Adapting and Surviving: Intra and Extra-Cellular Remodeling in Drug-Resistant Gastric Cancer Cells

 , , , , , ,
, , , , , , {kind=link}
Abstract
:1. Introduction
2. Role of Microenvironment in Tumor Growth and Chemo-Resistance
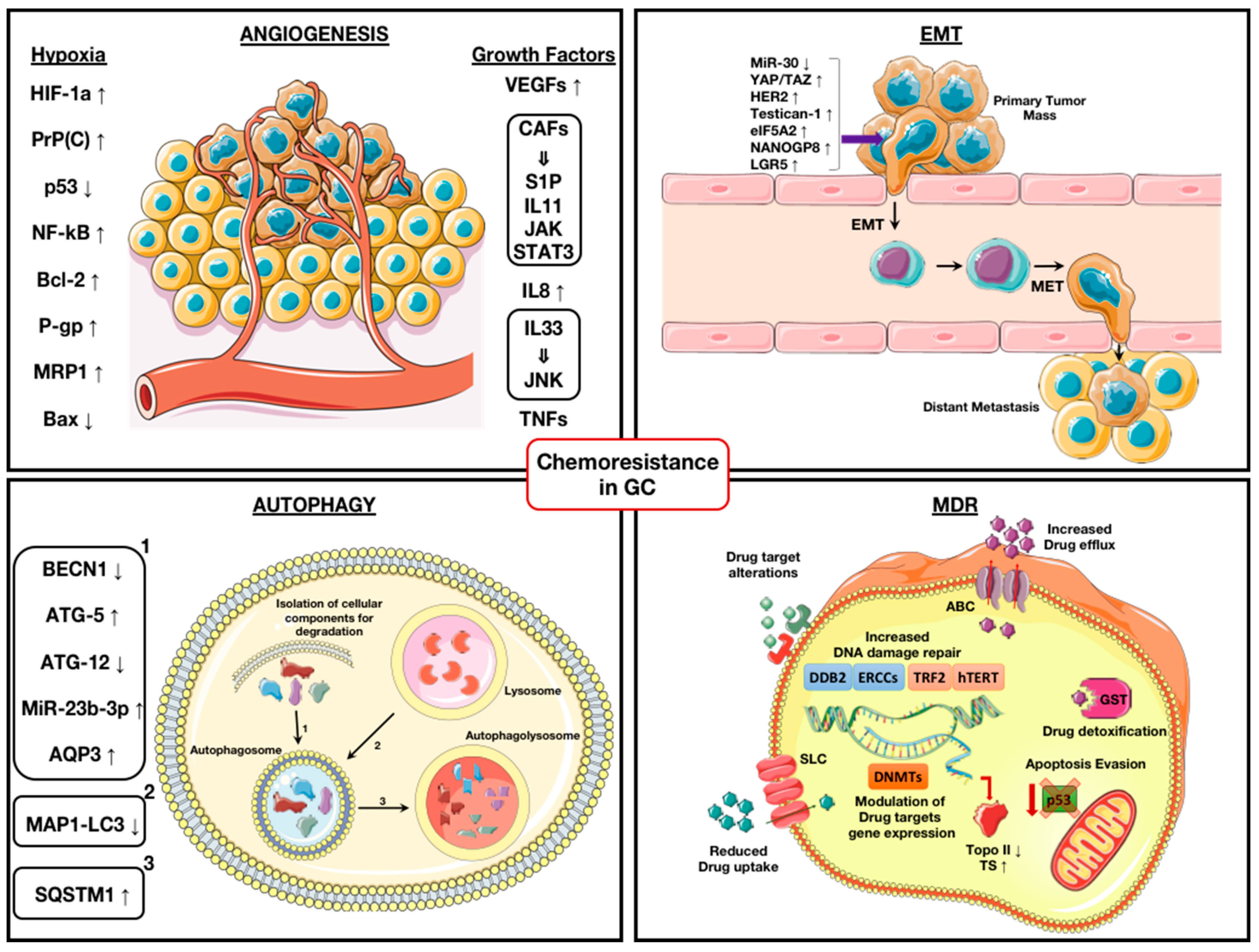
2.1. Angiogenesis and Hypoxia
2.2. Angiogenesis and Growth Factors
3. Role of Microenvironment in Tumor Growth and Chemo-resistance
4. Role of Autophagy in GC Chemo-Resistance
5. Multidrug Resistance (MDR) Mechanisms in GC
5.1. Role of ATP-Binding Cassette (ABC) Transporters
5.2. Increased Drug Detoxification
5.3. Counteracting Drug-Induced DNA Damage Response
5.4. Compensation of Drug Activity by Modulation of Targets Expression
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GC | Gastric cancer |
| EMT | Epithelial-mesenchymal transition |
| TME | Tumor microenvironment |
| ECM | Extracellular matrix |
| CDDP | Cisplatin |
| 5-FU | F-fluorouracil |
| P-gp | P-glycoprotein |
| VEGF | Vascular endothelial growth factor |
| CAFs | Cancer-associated fibroblasts |
| ILs | Interleukins |
| CSCs | Cancer stem cells |
| MET | Mesenchymal-epithelial transition |
| DOX | Doxorubicin |
| MDR | Multidrug resistance |
| ABC | ATP-binding cassette transporters |
| HCPT | Hydroxycamptothecin |
| SLCs | Solute carrier transporters |
| GSH | Glutathione |
| Topo II | Topoisomerase II |
References
- Sitarz, R.; Skierucha, M.; Mielko, J.; Offerhaus, G.J.A.; Maciejewski, R.; Polkowski, W.P. Gastric cancer: Epidemiology, prevention, classification, and treatment. Cancer Manag. Res. 2018, 10, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Marqués-Lespier, J.M.; González-Pons, M.; Cruz-Correa, M. Current perspectives on gastric cancer. Gastroenterol. Clin. N. Am. 2016, 45, 413–428. [Google Scholar] [CrossRef]
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends—An Update. Cancer Epidemiol. Biomarkers Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Orditura, M.; Galizia, G.; Sforza, V.; Gambardella, V.; Fabozzi, A.; Laterza, M.M.; Andreozzi, F.; Ventriglia, J.; Savastano, B.; Mabilia, A.; et al. Treatment of gastric cancer. World J. Gastroenterol. 2014, 20, 1635–1649. [Google Scholar] [CrossRef] [PubMed]
- Dicken, B.J.; Bigam, D.L.; Cass, C.; Mackey, J.R.; Joy, A.A.; Hamilton, S.M. Gastric adenocarcinoma: Review and considerations for future directions. Ann. Surg. 2005, 241, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Quadri, H.S.; Hong, Y.K.; Al-Refaie, W.B. Approach to the surgical management of resectable gastric cancer. Clin. Adv. Hematol. Oncol. 2016, 14, 249–257. [Google Scholar]
- Hamamoto, Y. Complications in advanced or recurrent gastric cancer patients with peritoneal metastasis during and after palliative systemic chemotherapy. Mol. Clin. Oncol. 2015, 3, 539–542. [Google Scholar] [CrossRef]
- Liu, D.; Lu, M.; Li, J.; Yang, Z.; Feng, Q.; Zhou, M.; Zhang, Z.; Shen, L. The patterns and timing of recurrence after curative resection for gastric cancer in China. World J. Surg. Oncol. 2016, 14, 305. [Google Scholar] [CrossRef]
- Shin, C.H.; Lee, W.Y.; Hong, S.W.; Chang, Y.G. Characteristics of gastric cancer recurrence five or more years after curative gastrectomy. Chin. J. Cancer Res. 2016, 28, 503–510. [Google Scholar] [CrossRef] [Green Version]
- Chon, S.H.; Berlth, F.; Plum, P.S.; Herbold, T.; Alakus, H.; Kleinert, R.; Moenig, S.P.; Bruns, C.J.; Hoelscher, A.H.; Meyer, H.J. Gastric cancer treatment in the world: Germany. Transl. Gastroenterol. Hepatol. 2017, 2, 53. [Google Scholar] [CrossRef]
- Kuo, C.Y.; Chao, Y.; Li, C.P. Update on treatment of gastric cancer. J. Chin. Med. Assoc. 2014, 77, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Yin, X.; Sheng, L.; Xu, S.; Dong, L.; Liu, L. Perioperative chemotherapy more of a benefit for overall survival than adjuvant chemotherapy for operable gastric cancer: An updated Meta-analysis. Sci. Rep. 2015, 5, 12850. [Google Scholar] [CrossRef] [PubMed]
- Jain, V.K.; Cunningham, D.; Rao, S. Chemotherapy for operable gastric cancer: Current perspectives. Indian J. Surg. Oncol. 2011, 2, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.G.; Sanders, A.J.; Katoh, M.; Ungefroren, H.; Gieseler, F.; Prince, M.; Thompson, S.K.; Zollo, M.; Spano, D.; Dhawan, P.; et al. Tissue invasion and metastasis: Molecular, biological and clinical perspectives. Semin. Cancer Biol. 2015, 35 Suppl, S244–S275. [Google Scholar] [CrossRef]
- Gao, J.P.; Xu, W.; Liu, W.T.; Yan, M.; Zhu, Z.G. Tumor heterogeneity of gastric cancer: From the perspective of tumor-initiating cell. World J. Gastroenterol. 2018, 24, 2567–2581. [Google Scholar] [CrossRef]
- Martin, T.A.; Ye, L.; Sanders, A.J.; Lane, J.; Jiang, W.G. Cancer Invasion and Metastasis: Molecular and Cellular Perspective, Madame Curie Bioscience Database—NCBI Bookshelf. 2013. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK164700/ (accessed on 20 June 2019).
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The different mechanisms of cancer drug resistance: A brief review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Lordick, F.; Allum, W.; Carneiro, F.; Mitry, E.; Tabernero, J.; Tan, P.; Van Cutsem, E.; van de Velde, C.; Cervantes, A. Unmet needs and challenges in gastric cancer: The way forward. Cancer Treat. Rev. 2014, 40, 692–700. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef]
- Lazăr, D.C.; Tăban, S.; Cornianu, M.; Faur, A.; Goldiş, A. New advances in targeted gastric cancer treatment. World J. Gastroenterol. 2016, 22, 6776–6799. [Google Scholar] [CrossRef]
- Cao, W.; Wei, W.; Zhan, Z.; Xie, D.; Xie, Y.; Xiao, Q. Regulation of drug resistance and metastasis of gastric cancer cells via the microRNA647-ANK2 axis. Int. J. Mol. Med. 2018, 41, 1958–1966. [Google Scholar] [CrossRef]
- Shi, W.J.; Gao, J.B. Molecular mechanisms of chemoresistance in gastric cancer. World J. Gastrointest. Oncol. 2016, 8, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Choi, I.J.; Kim, C.G.; Kim, H.S.; Oshima, A.; Michalowski, A.; Green, J.E. A gene expression signature of acquired chemoresistance to cisplatin and fluorouracil combination chemotherapy in gastric cancer patients. PLoS ONE 2011, 6, e16694. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.W.; Lim, J.B. Role of the tumor microenvironment in the pathogenesis of gastric carcinoma. World J. Gastroenterol. 2014, 20, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Chang, Y.H.; Heo, Y.J.; Kim, S.; Kim, N.K.; Park, J.O.; Kang, W.K.; Lee, J.; Kim, K.M. Four distinct immune microenvironment subtypes in gastric adenocarcinoma with special reference to microsatellite instability. ESMO Open 2018, 3, e000326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testerman, T.L.; Morris, J. Beyond the stomach: An updated view of Helicobacter pylori pathogenesis, diagnosis, and treatment. World J. Gastroenterol. 2014, 20, 12781–12808. [Google Scholar] [CrossRef]
- Qu, Y.; Dang, S.; Hou, P. Gene methylation in gastric cancer. Clin. Chim. Acta 2013, 424, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selaru, F.M.; David, S.; Meltzer, S.J.; Hamilton, J.P. Epigenetic events in gastrointestinal cancer. Am. J. Gastroenterol. 2009, 104, 1910–1912. [Google Scholar] [CrossRef]
- Yang, Q.; Zhu, C.; Zhang, Y.; Wang, Y.; Wang, Y.; Zhu, L.; Yang, X.; Li, J.; Nie, H.; Jiang, S.; et al. Molecular analysis of gastric cancer identifies genomic markers of drug sensitivity in Asian gastric cancer. J. Cancer 2018, 9, 2973–2980. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting tumor microenvironment for cancer therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.A.M.A.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The role of tumor microenvironment in chemoresistance: To survive, keep your enemies closer. Int. J. Mol. Sci. 2017, 18, 1568. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, X. Tipping Tumor Microenvironment against Drug Resistance. J. Oncol. Transl. Res. 2017, 1, 106. [Google Scholar] [CrossRef]
- Jo, Y.; Choi, N.; Kim, K.; Koo, H.J.; Choi, J.; Kim, H.N. Chemoresistance of Cancer Cells: Requirements of Tumor Microenvironment-mimicking In Vitro Models in Anti-cancer Drug Development. Theranostics 2018, 8, 5259–5275. [Google Scholar] [CrossRef] [PubMed]
- Mumenthaler, S.M.; Foo, J.; Choi, N.C.; Heise, N.; Leder, K.; Agus, D.B.; Pao, W.; Michor, F.; Mallick, P. The impact of microenvironmental heterogeneity on the evolution of drug resistance in cancer cells. Cancer Inform. 2015, 14, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Affo, S.; Yu, L.X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. 2017, 12, 153–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.W.; Mooney, D.J. Extracellular matrix stiffness causes systematic variations in proliferation and chemosensitivity in myeloid leukemias. Proc. Natl. Acad. Sci. USA 2016, 113, 12126–12131. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.; Koh, I.; Lee, J.E.; Lim, J.Y.; Cheong, J.H.; Kim, P. Increased extracellular matrix density disrupts E-cadherin/β-catenin complex in gastric cancer cells. Biomater. Sci. 2018, 6, 2704–2713. [Google Scholar] [CrossRef]
- Ji, R.; Zhang, B.; Zhang, X.; Xue, J.; Yuan, X.; Yan, Y.; Wang, M.; Zhu, W.; Qian, H.; Xu, W. Exosomes derived from human mesenchymal stem cells confer drug resistance in gastric cancer. Cell Cycle 2015, 14, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.; Koh, I.; Lee, S.J.; Cheong, J.H.; Kim, P. Droplet-based microtumor model to assess cell-ECM interactions and drug resistance of gastric cancer cells. Sci. Rep. 2017, 7, 41541. [Google Scholar] [CrossRef]
- Nienhüser, H.; Schmidt, T. Angiogenesis and Anti-angiogenic Therapy in Gastric Cancer. Int. J. Mol. Sci. 2017, 19, 43. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Michiels, C.; Tellier, C.; Feron, O. Cycling hypoxia: A key feature of the tumor microenvironment. Biochim. Biophys. Acta 2016, 1866, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Mayer, A. Hypoxia in tumors: Pathogenesis-related classification, characterization of hypoxia subtypes, and associated biological and clinical implications. Adv. Exp. Med. Biol. 2014, 812, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl) 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.A.; Pritchard, S.A.; Welch, I.M.; Price, P.M.; West, C.M. Is the hypoxia-inducible factor pathway important in gastric cancer? Eur. J. Cancer 2005, 41, 2792–2805. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, Y.; Miyazaki, K. The Critical Impact of HIF-1a on Gastric Cancer Biology. Cancers (Basel) 2013, 5, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skvortsova, I.; Skvortsov, S.; Haidenberger, A.; Devries, A.; Nevinny-Stickel, M.; Saurer, M.; Lukas, P.; Seppi, T. Effects of paclitaxel and docetaxel on EGFR-expressing human carcinoma cells under normoxic versus hypoxic conditions in vitro. J. Chemother. 2004, 16, 372–380. [Google Scholar] [CrossRef]
- Liang, J.; Bai, F.; Luo, G.; Wang, J.; Liu, J.; Ge, F.; Pan, Y.; Yao, L.; Du, R.; Li, X.; et al. Hypoxia induced overexpression of PrP(C) in gastric cancer cell lines. Cancer Biol. Ther. 2007, 6, 769–774. [Google Scholar] [CrossRef]
- Rohwer, N.; Dame, C.; Haugstetter, A.; Wiedenmann, B.; Detjen, K.; Schmitt, C.A.; Cramer, T. Hypoxia-inducible factor 1alpha determines gastric cancer chemosensitivity via modulation of p53 and NF-kappaB. PLoS ONE 2010, 5, e12038. [Google Scholar] [CrossRef]
- Nakamura, J.; Kitajima, Y.; Kai, K.; Mitsuno, M.; Ide, T.; Hashiguchi, K.; Hiraki, M.; Miyazaki, K. Hypoxia-inducible factor-1alpha expression predicts the response to 5-fluorouracil-based adjuvant chemotherapy in advanced gastric cancer. Oncol. Rep. 2009, 22, 693–699. [Google Scholar] [CrossRef]
- Liu, L.; Ning, X.; Sun, L.; Zhang, H.; Shi, Y.; Guo, C.; Han, S.; Liu, J.; Sun, S.; Han, Z.; et al. Hypoxia-inducible factor-1 alpha contributes to hypoxia-induced chemoresistance in gastric cancer. Cancer Sci. 2008, 99, 121–128. [Google Scholar] [CrossRef]
- Yashiro, M. Gastric cancer stem cells and resistance to cancer therapy. Chemotherapy (Los Angel) 2014, 3. [Google Scholar] [CrossRef]
- Abdel-Rahman, O. Targeting vascular endothelial growth factor (VEGF) pathway in gastric cancer: Preclinical and clinical aspects. Crit. Rev. Oncol. Hematol. 2015, 93, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Partyka, R.; Gonciarz, M.; Jałowiecki, P.; Kokocińska, D.; Byrczek, T. VEGF and metalloproteinase 2 (MMP 2) expression in gastric cancer tissue. Med. Sci. Monit. 2012, 18, BR130–BR134. [Google Scholar] [CrossRef] [Green Version]
- Ozdemir, F.; Akdogan, R.; Aydin, F.; Reis, A.; Kavgaci, H.; Gul, S.; Akdogan, E. The effects of VEGF and VEGFR-2 on survival in patients with gastric cancer. J. Exp. Clin. Cancer Res. 2006, 25, 83–88. [Google Scholar] [PubMed]
- Schimanski, C.C.; Schlaegel, F.; Jordan, M.; Moehler, M.; Sgourakis, G.; Drescher, D.G.; Galle, P.R.; Lang, H.; Gockel, I. VEGF-D correlates with metastatic disease in gastric cancer patients undergoing surgery. World J. Surg. 2011, 35, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Kim, I.K.; Park, S.M.; Baek, K.E.; Nam, I.K.; Park, S.H.; Ryu, K.J.; Choi, J.; Ryu, J.; Hong, S.C.; et al. VEGF-C mediates RhoGDI2-induced gastric cancer cell metastasis and cisplatin resistance. Int. J. Cancer 2014, 135, 1553–1563. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J.; Azuma, A.; Miura, Y.; Orimo, A. Activated fibroblast program orchestrates tumor initiation and progression; molecular mechanisms and the associated therapeutic strategies. Int. J. Mol. Sci. 2019, 20, 2256. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Bu, L.; Baba, H.; Yoshida, N.; Miyake, K.; Yasuda, T.; Uchihara, T.; Tan, P.; Ishimoto, T. Biological heterogeneity and versatility of cancer-associated fibroblasts in the tumor microenvironment. Oncogene 2019, 38, 4887–4901. [Google Scholar] [CrossRef]
- Guo, X.; Oshima, H.; Kitmura, T.; Taketo, M.M.; Oshima, M. Stromal fibroblasts activated by tumor cells promote angiogenesis in mouse gastric cancer. J. Biol. Chem. 2008, 283, 19864–19871. [Google Scholar] [CrossRef]
- Ma, J.; Song, X.; Xu, X.; Mou, Y. Cancer-Associated Fibroblasts Promote the Chemo-resistance in Gastric Cancer through Secreting IL-11 Targeting JAK/STAT3/Bcl2 Pathway. Cancer Res. Treat. 2019, 51, 194–210. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Zhu, J.; Chen, S.; Li, T.; Ma, J.; Guo, S.; Hu, J.; Yue, T.; Zhang, J.; Wang, P.; et al. Activated gastric cancer-associated fibroblasts contribute to the malignant phenotype and 5-FU resistance via paracrine action in gastric cancer. Cancer Cell Int. 2018, 18, 104. [Google Scholar] [CrossRef]
- Kuai, W.X.; Wang, Q.; Yang, X.Z.; Zhao, Y.; Yu, R.; Tang, X.J. Interleukin-8 associates with adhesion, migration, invasion and chemosensitivity of human gastric cancer cells. World J. Gastroenterol. 2012, 18, 979–985. [Google Scholar] [CrossRef]
- Ye, X.L.; Zhao, Y.R.; Weng, G.B.; Chen, Y.C.; Wei, X.N.; Shao, J.P.; Ji, H. IL-33-induced JNK pathway activation confers gastric cancer chemotherapy resistance. Oncol. Rep. 2015, 33, 2746–2752. [Google Scholar] [CrossRef]
- Huang, S.; Chen, M.; Shen, Y.; Shen, W.; Guo, H.; Gao, Q.; Zou, X. Inhibition of activated Stat3 reverses drug resistance to chemotherapeutic agents in gastric cancer cells. Cancer Lett. 2012, 315, 198–205. [Google Scholar] [CrossRef]
- Yang, Z.; Guo, L.; Liu, D.; Sun, L.; Chen, H.; Deng, Q.; Liu, Y.; Yu, M.; Ma, Y.; Guo, N.; et al. Acquisition of resistance to trastuzumab in gastric cancer cells is associated with activation of IL-6/STAT3/Jagged-1/Notch positive feedback loop. Oncotarget 2015, 6, 5072–5087. [Google Scholar] [CrossRef]
- Kang, Y.; Hu, W.; Bai, E.; Zheng, H.; Liu, Z.; Wu, J.; Jin, R.; Zhao, C.; Liang, G. Curcumin sensitizes human gastric cancer cells to 5-fluorouracil through inhibition of the NF-κB survival-signaling pathway. Onco Targets Ther. 2016, 9, 7373–7384. [Google Scholar] [CrossRef]
- Kwon, O.H.; Kim, J.H.; Kim, S.Y.; Kim, Y.S. TWEAK/Fn14 signaling mediates gastric cancer cell resistance to 5-fluorouracil via NF-κB activation. Int. J. Oncol. 2014, 44, 583–590. [Google Scholar] [CrossRef]
- Zhi, X.; Tao, J.; Xiang, G.; Cao, H.; Liu, Z.; Yang, K.; Lv, C.; Ni, S. APRIL induces cisplatin resistance in gastric cancer cells via activation of the NF-κB pathway. Cell Physiol. Biochem. 2015, 35, 571–585. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; Dos Santos, L.V.; Aprile, G.; Ferry, D.R.; et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef]
- Wang, Z.; Dabrosin, C.; Yin, X.; Fuster, M.M.; Arreola, A.; Rathmell, W.K.; Generali, D.; Nagaraju, G.P.; El-Rayes, B.; Ribatti, D.; et al. Broad targeting of angiogenesis for cancer prevention and therapy. Semin. Cancer Biol. 2015, 35 Suppl, S224–S243. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Caiazza, C.; Mallardo, M. The Roles of miR-25 and its Targeted Genes in Development of Human Cancer. Microrna 2016, 5, 113–119. [Google Scholar] [CrossRef]
- Ruggieri, V.; Russi, S.; Zoppoli, P.; La Rocca, F.; Angrisano, T.; Falco, G.; Calice, G.; Laurino, S. The Role of MicroRNAs in the Regulation of Gastric Cancer Stem Cells: A Meta-Analysis of the Current Status. J. Clin. Med. 2019, 8, 639. [Google Scholar] [CrossRef]
- Peng, Z.; Wang, C.X.; Fang, E.H.; Wang, G.B.; Tong, Q. Role of epithelial-mesenchymal transition in gastric cancer initiation and progression. World J. Gastroenterol. 2014, 20, 5403–5410. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Wik, E.; Ræder, M.B.; Krakstad, C.; Trovik, J.; Birkeland, E.; Hoivik, E.A.; Mjos, S.; Werner, H.M.J.; Mannelqvist, M.; Stefansson, I.M.; et al. Lack of estrogen receptor-α is associated with epithelial-mesenchymal transition and PI3K alterations in endometrial carcinoma. Clin. Cancer Res. 2013, 19, 1094–1105. [Google Scholar] [CrossRef]
- Oishi, N.; Kumar, M.R.; Roessler, S.; Ji, J.; Forgues, M.; Budhu, A.; Zhao, X.; Andersen, J.B.; Ye, Q.H.; Jia, H.L.; et al. Transcriptomic profiling reveals hepatic stem-like gene signatures and interplay of miR-200c and epithelial-mesenchymal transition in intrahepatic cholangiocarcinoma. Hepatology 2012, 56, 1792–1803. [Google Scholar] [CrossRef]
- Taube, J.H.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.T.; Gupta, P.B.; Evans, K.W.; et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Mitra, A.; Mishra, L.; Li, S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget 2015, 6, 10697–10711. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.L.; Zhang, X.H.; Zhang, X.; Chu, J.K. MiR-30a increases cisplatin sensitivity of gastric cancer cells through suppressing epithelial-to-mesenchymal transition (EMT). Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 1733–1739. [Google Scholar]
- Ge, L.; Li, D.-S.; Chen, F.; Feng, J.D.; Li, B.; Wang, T.J. TAZ overexpression is associated with epithelial-mesenchymal transition in cisplatin-resistant gastric cancer cells. Int. J. Oncol. 2017, 51, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.P.; Han, S.W.; Song, S.H.; Jeong, E.G.; Lee, M.Y.; Hwang, D.; Im, S.A.; Bang, Y.J.; Kim, T.Y. Testican-1-mediated epithelial-mesenchymal transition signaling confers acquired resistance to lapatinib in HER2-positive gastric cancer. Oncogene 2014, 33, 3334–3341. [Google Scholar] [CrossRef]
- Huang, D.; Duan, H.; Huang, H.; Tong, X.; Han, Y.; Ru, G.; Qu, L.; Shou, C.; Zhao, Z. Cisplatin resistance in gastric cancer cells is associated with HER2 upregulation-induced epithelial-mesenchymal transition. Sci. Rep. 2016, 6, 20502. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Xu, Z.; Lv, H.; Wang, Y.; Wang, L.; Ni, Y.; Wang, X.; Hu, C.; Chen, S.; Teng, F.; et al. eIF5A2 regulates the resistance of gastric cancer cells to cisplatin via induction of EMT. Am. J. Transl. Res. 2018, 10, 4269–4279. [Google Scholar]
- Grygielewicz, P.; Dymek, B.; Bujak, A.; Gunerka, P.; Stanczak, A.; Lamparska-Przybysz, M.; Wieczorek, M.; Dzwonek, K.; Zdzalik, D. Epithelial-mesenchymal transition confers resistance to selective FGFR inhibitors in SNU-16 gastric cancer cells. Gastric Cancer 2016, 19, 53–62. [Google Scholar] [CrossRef]
- Han, R.; Xiong, J.; Xiao, R.; Altaf, E.; Wang, J.; Liu, Y.; Xu, H.; Ding, Q.; Zhang, Q. Activation of β-catenin signaling is critical for doxorubicin-induced epithelial-mesenchymal transition in BGC-823 gastric cancer cell line. Tumour Biol. 2013, 34, 277–284. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Ryu, H.S.; Park, D.J.; Kim, H.H.; Kim, W.H.; Lee, H.S. Combination of epithelial-mesenchymal transition and cancer stem cell-like phenotypes has independent prognostic value in gastric cancer. Hum. Pathol. 2012, 43, 520–528. [Google Scholar] [CrossRef]
- Ma, X.; Wang, B.; Wang, X.; Luo, Y.; Fan, W. NANOGP8 is the key regulator of stemness, EMT, Wnt pathway, chemoresistance, and other malignant phenotypes in gastric cancer cells. PLoS ONE 2018, 13, e0192436. [Google Scholar] [CrossRef]
- Wang, B.; Chen, Q.; Cao, Y.; Ma, X.; Yin, C.; Jia, Y.; Zang, A.; Fan, W. LGR5 Is a Gastric Cancer Stem Cell Marker Associated with Stemness and the EMT Signature Genes NANOG, NANOGP8, PRRX1, TWIST1, and BMI1. PLoS ONE 2016, 11, e0168904. [Google Scholar] [CrossRef]
- Feng, S.; Zheng, Z.; Feng, L.; Yang, L.; Chen, Z.; Lin, Y.; Gao, Y.; Chen, Y. Proton pump inhibitor pantoprazole inhibits the proliferation, self-renewal and chemoresistance of gastric cancer stem cells via the EMT/β-catenin pathways. Oncol. Rep. 2016, 36, 3207–3214. [Google Scholar] [CrossRef]
- Shipitsin, M.; Campbell, L.L.; Argani, P.; Weremowicz, S.; Bloushtain-Qimron, N.; Yao, J.; Nikolskaya, T.; Serebryiskaya, T.; Beroukhim, R.; Hu, M.; et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007, 11, 259–273. [Google Scholar] [CrossRef]
- Chen, H.T.; Liu, H.; Mao, M.J.; Tan, Y.; Mo, X.Q.; Meng, X.J.; Cao, M.T.; Zhong, C.Y.; Liu, Y.; Shan, H.; et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol. Cancer 2019, 18, 101. [Google Scholar] [CrossRef]
- Gugnoni, M.; Sancisi, V.; Manzotti, G.; Gandolfi, G.; Ciarrocchi, A. Autophagy and epithelial-mesenchymal transition: An intricate interplay in cancer. Cell Death Dis. 2016, 7, e2520. [Google Scholar] [CrossRef]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Yoshida, G.J. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: From pathophysiology to treatment. J. Hematol. Oncol. 2017, 10, 67. [Google Scholar] [CrossRef]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef]
- Cao, Y.; Luo, Y.; Zou, J.; Ouyang, J.; Cai, Z.; Zeng, X.; Ling, H.; Zeng, T. Autophagy and its role in gastric cancer. Clin. Chim. Acta 2019, 489, 10–20. [Google Scholar] [CrossRef]
- Qian, H.; Yang, Y. Functional role of autophagy in gastric cancer. Oncotarget 2016, 7, 17641–17651. [Google Scholar] [CrossRef]
- Al-Akra, L.; Bae, D.H.; Leck, L.L.; Richardson, D.R.; Jansson, P.J. The biochemical and molecular mechanisms involved in the role of micro-tumor environment stress in development of drug resistance. Biochim. Biophys. Acta Gen. Subj. 2019. [Google Scholar] [CrossRef]
- Ge, J.; Chen, Z.; Huang, J.; Chen, J.; Yuan, W.; Deng, Z.; Chen, Z. Upregulation of autophagy-related gene-5 (ATG-5) is associated with chemoresistance in human gastric cancer. PLoS ONE 2014, 9, e110293. [Google Scholar] [CrossRef]
- Pei, G.; Luo, M.; Ni, X.; Wu, J.; Wang, S.; Ma, Y.; Yu, J. Autophagy Facilitates Metadherin-Induced Chemotherapy Resistance Through the AMPK/ATG5 Pathway in Gastric Cancer. Cell. Physiol. Biochem. 2018, 46, 847–859. [Google Scholar] [CrossRef]
- An, Y.; Zhang, Z.; Shang, Y.; Jiang, X.; Dong, J.; Yu, P.; Nie, Y.; Zhao, Q. miR-23b-3p regulates the chemoresistance of gastric cancer cells by targeting ATG12 and HMGB2. Cell Death Dis. 2015, 6, e1766. [Google Scholar] [CrossRef]
- Dong, X.; Wang, Y.; Zhou, Y.; Wen, J.; Wang, S.; Shen, L. Aquaporin 3 facilitates chemoresistance in gastric cancer cells to cisplatin via autophagy. Cell Death Discov. 2016, 2, 16087. [Google Scholar] [CrossRef]
- Ye, H.; Chai, X.; Wang, X.; Zheng, Q.; Zheng, D.; Wu, F.; Zheng, C.; Chen, P. Autophagy flux inhibition augments gastric cancer resistance to the anti-human epidermal growth factor receptor 2 antibody trastuzumab. Oncol. Lett. 2018, 15, 4143–4150. [Google Scholar] [CrossRef]
- Zhao, Y.; You, H.; Liu, F.; An, H.; Shi, Y.; Yu, Q.; Fan, D. Differentially expressed gene profiles between multidrug resistant gastric adenocarcinoma cells and their parental cells. Cancer Lett. 2002, 185, 211–218. [Google Scholar] [CrossRef]
- Gürel, S.; Yerci, O.; Filiz, G.; Dolar, E.; Yilmazlar, T.; Nak, S.G.; Gülten, M.; Zorluoğlu, A.; Memik, F. High expression of multidrug resistance-1 (MDR-1) and its relationship with multiple prognostic factors in gastric carcinomas in patients in Turkey. J. Int. Med. Res. 1999, 27, 79–84. [Google Scholar] [CrossRef]
- de Oliveira, J.; Felipe, A.V.; Neto, R.A.; Oshima, C.T.; de Souza Silva, M.; Forones, N.M. Association between ABCB1 immunohistochemical expression and overall survival in gastric cancer patients. Asian Pac. J. Cancer Prev. 2014, 15, 6935–6938. [Google Scholar] [CrossRef]
- Choi, J.H.; Lim, H.Y.; Joo, H.J.; Kim, H.S.; Yi, J.W.; Kim, H.C.; Cho, Y.K.; Kim, M.W.; Lee, K.B. Expression of multidrug resistance-associated protein1,P-glycoprotein, and thymidylate synthase in gastric cancer patients treated with 5-fluorouracil and doxorubicin-based adjuvant chemotherapy after curative resection. Br. J. Cancer 2002, 86, 1578–1585. [Google Scholar] [CrossRef] [Green Version]
- Kellner, U.; Hutchinson, L.; Seidel, A.; Lage, H.; Danks, M.K.; Dietel, M.; Kaufmann, S.H. Decreased drug accumulation in a mitoxantrone-resistant gastric carcinoma cell line in the absence of P-glycoprotein. Int. J. Cancer 1997, 71, 817–824. [Google Scholar] [CrossRef]
- Tomonaga, M.; Oka, M.; Narasaki, F.; Fukuda, M.; Nakano, R.; Takatani, H.; Ikeda, K.; Terashi, K.; Matsuo, I.; Soda, H.; et al. The multidrug resistance-associated protein gene confers drug resistance in human gastric and colon cancers. Jpn. J. Cancer Res. 1996, 87, 1263–1270. [Google Scholar] [CrossRef]
- Chung, H.C.; Gong, S.J.; Yoo, N.C.; Noh, S.H.; Kim, J.H.; Roh, J.K.; Min, J.S.; Kim, B.S.; Lee, K.B. P-glycoprotein as an intermediate end point of drug resistance to neoadjuvant chemotherapy in locally advanced gastric cancer. Yonsei Med. J. 1996, 37, 397–404. [Google Scholar] [CrossRef]
- Hou, G.; Yuan, X.; Li, Y.; Hou, G.; Liu, X. Cardamonin, a natural chalcone, reduces 5-fluorouracil resistance of gastric cancer cells through targeting Wnt/β-catenin signal pathway. Invest. New Drugs 2019. [Google Scholar] [CrossRef]
- Jeddi, F.; Soozangar, N.; Sadeghi, M.R.; Somi, M.H.; Shirmohamadi, M.; Eftekhar-Sadat, A.T.; Samadi, N. Nrf2 overexpression is associated with P-glycoprotein upregulation in gastric cancer. Biomed. Pharmacother. 2018, 97, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.W.; Xu, L.; Hao, J.H.; Qin, C.Y.; Liu, H. Expression of P-glycoprotein and multidrug resistance-associated protein is associated with multidrug resistance in gastric cancer. J. Int. Med. Res. 2010, 38, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Lacueva, J.; Perez-Ramos, M.; Soto, J.L.; Oliver, I.; Andrada, E.; Medrano, J.; Perez-Vazquez, T.; Arroyo, A.; Carrato, A.; Ferragut, J.A.; et al. Multidrug resistance-associated protein (MRP1) gene is strongly expressed in gastric carcinomas. Analysis by immunohistochemistry and real-time quantitative RT-PCR. Histopathology 2005, 46, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xing, X.; Shan, F.; Li, S.; Li, Z.; Xiao, A.; Xing, Z.; Xue, K.; Li, Z.; Hu, Y.; et al. ABCC2-24C > T polymorphism is associated with the response to platinum/5-Fu-based neoadjuvant chemotherapy and better clinical outcomes in advanced gastric cancer patients. Oncotarget 2016, 7, 55449–55457. [Google Scholar] [CrossRef] [PubMed]
- Mackey, J.R.; Mani, R.S.; Selner, M.; Mowles, D.; Young, J.D.; Belt, J.A.; Crawford, C.R.; Cass, C.E. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998, 58, 4349–4357. [Google Scholar]
- Kobayashi, Y.; Ohshiro, N.; Sakai, R.; Ohbayashi, M.; Kohyama, N.; Yamamoto, T. Transport mechanism and substrate specificity of human organic anion transporter 2 (hOat2 [SLC22A7]). J. Pharm. Pharmacol. 2005, 57, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Shimakata, T.; Kamoshida, S.; Kawamura, J.; Ogane, N.; Kameda, Y.; Yanagita, E.; Itoh, T.; Takeda, R.; Naka, A.; Sakamaki, K.; et al. Immunohistochemical expression profiles of solute carrier transporters in alpha-fetoprotein-producing gastric cancer. Histopathology 2016, 69, 812–821. [Google Scholar] [CrossRef]
- McIlwain, C.C.; Townsend, D.M.; Tew, K.D. Glutathione S-transferase polymorphisms: Cancer incidence and therapy. Oncogene 2006, 25, 1639–1648. [Google Scholar] [CrossRef]
- Peters, W.H.; Wormskamp, N.G.; Thies, E. Expression of glutathione S-transferases in normal gastric mucosa and in gastric tumors. Carcinogenesis 1990, 11, 1593–1596. [Google Scholar] [CrossRef]
- Satta, T.; Isobe, K.; Yamauchi, M.; Nakashima, I.; Takagi, H. Expression of MDR1 and glutathione S transferase-pi genes and chemosensitivities in human gastrointestinal cancer. Cancer 1992, 69, 941–946. [Google Scholar] [CrossRef]
- Monden, N.; Abe, S.; Sutoh, I.; Hishikawa, Y.; Kinugasa, S.; Nagasue, N. Prognostic significance of the expressions of metallothionein, glutathione-S-transferase-pi, and P-glycoprotein in curatively resected gastric cancer. Oncology 1997, 54, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Geng, M.; Wang, L.; Chen, X.; Cao, R.; Li, P. The association between chemosensitivity and Pgp, GST-π and Topo II expression in gastric cancer. Diagn. Pathol. 2013, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Schipper, D.L.; Wagenmans, M.J.; Peters, W.H.; Wils, J.A.; Wagener, D.J. Glutathione S-transferases and iododeoxyuridine labelling index during chemotherapy of gastric cancer. Anticancer Res. 2000, 20, 1705–1710. [Google Scholar] [PubMed]
- Kodera, Y.; Akiyama, S.; Isobe, K.; Kondo, K.; Ito, K.; Yamauchi, M.; Takagi, H. Expression of pi-glutathione S-transferase gene (GSTP1) in gastric cancer: Lack of correlation with resistance against cis-diamminedichloroplatinum (II). Eur. J. Cancer 1994, 30A, 2158–2162. [Google Scholar] [CrossRef]
- Nagano, O.; Okazaki, S.; Saya, H. Redox regulation in stem-like cancer cells by CD44 variant isoforms. Oncogene 2013, 32, 5191–5198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, G.J. The heterogeneity of cancer stem-like cells at the invasive front. Cancer Cell Int. 2017, 17, 23. [Google Scholar] [CrossRef] [Green Version]
- Hirata, K.; Suzuki, H.; Imaeda, H.; Matsuzaki, J.; Tsugawa, H.; Nagano, O.; Asakura, K.; Saya, H.; Hibi, T. CD44 variant 9 expression in primary early gastric cancer as a predictive marker for recurrence. Br. J. Cancer 2013, 109, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 2011, 19, 387–400. [Google Scholar] [CrossRef]
- Shitara, K.; Doi, T.; Nagano, O.; Imamura, C.K.; Ozeki, T.; Ishii, Y.; Tsuchihashi, K.; Takahashi, S.; Nakajima, T.E.; Hironaka, S.; et al. Dose-escalation study for the targeting of CD44v+ cancer stem cells by sulfasalazine in patients with advanced gastric cancer (EPOC1205). Gastric Cancer 2017, 20, 341–349. [Google Scholar] [CrossRef]
- Shitara, K.; Doi, T.; Nagano, O.; Fukutani, M.; Hasegawa, H.; Nomura, S.; Sato, A.; Kuwata, T.; Asai, K.; Einaga, Y.; et al. Phase 1 study of sulfasalazine and cisplatin for patients with CD44v-positive gastric cancer refractory to cisplatin (EPOC1407). Gastric Cancer 2017, 20, 1004–1009. [Google Scholar] [CrossRef]
- Matsuhashi, N.; Saio, M.; Matsuo, A.; Sugiyama, Y.; Saji, S. Apoptosis induced by 5-fluorouracil, cisplatin and paclitaxel are associated with p53 gene status in gastric cancer cell lines. Int. J. Oncol. 2005, 26, 1563–1567. [Google Scholar] [CrossRef] [PubMed]
- Le, T.V.T.; Seo, Y.; Ryu, C.J.; Lee, H.R.; Park, H.J. Increased expression of p27 is associated with the cisplatin resistance in gastric cancer cell line YCC-3. Arch. Pharm. Res. 2010, 33, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.X.; Zheng, L.H.; Liu, S.Y.; He, X.H. rAd-p53 enhances the sensitivity of human gastric cancer cells to chemotherapy. World J. Gastroenterol. 2011, 17, 4289–4297. [Google Scholar] [CrossRef] [PubMed]
- Cascinu, S.; Graziano, F.; Del Ferro, E.; Staccioli, M.P.; Ligi, M.; Carnevali, A.; Muretto, P.; Catalano, G. Expression of p53 protein and resistance to preoperative chemotherapy in locally advanced gastric carcinoma. Cancer 1998, 83, 1917–1922. [Google Scholar] [CrossRef]
- Xu, H.Y.; Xu, W.L.; Wang, L.Q.; Chen, M.B.; Shen, H.L. Relationship between p53 status and response to chemotherapy in patients with gastric cancer: A meta-analysis. PLoS ONE 2014, 9, e95371. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lu, H.; Lv, M.; Wang, Q.; Sun, Y. Parthenolide facilitates apoptosis and reverses drug-resistance of human gastric carcinoma cells by inhibiting the STAT3 signaling pathway. Oncol. Lett. 2018, 15, 3572–3579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Song, C.; Wang, J.; Tang, H.; Peng, Z.; Lu, S. HOXA13 contributes to gastric carcinogenesis through DHRS2 interacting with MDM2 and confers 5-FU resistance by a p53-dependent pathway. Mol. Carcinog. 2018, 57, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, L.; Huang, C. Downregulation of Inhibition of Apoptosis-Stimulating Protein of p53 (iASPP) Suppresses Cisplatin-Resistant Gastric Carcinoma In Vitro. Med. Sci. Monit. 2017, 23, 5542–5549. [Google Scholar] [CrossRef] [Green Version]
- Mori, J.; Tanikawa, C.; Ohnishi, N.; Funauchi, Y.; Toyoshima, O.; Ueda, K.; Matsuda, K. EPSIN 3, A novel p53 target, regulates the apoptotic pathway and gastric carcinogenesis. Neoplasia 2017, 19, 185–195. [Google Scholar] [CrossRef]
- Subhash, V.V.; Tan, S.H.; Tan, W.L.; Yeo, M.S.; Xie, C.; Wong, F.Y.; Kiat, Z.Y.; Lim, R.; Yong, W.P. GTSE1 expression represses apoptotic signaling and confers cisplatin resistance in gastric cancer cells. BMC Cancer 2015, 15, 550. [Google Scholar] [CrossRef]
- Endo, F.; Nishizuka, S.S.; Kume, K.; Ishida, K.; Katagiri, H.; Ishida, K.; Sato, K.; Iwaya, T.; Koeda, K.; Wakabayashi, G. A compensatory role of NF-κB to p53 in response to 5-FU-based chemotherapy for gastric cancer cell lines. PLoS ONE 2014, 9, e90155. [Google Scholar] [CrossRef] [PubMed]
- Zoppoli, P.; Calice, G.; Laurino, S.; Ruggieri, V.; La Rocca, F.; La Torre, G.; Ciuffi, M.; Amendola, E.; De Vita, F.; Petrillo, A.; et al. TRPV2 calcium channel gene expression and outcomes in gastric cancer patients: A clinically relevant association. J. Clin. Med. 2019, 8, 662. [Google Scholar] [CrossRef] [PubMed]
- Tanida, S.; Mizoshita, T.; Ozeki, K.; Tsukamoto, H.; Kamiya, T.; Kataoka, H.; Sakamuro, D.; Joh, T. Mechanisms of Cisplatin-Induced Apoptosis and of Cisplatin Sensitivity: Potential of BIN1 to Act as a Potent Predictor of Cisplatin Sensitivity in Gastric Cancer Treatment. Int. J. Surg. Oncol. 2012, 2012, 862879. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, M.; Sato, Y.; Ohnuma, H.; Takayama, T.; Sagawa, T.; Nobuoka, T.; Harada, K.; Miyamoto, H.; Sato, Y.; Takahashi, Y.; et al. A phase II study of neoadjuvant combination chemotherapy with docetaxel, cisplatin, and S-1 for locally advanced resectable gastric cancer: Nucleotide excision repair (NER) as potential chemoresistance marker. Cancer Chemother. Pharmacol. 2013, 71, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Ning, H.; Li, T.; Zhao, L.; Li, T.; Li, J.; Liu, J.; Liu, Z.; Fan, D. TRF2 promotes multidrug resistance in gastric cancer cells. Cancer Biol. Ther. 2006, 5, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, P.F.; Geng, M.; Cao, Y.C.; Yin, Y.C. Correlation between chemosensitivity to anticancer drugs and telomerase reverse transcriptase mRNA expression in gastric cancer. Diagn. Pathol. 2013, 8, 33. [Google Scholar] [CrossRef]
- Fattahi, S.; Golpour, M.; Amjadi-Moheb, F.; Sharifi-Pasandi, M.; Khodadadi, P.; Pilehchian-Langroudi, M.; Ashrafi, G.H.; Akhavan-Niaki, H. DNA methyltransferases and gastric cancer: Insight into targeted therapy. Epigenomics 2018, 10, 1477–1497. [Google Scholar] [CrossRef]
- Gao, Y.; Cui, J.; Xi, H.; Cai, A.; Shen, W.; Li, J.; Zhang, K.; Wei, B.; Chen, L. Association of thymidylate synthase expression and clinical outcomes of gastric cancer patients treated with fluoropyrimidine-based chemotherapy: A meta-analysis. Onco Targets Ther. 2016, 9, 1339–1350. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russi, S.; Verma, H.K.; Laurino, S.; Mazzone, P.; Storto, G.; Nardelli, A.; Zoppoli, P.; Calice, G.; La Rocca, F.; Sgambato, A.; et al. Adapting and Surviving: Intra and Extra-Cellular Remodeling in Drug-Resistant Gastric Cancer Cells. Int. J. Mol. Sci. 2019, 20, 3736. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153736
Russi S, Verma HK, Laurino S, Mazzone P, Storto G, Nardelli A, Zoppoli P, Calice G, La Rocca F, Sgambato A, et al. Adapting and Surviving: Intra and Extra-Cellular Remodeling in Drug-Resistant Gastric Cancer Cells. International Journal of Molecular Sciences. 2019; 20(15):3736. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153736
Chicago/Turabian StyleRussi, Sabino, Henu Kumar Verma, Simona Laurino, Pellegrino Mazzone, Giovanni Storto, Anna Nardelli, Pietro Zoppoli, Giovanni Calice, Francesco La Rocca, Alessandro Sgambato, and et al. 2019. "Adapting and Surviving: Intra and Extra-Cellular Remodeling in Drug-Resistant Gastric Cancer Cells" International Journal of Molecular Sciences 20, no. 15: 3736. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153736