Comparative Analysis of the Characteristics of Triterpenoid Transcriptome from Different Strains of Wolfiporia cocos

Abstract
:1. Introduction
2. Results
2.1. Differences in Total Triterpenoid Content between H and L
2.2. RNA Sequencing and Assembly Results of Transcriptomes of H and L
2.3. Principal Component Analysis for Correction of Differentially Expressed Genes (DEGs)
2.4. Verification of Transcriptome Sequencing Results by RT-qPCR Experiment
2.5. Gene Expression Pattern and DEG Function Enrichment Analysis in H and L
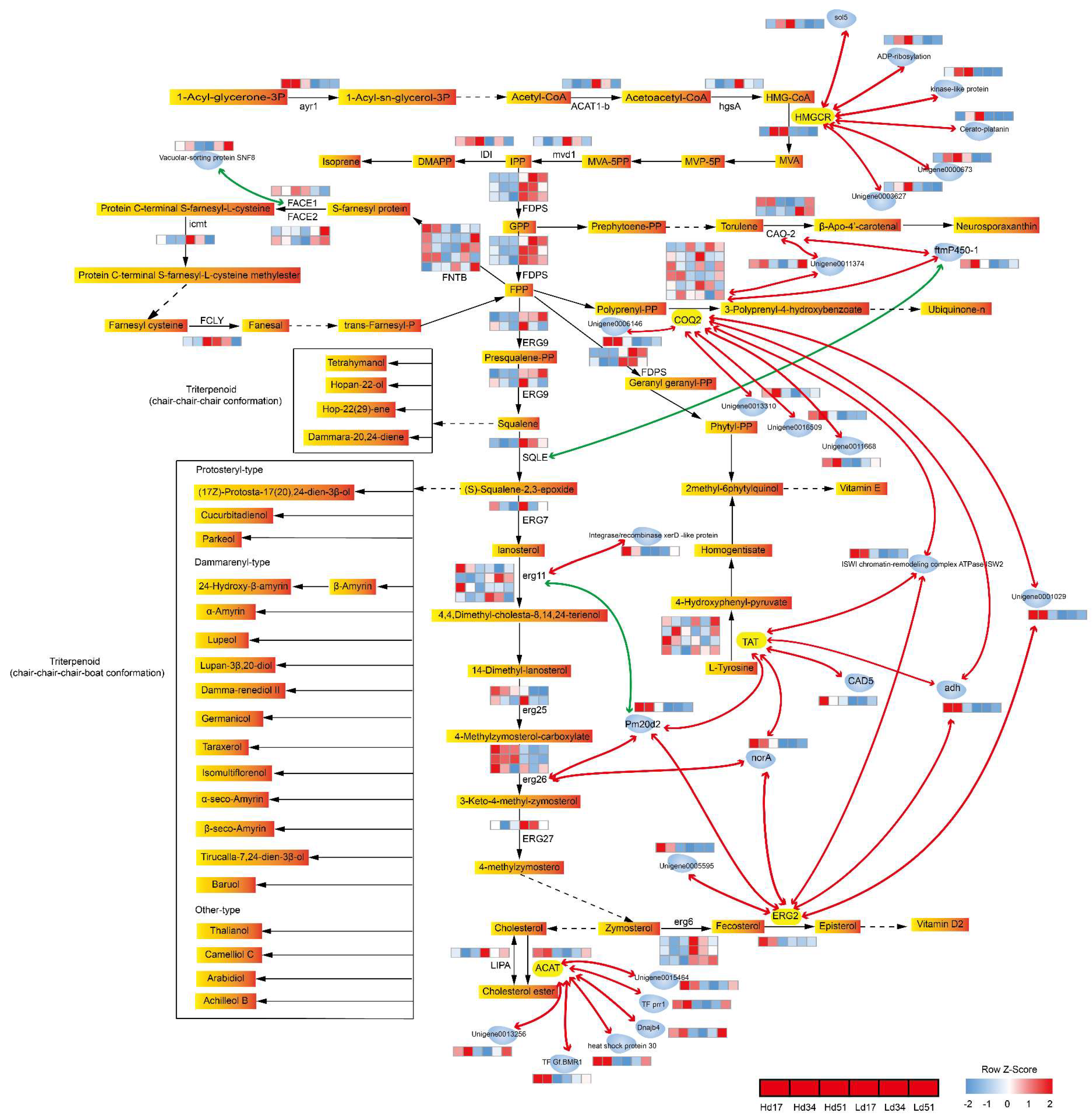
2.6. Correlation Analysis of Genes Related to Triterpenoid Biosynthesis in W. cocos
3. Discussion
4. Materials and Methods
4.1. Biomaterials and Culture Methods
4.2. Colorimetry Measurement of Total Triterpenoid
4.3. RNA Extraction and Quantification Analysis
4.4. Construction and Sequencing of cDNA Library
4.5. Sequence Assembly and Functional Annotations
4.6. Unigene Expression Differential Analysis
4.7. RT-qPCR Validation
4.8. Expression Trend Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| H | high-yielding strains DZAC-Wp-H-29 |
| L | low-yielding strains DZAC-Wp-L-123 |
| HMGCR | hydroxymethyl glutaryl- coenzyme A reductase |
| FDPS | farnesyl-diphosphate synthase |
| COQ2 | 4-hydroxybenzoate polyprenyltransferase |
| ERG2 | C-8 sterol isomerase |
| ACAT | sterol O-acyltransferase |
| TAT | tyrosine aminotransferase |
| CAO2 | carotenoid oxygenase |
| erg26 | sterol-4alpha-carboxylate 3-dehydrogenase |
| Pm20d2 | peptidase M20 domain-containing protein 2 |
| norA | aryl-alcohol dehydrogenase |
| adh | GroES-like protein |
| ftmP450-1 | cytochrome P450 |
| GHs | glycosyl hydrolases |
| CEs | carbohydrate esterases |
| AAs | auxiliary activities |
| CBMs | carbohydrate - binding modules |
| GTs | glycosyltransferases |
| PLs | polysaccharide lyases |
| SSR | simple sequence repetition |
| MVD | diphosphomevalonate decarboxylase |
| ERG7 | lanosterol synthase |
| LSD | least-significant difference |
| DEGs | differentially expressed genes |
| RPKM | Read Per kb per Million Reads |
| PCA | principal component analysis |
| FDR | false discovery rate |
| qPCR | real-time quantitative polymerase chain reaction |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| HMG-CoA | 3-hydroxy-3-methylglutaryl coenzyme A |
| CAD5 | heat shock protein |
| CoQ | ubiquinone |
| erg11 | lanosterol 14-alpha-demethylase |
| SQLE | squalene monooxygenase |
| FPP | farnesyl diphosphate |
| IPP | isopentenyl diphosphate |
| DMAPP | dimethylallyl diphosphate |
| ERG9 | farnesyl-diphosphate farnesyltransferase |
| RIN | RNA integrity number |
| Nr | non-redundant protein |
| NAD+ | oxidized form of nicotinamide adenine dinucleoside |
| NADP+ | oxidized form of nicotinamide adenine dinucleotide phosphate |
| FAD | flavin-adenine dinucleotide |
| PDA | potato dextrose agar |
| COG/KOG | Cluster of Orthologous Groups of proteins |
| NCBI | the National Center for Biotechnology Information |
References
- Zhao, J.D. Flora Fungorum Sinicorum; Polyporaceae Consilio Florarum Cryptogamarum Sinicarum Academiae Sinicae, Ed.; Science Press: Beijing, China, 1998; Volume 3, pp. 411–414. [Google Scholar]
- Liu, S.C.; Wu, Q.; Xing, P.; Xia, Z.L. Research progress of poria cocos germplasm resources. Edible Med. Mushrooms 2017, 25, 171–175. [Google Scholar]
- National Pharmacopoeia Commission (Ed.) Chinese Pharmacopoeia; China Medicine Science and Technology Press: Beijing, China, 2015; pp. 240–241. [Google Scholar]
- Zhang, H.K. National Chinese Patent Medicine Product Collection; Shanxi Science and Technology Press: Taiyuan, China, 1989. [Google Scholar]
- Yang, D.F. Study on Poria cocos Biscuits. J. Shanxi Agric. Sci. 2018, 46, 823–825. [Google Scholar] [CrossRef]
- Yu, H.H. Quality Characteristics and Antioxidant Activity of Cookies added with Baekbokrung(Poria cocos Wolf) Powder. Korean J. Hum. Ecol. 2014, 23, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.J.; Bae, S.Y.; Son, R.H.; Lee, Y.H. The Inhibitory Effects of Poria cocos Bark Extract on Melanogenesis. J. Soc. Cosmet. Sci. Korea 2009, 35, 243–250. [Google Scholar]
- Gao, Y.Q.; Yan, H.; Jin, R.R.; Lei, P. Antiepileptic activity of total triterpenes isolated from Poria cocos is mediated by suppression of aspartic and glutamic acids in the brain. Pharm. Biol. 2016, 54, 2528–2535. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Lee, S.; Shim, S.H.; Lee, H.J.; Choi, Y.; Jang, T.S.; Kim, K.H.; Kang, K.S. Protective effect of lanostane triterpenoids from the sclerotia of Poria cocos Wolf against cisplatin-induced apoptosis in LLC-PK1 cells. Bioorg. Med. Chem. Lett. 2017, 27, 2881–2885. [Google Scholar] [CrossRef]
- Kikuchi, T.; Uchiyama, E.; Ukiya, M.; Tabata, K.; Kimura, Y.; Suzuki, T.; Akihisa, T. Cytotoxic and Apoptosis-Inducing Activities of Triterpene Acids from Poria cocos. J. Nat. Prod. 2011, 74, 137–144. [Google Scholar] [CrossRef]
- Hu, G.S.; Huang, C.G.; Zhang, Y.; Xiao, W.; Jia, J.M. Accumulation of biomass and four triterpenoids in two-stage cultured Poria cocos mycelia and diuretic activity in rats. Chin. J. Nat. Med. 2017, 15, 265–270. [Google Scholar] [CrossRef]
- Dong, H.J.; Xue, Z.Z.; Geng, Y.L.; Wang, X.; Yang, B. Lanostane triterpenes isolated from epidermis of Poria cocos. Phytochem. Lett. 2017, 22, 102–106. [Google Scholar] [CrossRef]
- Lai, K.H.; Lu, M.C.; Du, Y.C.; El-Shazly, M.; Wu, T.Y.; Hsu, Y.M.; Henz, A.; Yang, J.C.; Backlund, A.; Chang, F.R.; et al. Cytotoxic Lanostanoids from Poria cocos. J. Nat. Prod. 2016, 79, 2805–2813. [Google Scholar] [CrossRef]
- Yimam, M.; Jiao, P.; Hong, M.; Jia, Q. A Standardized Composition from Extracts of Myristica Fragrans, Astragalus Membranaceus, and Poria Cocos Protects Liver from Acute Ethanol Insult. J. Med. Food 2016, 19, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.H.; Backlund, A.; Lu, M.C.; Du, Y.C.; El-Shazly, M.; Wu, T.Y.; Hsu, Y.M.; Henz, A.; Chang, F.R.; Wu, Y.C. Antileukemic lanostanoids from Poria cocos. Planta Med. 2015, 81, 1418. [Google Scholar] [CrossRef]
- Deng, J.S.; Huang, S.S.; Lin, T.H.; Lee, M.M.; Kuo, C.C.; Sung, P.J.; Hou, W.C.; Huang, G.J.; Kuo, Y.H. Analgesic and Anti-Inflammatory Bioactivities of Eburicoic Acid and Dehydroeburicoic Acid Isolated from Antrodia camphorata on the Inflammatory Mediator Expression in Mice. J. Agric. Food Chem. 2013, 61, 5064–5071. [Google Scholar] [CrossRef] [PubMed]
- Rios, J.L. Chemical Constituents and Pharmacological Properties of Poria cocos. Planta Med. 2011, 77, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.P.; Hu, B.X.; Wei, W.; Xiong, Y.; Zhu, W.J.; Peng, F.; Yu, Y.; Zheng, Y.L.; Chen, P. De Novo Analysis of Wolfiporia cocos Transcriptome to Reveal the Differentially Expressed Carbohydrate-Active Enzymes (CAZymes) Genes During the Early Stage of Sclerotial Growth. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Guo, J.Y.; Ma, Y.P.; Zhou, M.C.; Wang, M.; Shu, S.H. Characterization and gene function analysis of SSR sequences in Poria cocos transcriptome. Chin. Tradit. Herb. Drugs 2015, 46, 3558–3563. [Google Scholar]
- Wu, Y.Y.; Zhu, W.J.; Wei, W.; Zhao, X.L.; Wang, Q.; Zeng, W.Y.; Zheng, Y.L.; Chen, P.; Zhang, S.P. De novo assembly and transcriptome analysis of sclerotial development in Wolfiporia cocos. Gene 2016, 588, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.H.; Chen, B.; Zhou, M.C.; Zhao, X.M.; Xia, H.Y.; Wang, M. De Novo Sequencing and Transcriptome Analysis of Wolfiporia cocos to Reveal Genes Related to Biosynthesis of Triterpenoids. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Liu, C.L.; Xie, X.X.; Liu, H.G.; Xu, L. Study on the optimal conditions for the determination of effective components of poria cocos by spectrophotometry. Asia Pac. Tradit. Med. 2014, 10, 17–19. [Google Scholar]
- Cock, P.J.A.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef]
- Li, R.; Yu, C.; Li, Y.; Lam, T.-W.; Yiu, S.-M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhang, Y.; Cui, H.H.; Liu, J.W.; Wu, Y.Q.; Cheng, Y.; Xu, H.X.; Huang, X.X.; Li, S.T.; Zhou, A.; et al. WEGO 2.0: A web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018, 46, W71–W75. [Google Scholar] [CrossRef]
- Friesen, J.A.; Rodwell, V.W. The 3-hydroxy-3-methylglutaryl coenzyme-A (HMG-CoA) reductases. Genome Biol. 2004, 5, 248. [Google Scholar] [CrossRef]
- Burg, J.S.; Espenshade, P.J. Regulation of HMG-CoA reductase in mammals and yeast. Prog. Lipid Res. 2011, 50, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Sturley, S.L. Molecular aspects of intracellular sterol esterification: The acyl coenzyme A: Cholesterol acyltransferase reaction. Curr. Opin. Lipidol. 1997, 8, 167–173. [Google Scholar] [CrossRef]
- Uelmen, P.J.; Oka, K.; Sullivan, M.; Chang, C.C.; Chang, T.Y.; Chan, L. Tissue-specific expression and cholesterol regulation of acylcoenzyme A:cholesterol acyltransferase (ACAT) in mice. Molecular cloning of mouse ACAT cDNA, chromosomal localization, and regulation of ACAT in vivo and in vitro. J. Biol. Chem. 1995, 270, 26192–26201. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Huh, H.Y.; Cadigan, K.M.; Chang, T.Y. Molecular cloning and functional expression of human acyl-coenzyme A: Cholesterol acyltransferase cDNA in mutant Chinese hamster ovary cells. J. Biol. Chem. 1993, 268, 20747–20755. [Google Scholar] [PubMed]
- Meng, Q.; Sun, P.; Wang, L.L.; Ma, X.Y.; Zhao, J.Y. Study on hemolysis of panax ginseng and panax quinquefolium. J. Norman Bethune Univ. Med. Sci. 1998, 24, 135–136. [Google Scholar]
- Zhang, Q.J.; Zhang, A.H.; Lei, F.J.; Wang, S.J.; Zhang, L.X. Allelopathic Effect of Crude Ginsenoside Extracts on Early Growth of Panax quinquefolium L. Acta Bot. Boreali Occident. Sin. 2011, 31, 576–582. [Google Scholar]
- Forsgren, M.; Attersand, A.; Lake, S.; Grunler, J.; Swiezewska, E.; Dallner, G.; Climent, I. Isolation and functional expression of human COQ2, a gene encoding a polyprenyl transferase involved in the synthesis of CoQ. Biochem. J. 2004, 382, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Christianson, D.W. Structural and Chemical Biology of Terpenoid Cyclases. Chem. Rev. 2017, 117, 11570–11648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunillera, N.; Arro, M.; Delourme, D.; Karst, F.; Boronat, A.; Ferrer, A. Arabidopsis thaliana contains two differentially expressed farnesyl-diphosphate synthase genes. J. Biol. Chem. 1996, 271, 7774–7780. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Ohara, K.; Yazaki, K.; Nozaki, K.; Uchida, N.; Kawamukai, M.; Nojiri, H.; Yamane, H. The AtPPT1 gene encoding 4-hydroxybenzoate polyprenyl diphosphate transferase in ubiquinone biosynthesis is required for embryo development in Arabidopsis thaliana. Plant Mol. Biol. 2004, 55, 567–577. [Google Scholar] [CrossRef]
- Lucas, M.E.; Ma, Q.; Cunningham, D.; Peters, J.; Cattanach, B.; Bard, M.; Elmore, B.K.; Herman, G.E. Identification of two novel mutations in the murine Nsdhl sterol dehydrogenase gene and development of a functional complementation assay in yeast. Mol. Genet. Metab. 2003, 80, 227–233. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999, 99, 138–148. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | No. of Raw Reads | No. of Clean Reads | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|
| Hd17-1 | 49,443,264 | 48,646,584 | 99.1 | 96.98 | 57.67 |
| Hd17-2 | 57,069,334 | 56,143,790 | 99.09 | 96.95 | 57.73 |
| Hd17-3 | 58,364,474 | 57,334,282 | 99.05 | 96.86 | 57.42 |
| Hd34-1 | 47,643,390 | 46,916,374 | 99.07 | 96.9 | 57.65 |
| Hd34-2 | 49,995,230 | 49,215,252 | 99.08 | 96.93 | 57.46 |
| Hd34-3 | 46,722,588 | 45,978,738 | 99.05 | 96.86 | 57.46 |
| Hd51-1 | 52,938,778 | 52,207,298 | 99.16 | 97.16 | 57.46 |
| Hd51-2 | 47,556,084 | 46,827,310 | 99.08 | 96.92 | 57.7 |
| Hd51-3 | 54,687,348 | 53,813,252 | 99.08 | 96.93 | 57.48 |
| Ld17-1 | 49,825,826 | 48,853,942 | 99.01 | 96.75 | 57.81 |
| Ld17-2 | 76,163,900 | 74,830,456 | 99.06 | 96.88 | 57.65 |
| Ld17-3 | 64,840,360 | 63,758,552 | 99.08 | 96.94 | 57.72 |
| Ld34-1 | 69,549,900 | 68,317,596 | 99.19 | 97.38 | 57.35 |
| Ld34-2 | 73,145,282 | 72,046,316 | 99.22 | 97.43 | 57.74 |
| Ld34-3 | 60,119,712 | 59,271,498 | 99.23 | 97.47 | 57.65 |
| Ld51-1 | 74,741,142 | 73,658,172 | 99.22 | 97.44 | 57.6 |
| Ld51-2 | 73,090,694 | 72,050,486 | 99.23 | 97.47 | 57.4 |
| Ld51-3 | 87,693,220 | 86,345,808 | 99.2 | 97.38 | 57.79 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, G.; Li, Z.; Zhao, Z. Comparative Analysis of the Characteristics of Triterpenoid Transcriptome from Different Strains of Wolfiporia cocos. Int. J. Mol. Sci. 2019, 20, 3703. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153703
Zeng G, Li Z, Zhao Z. Comparative Analysis of the Characteristics of Triterpenoid Transcriptome from Different Strains of Wolfiporia cocos. International Journal of Molecular Sciences. 2019; 20(15):3703. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153703
Chicago/Turabian StyleZeng, Guiping, Zhong Li, and Zhi Zhao. 2019. "Comparative Analysis of the Characteristics of Triterpenoid Transcriptome from Different Strains of Wolfiporia cocos" International Journal of Molecular Sciences 20, no. 15: 3703. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153703