ESRP1 Induces Cervical Cancer Cell G1-Phase Arrest Via Regulating Cyclin A2 mRNA Stability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
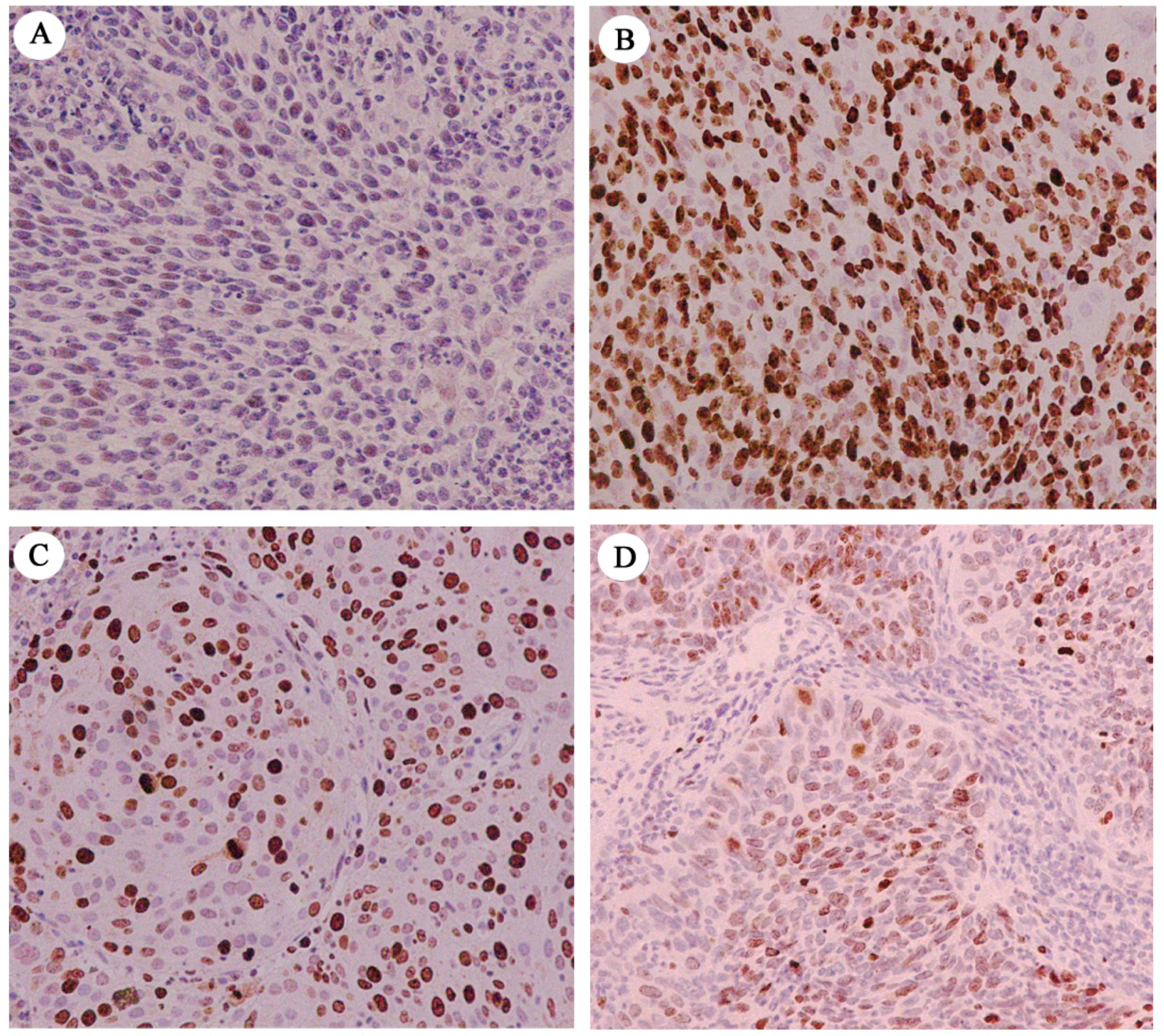
2.1. Immunohistochemical Assessment of ESRP1 Levels in Human Cervical Carcinoma Tissues
2.2. ESRP1 Overexpression Inhibits Cervical Carcinoma Cell Proliferation Via Inducing G1-Phase Arrest
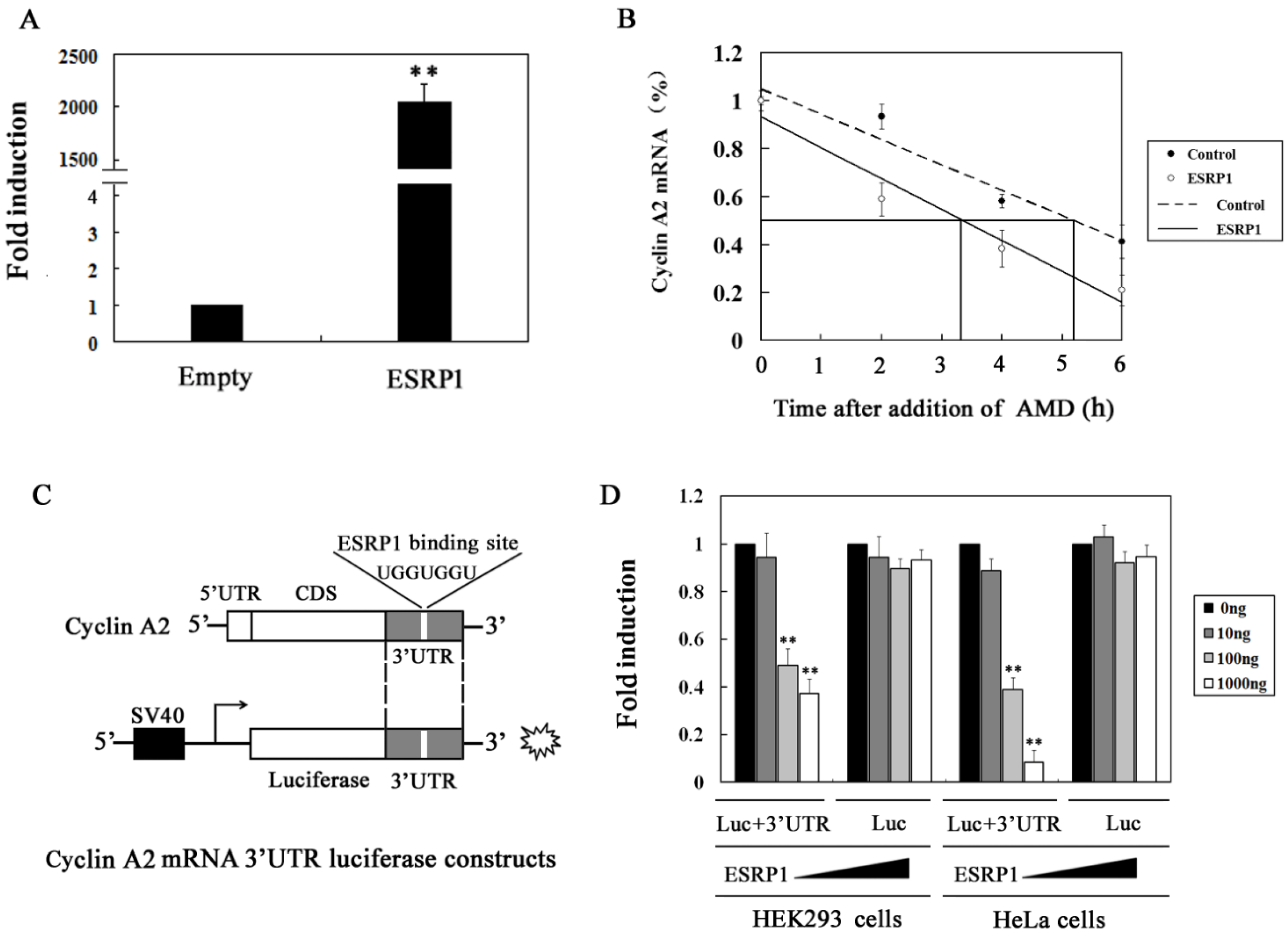
2.3. ESRP1 Regulates the Stability of the Cyclin A2 mRNA Via 3′UTR Interactions
2.4. ESRP1 Binds to the 3′UTR of Cyclin A2 mRNA
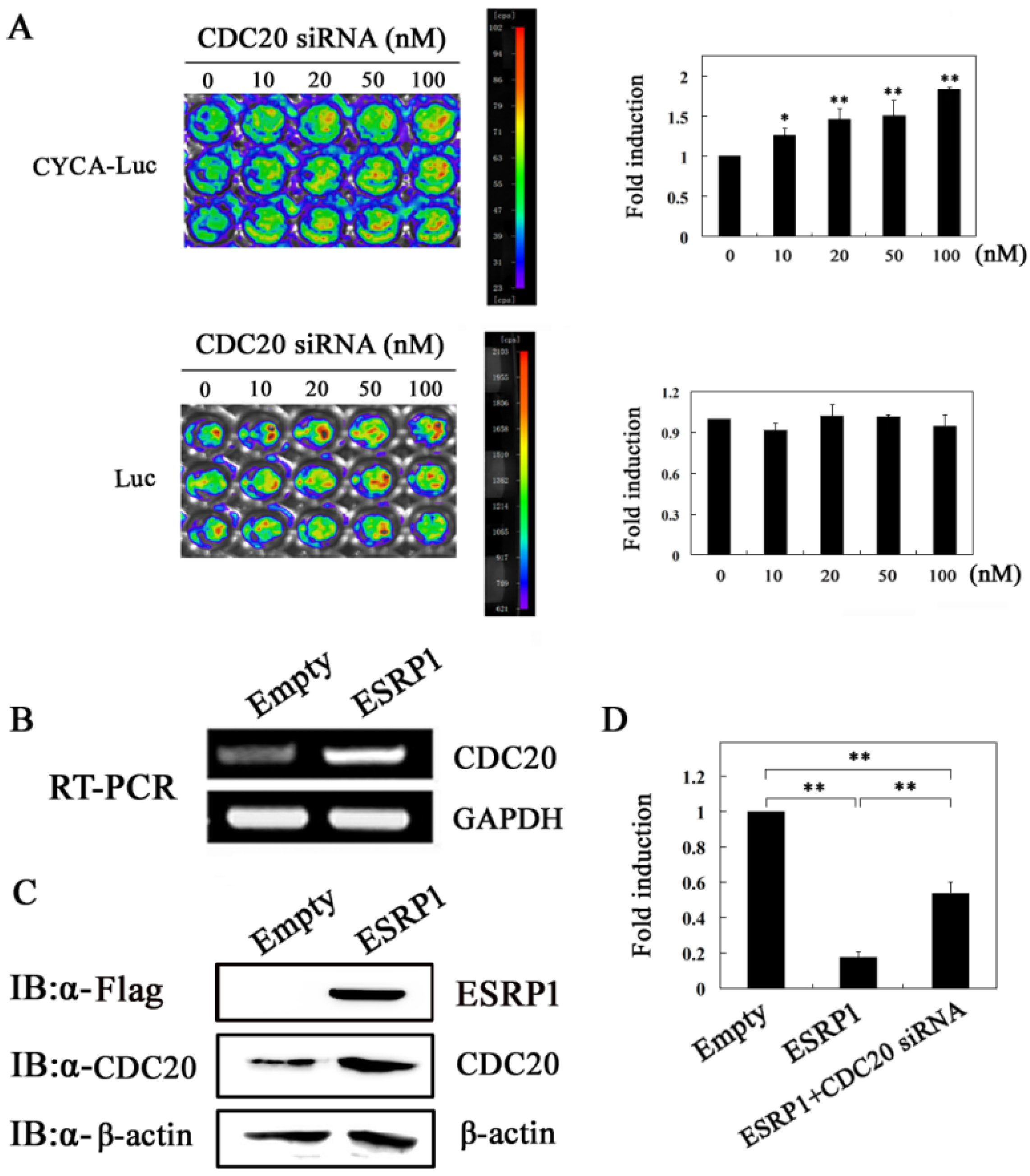
2.5. ESRP1 Downregulates Cyclin A2 Expression Partially Dependent on CDC20
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Tissue Sample Collection
4.2. Plasmids, Small Interfering RNA, and Transfection
4.3. RNA Isolation, Conventional RT-PCR, and Quantitative Real-Time RT-PCR (qRT-PCR)
4.4. Assessment of Cell Proliferation
4.5. Immunoblotting, Luciferase Assay, and Cell Cycle Analysis
4.6. Electrophoretic Mobility Shift Assay (EMSA)
4.7. RNA Immunoprecipitation Assay
4.8. Immunohistochemistry
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yi, Y.; Liu, Y.; Wu, W.; Wu, K.; Zhang, W. The role of miR-106p-5p in cervical cancer: From expression to molecular mechanism. Cell Death Discov. 2018, 4, 94. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Zhang, Y.; Yin, X.; Di, W. CDK7 inhibitor suppresses tumor progression through blocking the cell cycle at the G2/M phase and inhibiting transcriptional activity in cervical cancer. Onco Targets Ther. 2019, 12, 2137–2147. [Google Scholar] [CrossRef] [PubMed]
- Altinok, A.; Gonze, D.; Lévi, F.; Goldbeter, A. An automaton model for the cell cycle. Interface Focus 2011, 1, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.; Pepperkok, R.; Verde, F.; Ansorge, W.; Draetta, G. Cyclin A is required at two points in the human cell cycle. EMBO J. 1992, 11, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Ming, P.; Cai, T.; Li, J.; Ning, Y.; Xie, S.; Tao, T.; Tang, F. A novel arylbenzofuran induces cervical cancer cell apoptosis and G1/S arrest through ERK-mediated Cdk2/cyclin-A signaling pathway. Oncotarget 2016, 7, 41843–41856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yam, C.H.; Fung, T.K.; Poon, R.Y. Cyclin A in cell cycle control and cancer. Cell. Mol. Life Sci. 2002, 59, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Balczon, R.C. Overexpression of cyclin A in human HeLa cells induces detachment of kinetochores and spindlepole/centrosome overproduction. Chromosoma 2001, 110, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, R.; Priyanka, P.; Kumar, P.; Ghosh, T.; Khan, M.M.; Nagarajan, P.; Saxena, S. The microRNAs miR-449a and miR-424 suppress osteosarcoma by targeting cyclin A2 expression. J. Biol. Chem. 2019, 294, 4381–4400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurth, L. Versatility of RNA-Binding Proteins in Cancer. Comp. Funct. Genom. 2012, 2012, 178525. [Google Scholar] [CrossRef]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Lal, A.; Mazan-Mamczarz, K.; Kawai, T.; Yang, X.; Martindale, J.L.; Gorospe, M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 2004, 23, 3092–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.J.; Zhang, J.; Chen, X. RNPC1 modulates the RNA-binding activity of, and cooperates with, HuR to regulate p21 mRNA stability. Nucleic Acids Res. 2010, 38, 2256–2267. [Google Scholar] [CrossRef] [PubMed]
- Sagnol, S.; Marchal, S.; Yang, Y.; Allemand, F.; de Santa Barbara, P. Epithelial Splicing Regulatory Protein 1 (ESRP1) is a new regulator of stomach smooth muscle development and plasticity. Dev. Biol. 2016, 414, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Turley, E.A.; Veiseh, M.; Radisky, D.C.; Bissell, M.J. Mechanisms of disease: Epithelial-mesenchymal transition-does cellular plasticity fuel neoplastic progression. Nat. Clin. Pract. Oncol. 2008, 5, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [Green Version]
- Leontieva, O.V.; Ionov, Y. RNA-binding motif protein 35A is a novel tumor suppressor for colorectal cancer. Cell Cycle 2009, 8, 490–497. [Google Scholar] [CrossRef] [Green Version]
- Vukelic, J.; Dobrila-Dintinjana, R.; Dekanic, A.; Marijic, B.; Cubranic, A.; Braut, T. The relevance of assessing the cell proliferation factor ki-67 in squamous cell carcinoma of the larynx. Biomed. Res. Int. 2019, 2019, 8142572. [Google Scholar] [CrossRef]
- Huang, S.; Lin, J.; Guo, N.; Zhang, M.; Yun, X.; Liu, S.; Zhou, J.; He, E.; Skog, S. Elevated serum thymidine kinase 1 predicts risk of pre/early cancerous progression. Asian Pac. J. Cancer Prev. 2011, 12, 497–505. [Google Scholar]
- Brown, R.L.; Reinke, L.M.; Damerow, M.S.; Perez, D.; Chodosh, L.A.; Yang, J.; Cheng, C. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J. Clin. Invest. 2011, 121, 1064–1074. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.H.; Zhao, R.J.; Li, R.H.; Guo, C.P.; Zhang, G.J. Bioluminescence imaging of DNA synthetic phase of cell cycle in living animals. PLoS ONE 2013, 8, e53291. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.M.; Yang, C.W.; Lee, Y.Z.; Chuang, T.H.; Wu, P.L.; Chao, Y.S.; Lee, S.J. Tylophorine arrests carcinoma cells at G1 phase by downregulating cyclin A2 expression. Biochem. Biophys. Res. Commun. 2009, 386, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Caldwell, M.C.; Lin, S.; Furneaux, H.; Gorospe, M. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J. 2000, 19, 2340–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bebee, T.W.; Park, J.W.; Sheridan, K.I.; Warzecha, C.C.; Cieply, B.W.; Rohacek, A.M.; Xing, Y.; Carstens, R.P. The splicing regulators Esrp1 and Esrp2 direct an epithelial splicing program essential for mammalian development. Elife 2015, 4, e08954. [Google Scholar] [CrossRef] [PubMed]
- Gross, F.; Bonaiuti, P.; Hauf, S.; Ciliberto, A. Implications of alternative routes to APC/C inhibition by the mitotic checkpoint complex. PLoS Comput. Biol. 2018, 14, e1006449. [Google Scholar] [CrossRef] [PubMed]
- Allan, L.A.; Skowyra, A.; Rogers, K.I.; Zeller, D.; Clarke, P.R. Atypical APC/C-dependent degradation of Mcl-1 provides an apoptotic timer during mitotic arrest. EMBO J. 2018, 37, e96831. [Google Scholar] [CrossRef]
- Hein, J.B.; Nilsson, J. Interphase APC/C-Cdc20 inhibition by cyclin A2-Cdk2 ensures efficient mitotic entry. Nat. Commun. 2016, 7, 10975. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Ueda, J.; Matsuda, Y.; Yamahatsu, K.; Uchida, E.; Naito, Z.; Korc, M.; Ishiwata, T. Epithelial splicing regulatory protein 1 is a favorable prognostic factor in pancreatic cancer that attenuates pancreatic metastases. Oncogene 2014, 33, 4485–4495. [Google Scholar] [CrossRef]
- Loukil, A.; Cheung, C.T.; Bendris, N.; Lemmers, B.; Peter, M.; Blanchard, J.M. Cyclin A2: At the crossroads of cell cycle and cell invasion. World J. Biol. Chem. 2015, 6, 346–350. [Google Scholar] [CrossRef]
- Ishii, H.; Saitoh, M.; Sakamoto, K.; Kondo, T.; Katoh, R.; Tanaka, S.; Motizuki, M.; Masuyama, K.; Miyazawa, K. Epithelial splicing regulatory proteins 1 (ESRP1) and 2 (ESRP2) suppress cancer cell motility via different mechanisms. J. Biol. Chem. 2014, 289, 27386–27399. [Google Scholar] [CrossRef] [PubMed]
- Warzecha, C.C.; Shen, S.; Xing, Y.; Carstens, R.P. The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol. 2009, 6, 546–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burguera, D.; Marquez, Y.; Racioppi, C.; Permanyer, J.; Torres-Méndez, A.; Esposito, R.; Albuixech-Crespo, B.; Fanlo, L.; D’Agostino, Y.; Gohr, A.; et al. Evolutionary recruitment of flexible Esrp-dependent splicing programs into diverse embryonic morphogenetic processes. Nat. Commun. 2017, 8, 1799. [Google Scholar] [CrossRef] [PubMed]
- Kuang, J.; Li, L.; Guo, L.; Su, Y.; Wang, Y.; Xu, Y.; Wang, X.; Meng, S.; Lei, L.; Xu, L.; et al. RNF8 promotes epithelial-mesenchymal transition of breast cancer cells. J. Exp. Clin. Cancer Res. 2016, 35, 88. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.M.; Han, J.; Lee, S.H.; Park, H.J.; Lee, H.J.; Choi, J.S.; Lee, Y.M.; Choi, Y.L.; Shin, Y.K.; Kwon, M.J. ESRP1 is overexpressed in ovarian cancer and promotes switching from mesenchymal to epithelial phenotype in ovarian cancer cells. Oncogenesis 2017, 6, e389. [Google Scholar] [CrossRef] [PubMed]
- Horie, K.; Yamamoto, H.; Karube, K.; Takebayashi, K.; Yoshino, H.; Yoshioka, H.; Watanabe, J. Cyclin A is a reliable proliferation marker in endometrial cancer cell lines. Oncol. Lett. 2019, 17, 4455–4462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiguchi, K.; Sakamoto, K.; Koinuma, D.; Semba, K.; Inoue, A.; Inoue, S.; Fujii, H.; Yamaguchi, A.; Miyazawa, K.; Miyazono, K.; et al. TGF-β drives epithelial-mesenchymal transition through δEF1-mediated downregulation of ESRP. Oncogene 2012, 31, 3190–3201. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wu, B.; Guo, J.; Luo, W.; Wu, D.; Yang, H.; Zhen, Y.; Yu, X.; Wang, H.; Zhou, Y.; et al. Elevated expression of CDK4 in lung cancer. J. Transl. Med. 2011, 9, 38. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.-H.; Jing, Y.-J.; Yu, J.-B.; Jin, Z.-S.; Li, Z.; He, T.-T.; Su, X.-Z. ESRP1 Induces Cervical Cancer Cell G1-Phase Arrest Via Regulating Cyclin A2 mRNA Stability. Int. J. Mol. Sci. 2019, 20, 3705. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153705
Chen Z-H, Jing Y-J, Yu J-B, Jin Z-S, Li Z, He T-T, Su X-Z. ESRP1 Induces Cervical Cancer Cell G1-Phase Arrest Via Regulating Cyclin A2 mRNA Stability. International Journal of Molecular Sciences. 2019; 20(15):3705. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153705
Chicago/Turabian StyleChen, Zhi-Hong, Ya-Jie Jing, Jian-Bo Yu, Zai-Shu Jin, Zhu Li, Ting-Ting He, and Xiu-Zhen Su. 2019. "ESRP1 Induces Cervical Cancer Cell G1-Phase Arrest Via Regulating Cyclin A2 mRNA Stability" International Journal of Molecular Sciences 20, no. 15: 3705. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153705