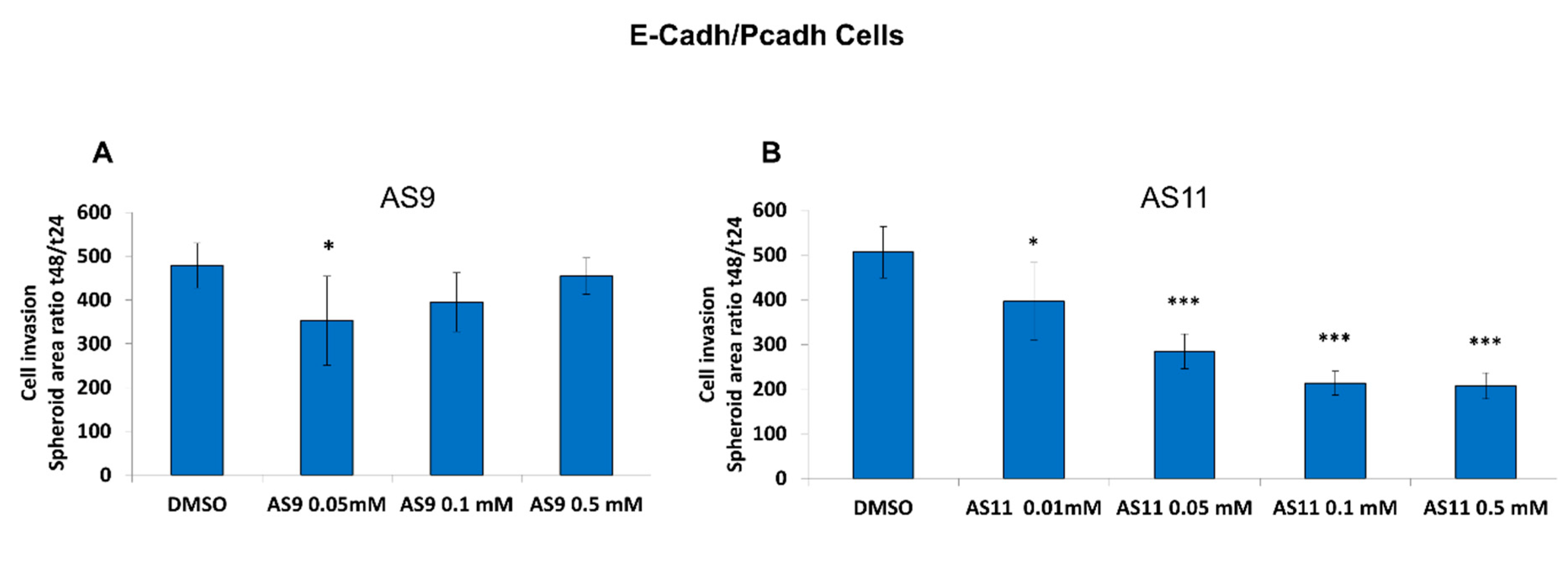
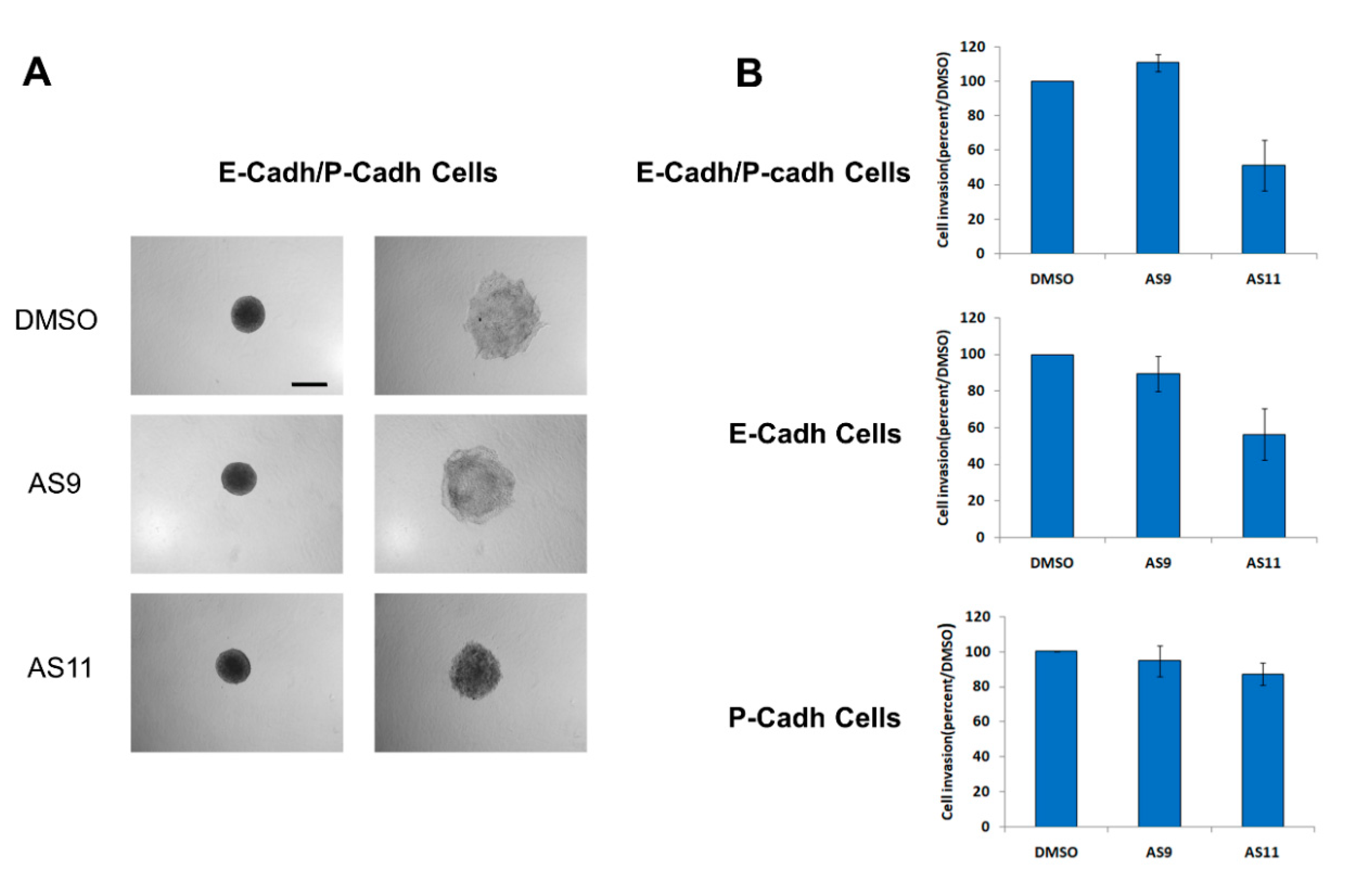
2.2. Cell Invasion
3. Discussion
4. Materials and Methods
4.1. Database Preparation
4.2. Protein Preparation
4.3. High-Throughput Docking (HTPD)
4.4. Ligands
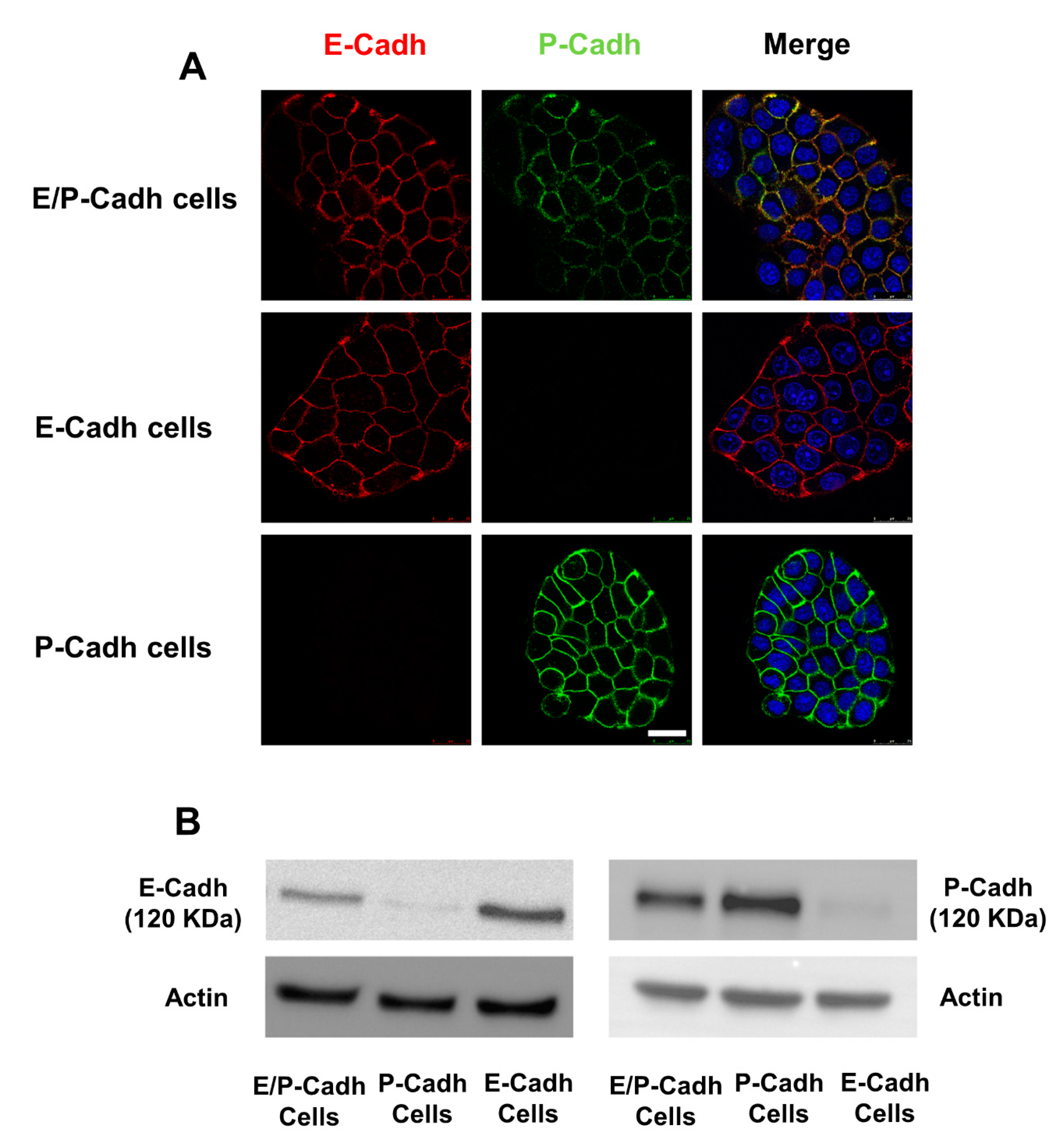
4.5. Cell Models
4.6. Cell–Cell Adhesion Assay
4.7. D-Invasion Assay
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| E-Cadh | E-cadherin |
| P-Cadh | P-cadherin |
| EMT | Epithelial-to-Mesenchymal Transition |
| DMSO | Dimethyl sulfoxide |
| DMEM | Dulbecco’s Modified Eagle Medium |
| PBS | Phosphate-buffered saline |
| BSA | Bovine serum albumin |
| EOC | Epithelial Ovarian Cancer |
| FCS | Fetal Calf Serum |
| STR | Short Tandem Repeats |
| VS | Virtual screening |
| shRNA | Short hairpin RNA |
| MD | Molecular Dynamics |
| PME | Particle mesh Ewald |
| RMSD | Root-Mean-Square Deviation |
| HTPD | High Throughput Docking |
| PDB | Protein Data Bank |
References
- Blaschuk, O.W.; Devemy, E. Cadherins as novel targets for anti-cancer therapy. Eur. J. Pharmacol. 2009, 625, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Berx, G.; Van Roy, F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a003129. [Google Scholar] [CrossRef] [PubMed]
- De Santis, G.; Miotti, S.; Mazzi, M.; Canevari, S.; Tomassetti, A. E-cadherin directly contributes to PI3K/AKT activation by engaging the PI3K-p85 regulatory subunit to adherens junctions of ovarian carcinoma cells. Oncogene 2009, 28, 1206–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paredes, J.; Figueiredo, J.; Albergaria, A.; Oliveira, P.; Carvalho, J.; Ribeiro, A.S.; Caldeira, J.; Costa, A.M.; Simões-Correia, J.; Oliveira, M.J.; et al. Epithelial E- and P-cadherins: Role and clinicalsignificance in cancer. Biochim. Biophys. Acta 2012, 1826, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Hirata, S.; Irie, A.; Senju, S.; Ikuta, Y.; Yokomine, K.; Harao, M.; Inoue, M.; Tsunoda, T.; Nakatsuru, S.; et al. Identification of a novel tumor-associated antigen, cadherin 3/P-cadherin, as a possible target for immunotherapy of pancreatic, gastric, and colorectal cancers. Clin. Cancer Res. 2008, 14, 6487–6495. [Google Scholar] [CrossRef] [PubMed]
- Sarrió, D.; Rodriguez-Pinilla, S.M.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008, 68, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Hazan, R.B.; Qiao, R.; Keren, R.; Badano, I.; Suyama, K. Cadherin switch in tumor progression. Ann. N. Y. Acad. Sci. 2004, 1014, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Taniuchi, K.; Nakagawa, H.; Hosokawa, M.; Nakamura, T.; Eguchi, H.; Ohigashi, H.; Ishikawa, O.; Katagiri, T.; Nakamura, Y. Overexpressed P-cadherin/CDH3 promotes motility of pancreatic cancer cells by interacting with p120ctn and activating rho-family GTPases. Cancer Res. 2005, 65, 3092–3099. [Google Scholar] [CrossRef]
- Zhang, C.C.; Yan, Z.; Zhang, Q.; Kuszpit, K.; Zasadny, K.; Qiu, M.; Painter, C.L.; Wong, A.; Kraynov, E.; Arango, M.E.; et al. PF-03732010: A fully human monoclonal antibody against P-cadherin with antitumor and antimetastatic activity. Clin. Cancer Res. 2010, 16, 5177–5188. [Google Scholar] [CrossRef]
- Assefnia, S.; Dakshanamurthy, S.; Guidry Auvil, J.M.; Hampel, C.; Anastasiadis, P.Z.; Kallakury, B.; Uren, A.; Foley, D.W.; Brown, M.L.; Shapiro, L.; et al. Cadherin-11 in poor prognosis malignancies and rheumatoid arthritis: Common target, common therapies. Oncotarget 2014, 5, 1458–1474. [Google Scholar] [CrossRef]
- Sfikakis, P.P.; Vlachogiannis, N.I.; Christopoulos, P.F. Cadherin-11 as a therapeutic target in chronic, inflammatory rheumatic diseases. Clin. Immunol. 2017, 176, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Park, E.; Han, S.-W.; Im, S.-A.; Kim, T.-Y.; Kim, W.-H.; Oh, D.Y.; Bang, Y.-J. Down-regulation of P-cadherin with PF-03732010 inhibits cell migration and tumor growth in gastric cancer. Investig. New Drugs 2012, 30, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Senoo, A.; Nagatoishi, S.; Moberg, A.; Babol, L.N.; Mitani, T.; Tashima, T.; Kudo, S.; Tsumoto, K. Inhibition of homophilic dimerization and disruption of cell adhesion by P-cadherin-specific small molecules from SPR-based assays. Chem. Commun. 2018, 54, 5350–5353. [Google Scholar] [CrossRef] [PubMed]
- Civera, M.; Vasile, F.; Potenza, D.; Colombo, C.; Parente, S.; Vettraino, C.; Prosdocimi, T.; Parisini, E.; Belivisi, L. Exploring E-cadherin-peptidomimetics interaction using NMR and computational studies. PLoS Comput. Biol. 2019, 15, e1007041. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, F.; Geert, B. The cell-cell adhesion molecule E-cadherin. Cell Mol. Life Sci. 2008, 65, 3756–3788. [Google Scholar] [CrossRef]
- Vendome, J.; Posy, S.; Jin, X.; Bahna, F.; Ahlsen, G.; Shapiro, L.; Honig, B. Molecular design principles underlying β-strand swapping in the adhesive dimerization of cadherins. Nat. Struct. Mol. Biol. 2011, 18, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Harrison, O.J.; Bahna, F.; Katsamba, P.S.; Jin, X.; Brasch, J.; Vendome, J.; Ahlsen, G.; Carroll, K.J.; Price, S.R.; Honig, B.; et al. Two-step adhesive binding by classical cadherins. Nat. Struct. Mol. Biol. 2010, 17, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Kudo, S.; Caaveiro, J.M.; Goda, S.; Nagatoishi, S.; Ishii, K.; Matsuura, T.; Sudou, Y.; Kodama, T.; Hamakubo, T.; Tsumoto, K. Identification and characterization of the X-dimer of human P-cadherin: Implications for homophilic cell adhesion. Biochemistry 2014, 53, 1742–1752. [Google Scholar] [CrossRef]
- Parisini, E.; Higgins, J.M.; Liu, J.; Brenner, M.B.; Wang, J. The crystal structure of human E-cadherin domains 1 and 2, and comparison with other cadherins in the context of adhesion mechanism. J. Mol. Biol. 2007, 373, 401–411. [Google Scholar] [CrossRef]
- Dalle Vedove, A.; Lucarelli, A.P.; Nardone, V.; Matino, A.; Parisini, E. The X-ray structure of human P-cadherin EC1-EC2 in a closed conformation provides insight into the type I cadherin dimerization pathway. Acta Crystallogr. F Struct. Biol. Commun. 2015, 71, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Priest, A.V.; Shafraz, O.; Sivasankar, S. Biophysical basis of cadherin mediated cell-cell adhesion. Exp. Cell Res. 2017, 358, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Manibog, K.; Sankar, K.; Kim, S.A.; Zhang, Y.; Jernigan, R.L.; Sivasankar, S. Molecular determinants of cadherin ideal bond formation: Conformation-dependent unbinding on a multidimensional landscape. Proc. Natl. Acad. Sci. USA. 2016, 113, E5711–E5720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manibog, K.; Li, H.; Rakshit, S.; Sivasankar, S. Resolving the molecular mechanism of cadherin catch bond formation. Nat. Commun. 2014, 5, 3941. [Google Scholar] [CrossRef] [PubMed]
- Sivasankar, S. Tuning the kinetics of cadherin adhesion. J. Investig. Dermatol. 2013, 133, 2318–2323. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Altorelli, N.L.; Bahna, F.; Honig, B.; Shapiro, L.; Palmer, A.G., III. Mechanism of E-cadherin dimerization probed by NMR relaxation dispersion. Proc. Natl. Acad. Sci. USA 2013, 110, 16462–16467. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.D.; Ciatto, C.; Chen, C.P.; Bahna, F.; Rajebhosale, M.; Arkus, N.; Schieren, I.; Jessell, T.M.; Honig, B.; Price, S.R.; et al. Type II cadherin ectodomain structures: Implications for classical cadherin specificity. Cell 2006, 124, 1255–1268. [Google Scholar] [CrossRef]
- Nardone, V.; Lucarelli, A.P.; Dalle Vedove, A.; Fanelli, R.; Tomassetti, A.; Belvisi, L.; Civera, M.; Parisini, E. Crystal structure of human E-cadherin-EC1EC2 in complex with a peptidomimetic competitive inhibitor of cadherin homophilic interaction. J. Med. Chem. 2016, 59, 5089–5094. [Google Scholar] [CrossRef]
- Doro, F.; Colombo, C.; Alberti, C.; Arosio, D.; Belvisi, L.; Casagrande, C.; Fanelli, R.; Manzoni, L.; Parisini, E.; Piarulli, U.; et al. Computational design of novel peptidomimetic inhibitors of cadherin homophilic interactions. Org. Biomol. Chem. 2015, 13, 2570–2573. [Google Scholar] [CrossRef] [Green Version]
- Blaschuk, O.W. Discovery and development of N-cadherin antagonists. Cell Tissue Res. 2012, 348, 309–313. [Google Scholar] [CrossRef]
- Perotti, A.; Sessa, C.; Mancuso, A.; Noberasco, C.; Cresta, S.; Locatelli, A.; Carcangiu, M.L.; Passera, K.; Braghetti, A.; Scaramuzza, D.; et al. Clinical and pharmacological phase I evaluation of Exherin (ADH-1), a selective anti-N-cadherin peptide in patients with N-cadherin-expressing solid tumours. Ann. Oncol. 2009, 20, 741–745. [Google Scholar] [CrossRef]
- Yarom, N.; Stewart, D.; Malik, R.; Wells, J.; Avruch, L.; Jonker, D.J. Phase I clinical trial of Exherin (ADH-1) in patients with advanced solid tumors. Curr. Clin. Pharmacol. 2013, 8, 81–88. [Google Scholar] [PubMed]
- Siret, C.; Dobric, A.; Martirosyan, A.; Terciolo, C.; Germain, S.; Bonier, R.; Dirami, T.; Dusetti, N.; Tomasini, R.; Rubis, M.; et al. Cadherin-1 and cadherin-3 cooperation determines the aggressiveness of pancreatic ductal adenocarcinoma. Br. J. Cancer 2018, 118, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Blaschuk, O.W.; Rowlands, T.M. Cadherins as modulators of angiogenesis and the structural integrity of blood vessels. Cancer Metastasis Rev. 2000, 19, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Vasudev, N.S.; Reynolds, A.R. Anti-angiogenic therapy for cancer: Current progress, unresolved questions and future directions. Angiogenesis 2014, 17, 471–494. [Google Scholar] [CrossRef] [PubMed]
- Corada, M.; Zanetta, L.; Orsenigo, F.; Breviario, F.; Lampugnani, M.G.; Bernasconi, S.; Liao, F.; Hicklin, D.J.; Bohlen, P.; Dejana, E. A monoclonal antibody to vascular endothelial-cadherin inhibits tumor angiogenesis without side effects on endothelial permeability. Blood 2002, 100, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Doody, J.F.; Overholser, J.; Finnerty, B.; Bassi, R.; Wu, Y.; Dejana, E.; Kussie, P.; Bohlen, P.; Hicklin, D.J. Selective targeting of angiogenic tumor vasculature by vascular endothelial-cadherin antibody inhibits tumor growth without affecting vascular permeability. Cancer Res. 2002, 62, 2567–2575. [Google Scholar]
- May, C.; Doody, J.F.; Abdullah, R.; Balderes, P.; Xu, X.; Chen, C.P.; Zhu, Z.; Shapiro, L.; Kussie, P.; Hicklin, D.J.; et al. Identification of a transiently exposed VE-cadherin epitope that allows for specific targeting of an antibody to the tumor neovasculature. Blood 2005, 105, 4337–4344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhardt, H.; Wolburg, H.; Redies, C. N-cadherin mediates pericytic-endothelial interaction during brain angiogenesis in the chicken. Dev. Dyn. 2000, 218, 472–479. [Google Scholar] [CrossRef]
- Chang, S.K.; Kohlgruber, A.C.; Mizoguchi, F.; Michelet, X.; Wolf, B.J.; Wei, K.; Lee, P.Y.; Lynch, L.; Duquette, D.; Ceperuelo-Mallafré, V.; et al. Stromal cell cadherin-11 regulates adipose tissue inflammation and diabetes. J. Clin. Investig. 2017, 127, 3300–3312. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.M.; Kiener, H.P.; Agarwal, S.K.; Noss, E.H.; Watts, G.F.; Chisaka, O.; Takeichi, M.; Brenner, M.B. Cadherin-11 in synovial lining formation and pathology in arthritis. Science 2007, 315, 1006–1010. [Google Scholar] [CrossRef]
- Chang, S.K.; Noss, E.H.; Chen, M.; Gu, Z.; Townsend, K.; Grenha, R.; Leon, L.; Lee, S.Y.; Lee, D.M.; Brenner, M.B. Cadherin-11 regulates fibroblast inflammation. Proc. Natl. Acad. Sci. USA 2011, 108, 8402–8407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertz, O.; Bozic, D.; Koch, A.W.; Fauser, C.; Brancaccio, A.; Engel, J. A new crystal structure, Ca2+ dependence and mutational analysis reveal molecular details of E-cadherin homoassociation. EMBO J. 1999, 18, 1738–1747. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. Amber 14; University of California: Oakland, CA, USA, 2014. [Google Scholar]
- Jorgensen, W.L. Revised TIPS for simulations of liquid water and aqueous solutions. J. Chem. Phys. 1982, 77, 4156–4163. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E., III. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Doshi, U.; Hamelberg, D. Reoptimization of the AMBER force field parameters for peptide bond (Omega) torsions using accelerated molecular dynamics. J. Phys. Chem. B 2009, 113, 16590–16595. [Google Scholar] [CrossRef]
- Duarte, F.; Bauer, P.; Barrozo, A.; Amrein, B.A.; Purg, M.; Aqvist, J.; Kamerlin, S.C. Force field independent metal parameters using a nonbonded dummy model. J. Phys. Chem. B 2014, 118, 4351–4362. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Company (ID#) | Internal Code | Chemical Structure |
|---|---|---|
| Asinex (AEM14687298) | AS2 |  |
| Asinex (BAS00093476) | AS8 |  |
| Asinex (BAS00132635) | AS9 |  |
| Asinex (BAS00602705) | AS11 |  |
| Life Chemicals (F2762-0527) | LC11 |  |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalle Vedove, A.; Falchi, F.; Donini, S.; Dobric, A.; Germain, S.; Di Martino, G.P.; Prosdocimi, T.; Vettraino, C.; Torretta, A.; Cavalli, A.; et al. Structure-Based Virtual Screening Allows the Identification of Efficient Modulators of E-Cadherin-Mediated Cell–Cell Adhesion. Int. J. Mol. Sci. 2019, 20, 3404. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20143404
Dalle Vedove A, Falchi F, Donini S, Dobric A, Germain S, Di Martino GP, Prosdocimi T, Vettraino C, Torretta A, Cavalli A, et al. Structure-Based Virtual Screening Allows the Identification of Efficient Modulators of E-Cadherin-Mediated Cell–Cell Adhesion. International Journal of Molecular Sciences. 2019; 20(14):3404. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20143404
Chicago/Turabian StyleDalle Vedove, Andrea, Federico Falchi, Stefano Donini, Aurelie Dobric, Sebastien Germain, Giovanni Paolo Di Martino, Tommaso Prosdocimi, Chiara Vettraino, Archimede Torretta, Andrea Cavalli, and et al. 2019. "Structure-Based Virtual Screening Allows the Identification of Efficient Modulators of E-Cadherin-Mediated Cell–Cell Adhesion" International Journal of Molecular Sciences 20, no. 14: 3404. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20143404