Comparative Analysis of two Sugarcane Ancestors Saccharum officinarum and S. spontaneum based on Complete Chloroplast Genome Sequences and Photosynthetic Ability in Cold Stress


Abstract
:1. Introduction
2. Results
2.1. Quality and Quantity of Raw Data and Clean Data Obtained from Sequencing
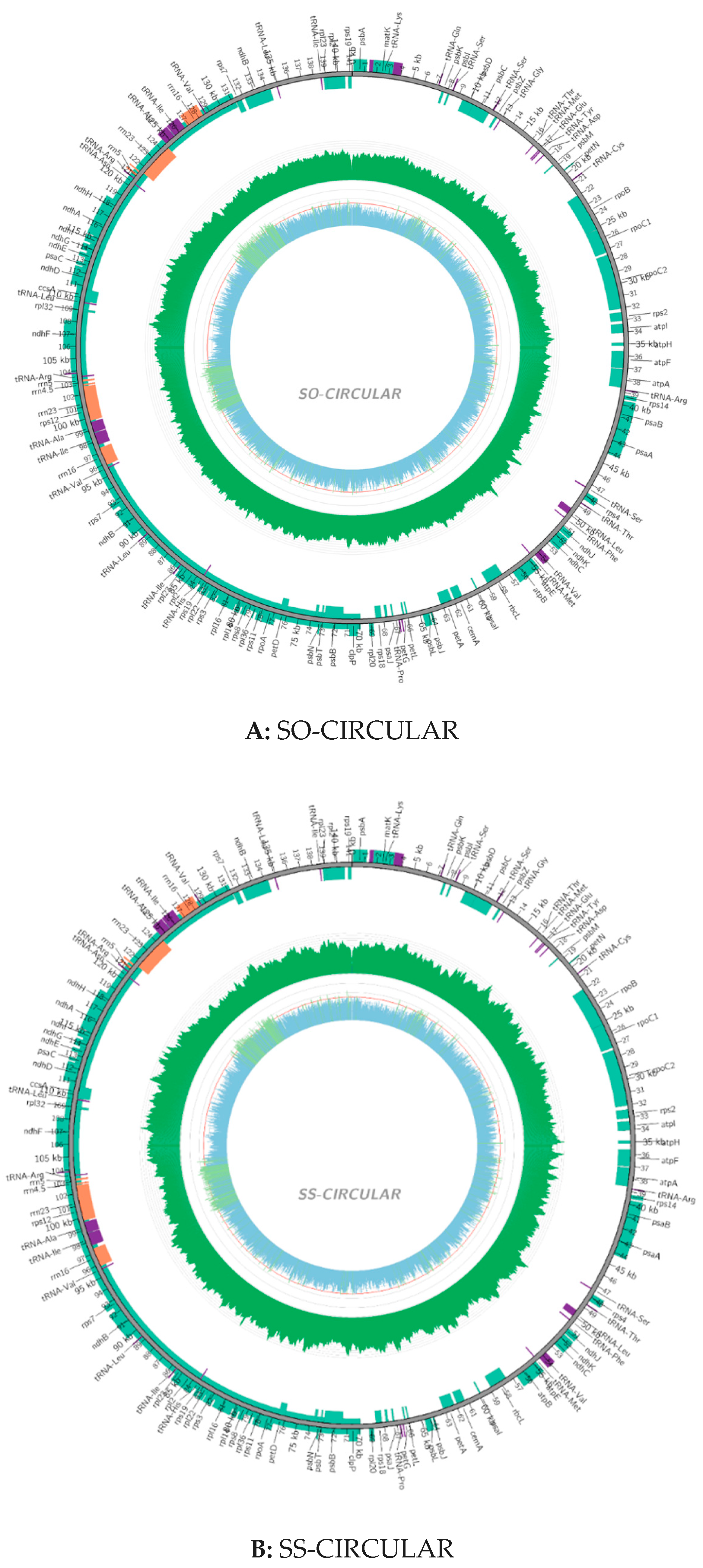
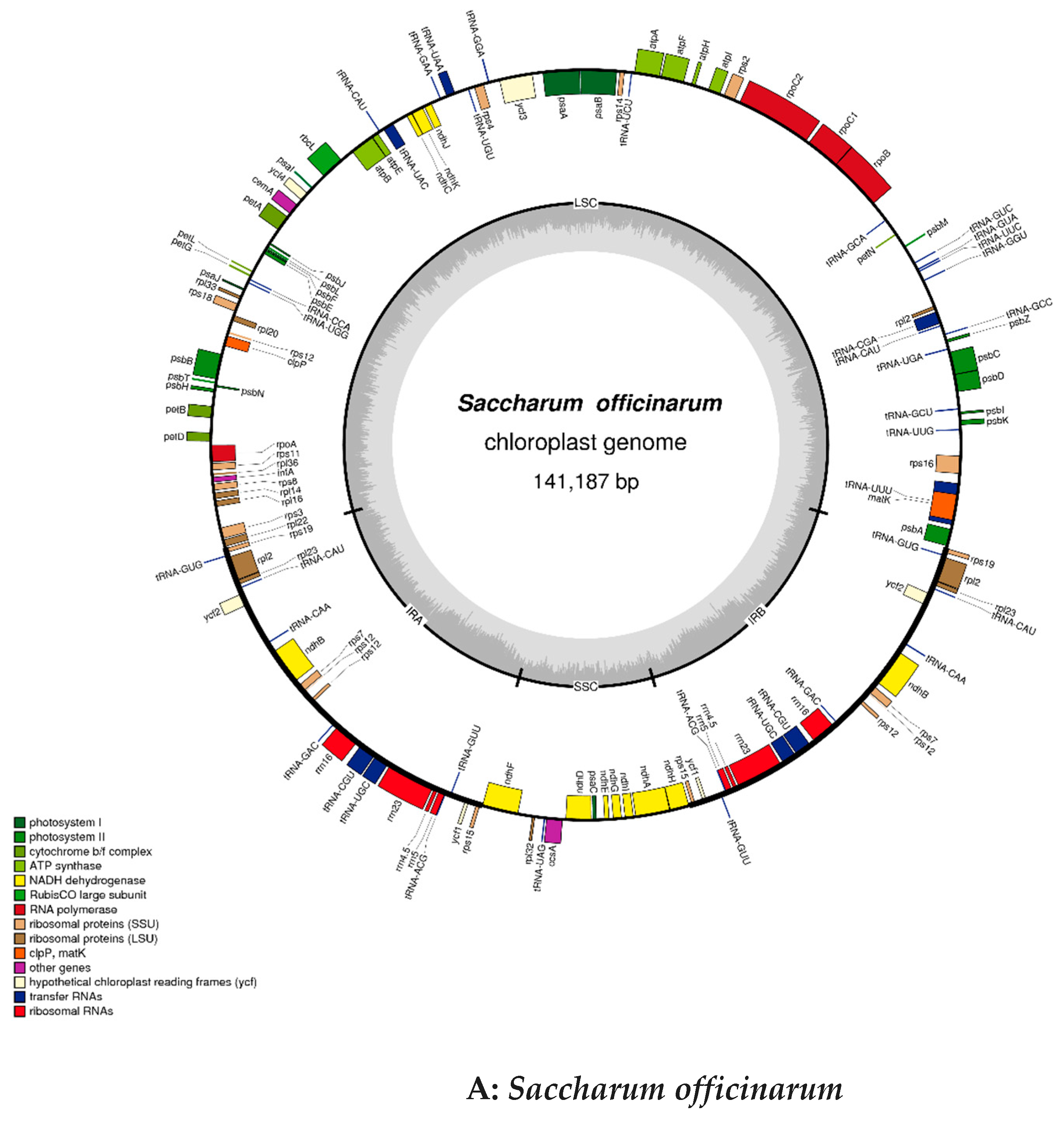
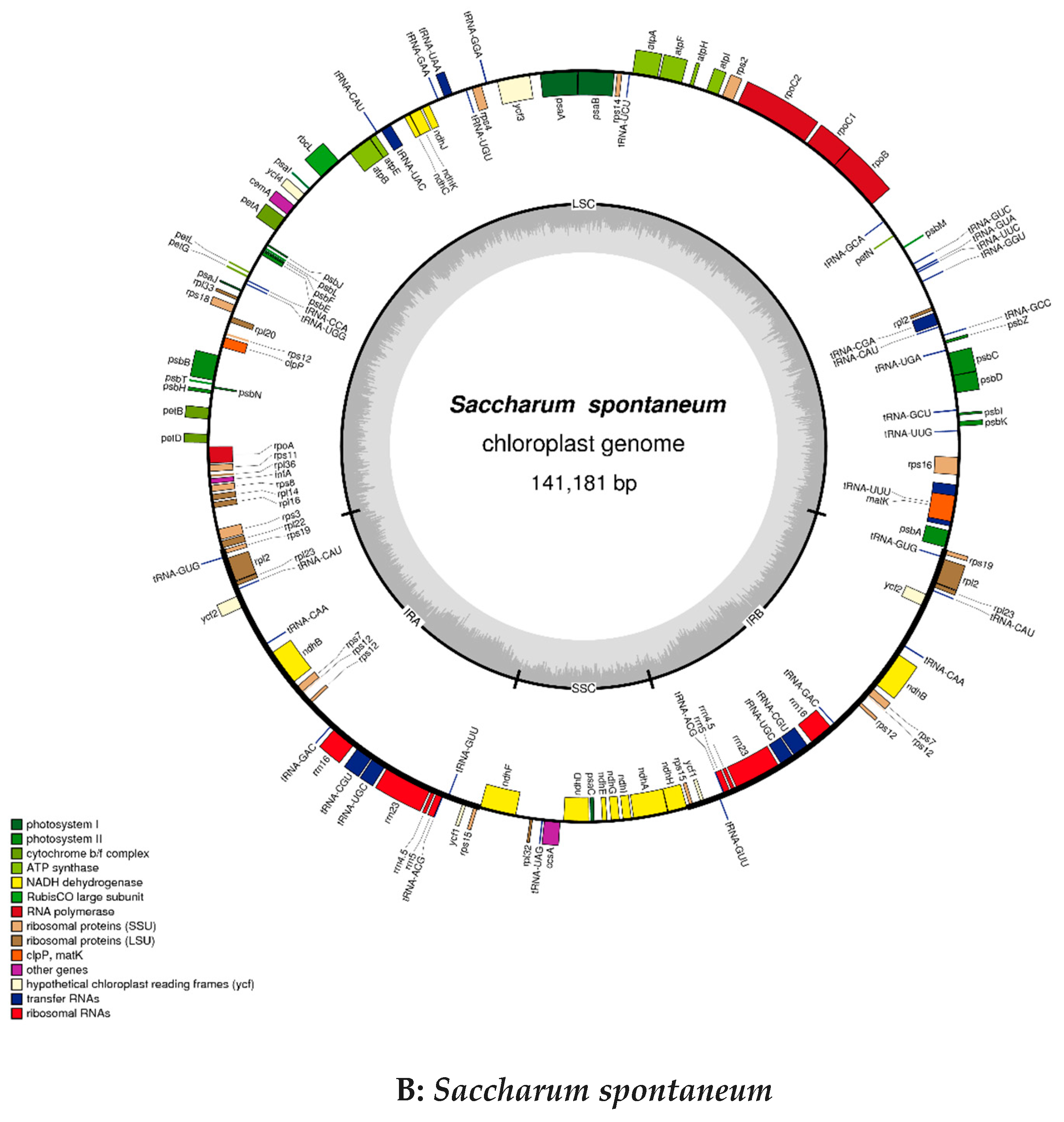
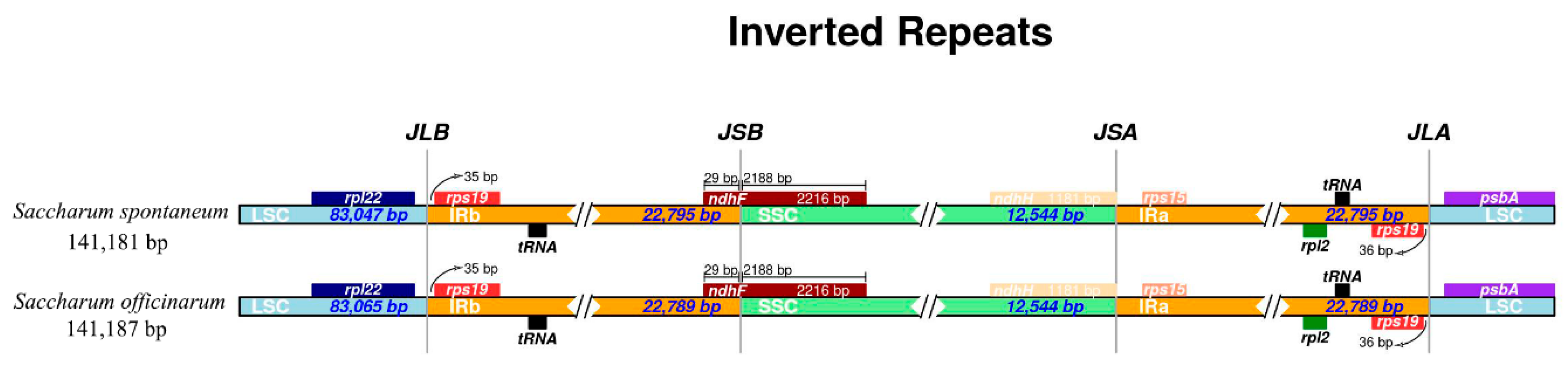
2.2. Chloroplast Genomic Structure and Gene Diversification
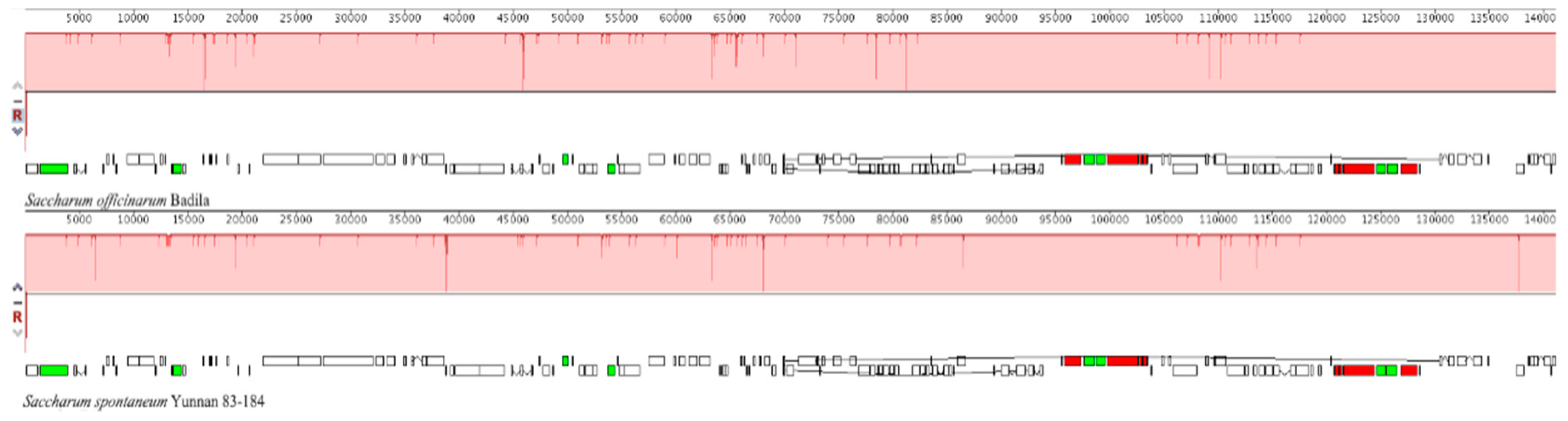
2.3. The Collinear Analysis
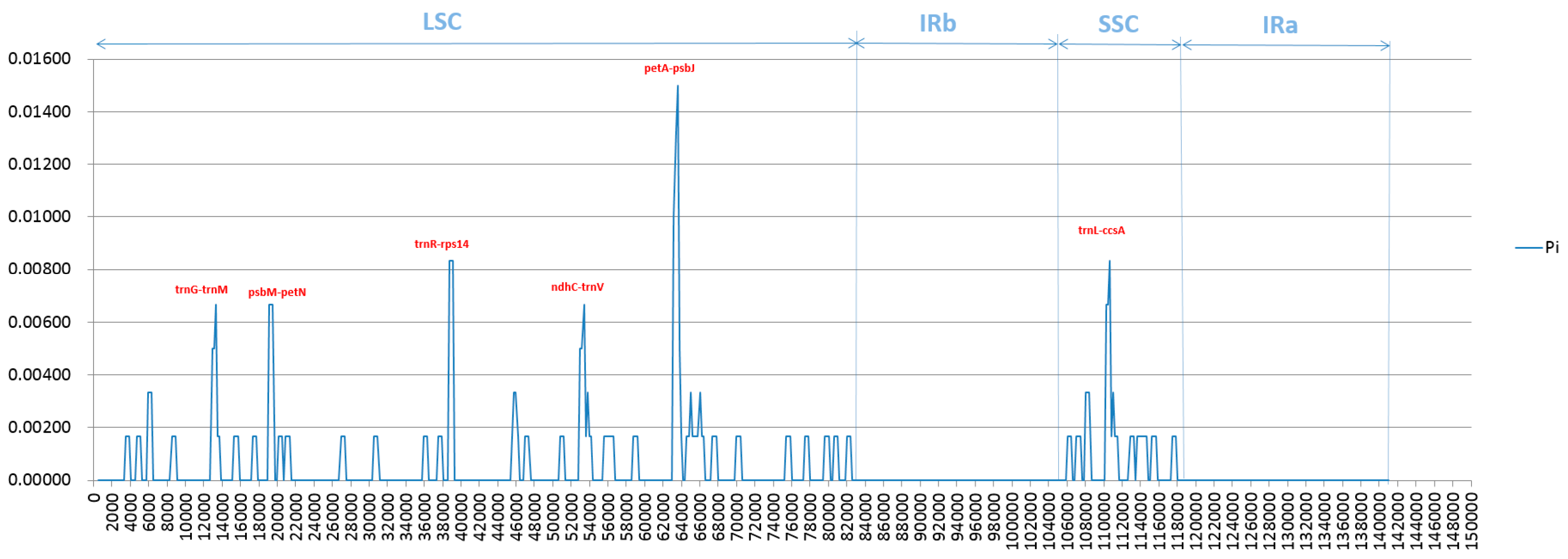
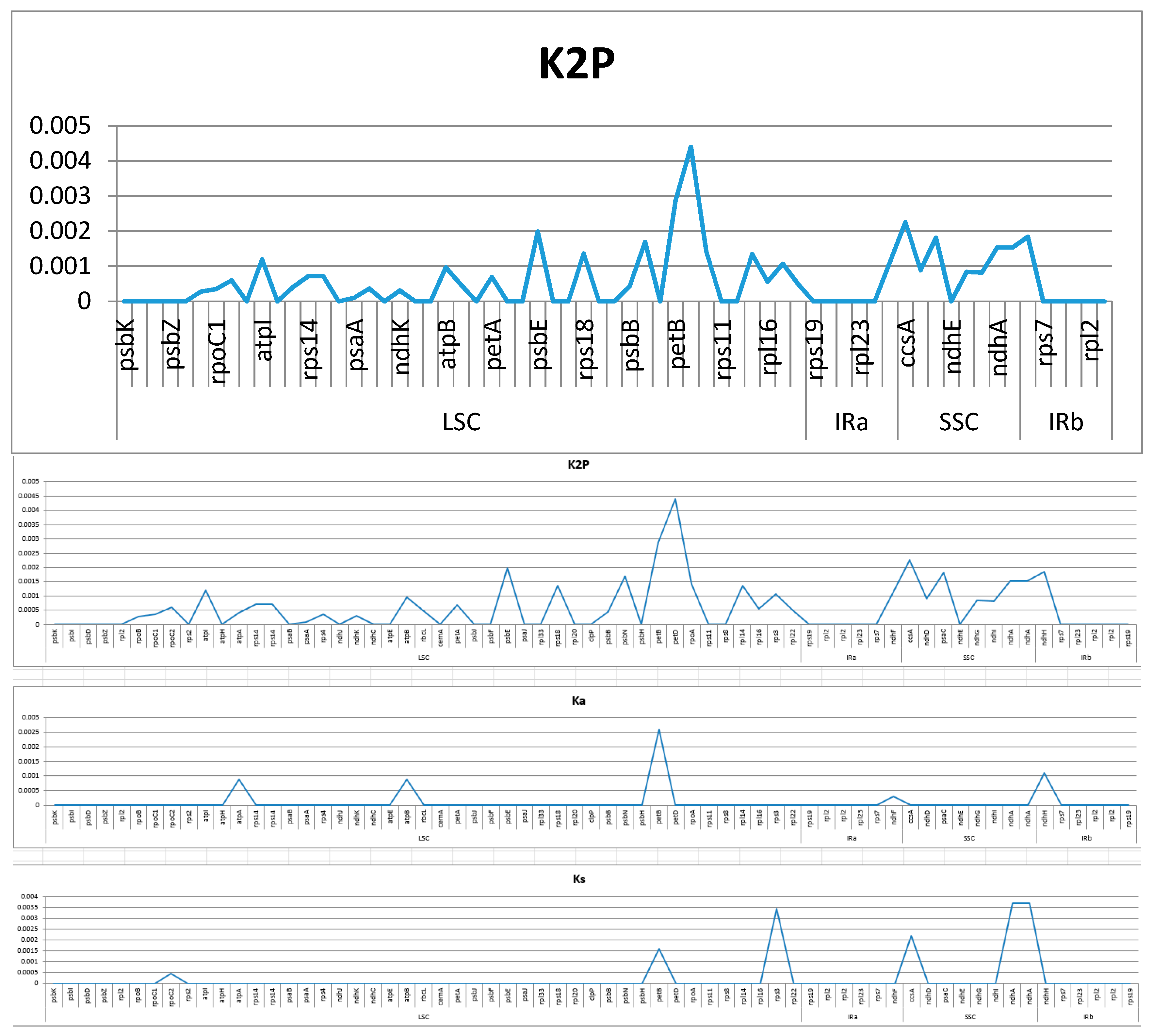
2.4. Analysis of Highly Variable Regions and Base Substitutions in CDS Genes
2.5. Coding Capacity of Protein Coding Genes (PCGs) and Relative Synonymous Codon Usage (RSCU)
2.6. Analysis on IR Junctions Proline, Serine, Glutamine, Glycine, Alanine and/or Asparagine
2.7. Repeat Structure Analysis
2.8. Photosynthetic Ability Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Material, Sample Collection and DNA Preparation
4.2. DNA Sequencing and Genome Assembly
4.3. Comparison of Complete cp Genomes, Genomic Annotation and Analysis
4.4. Repeat Structure Analysis
4.5. Photosynthetic Parameter Measurement and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; Depamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.; Peery, R.; Herman, S.J.; et al. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 2005, 395, 348–384. [Google Scholar] [PubMed]
- Curci, P.L.; De Paola, D.; Danzi, D.; Vendramin, G.G.; Sonnante, G. Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other asteraceae. PLoS ONE 2015, 10, e0120589. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Y.F.; Zang, M.Y.; Li, M.Z.; Fang, Y.M. Complete chloroplast genome sequence and phylogenetic analysis of Quercus acutissima. Int. J. Mol. Sci. 2018, 19, 2443. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D. Comparative organization of chloroplast genomes. Annu. Rev. Genet. 1985, 19, 325–354. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.H.; Abe, J.; Sakai, M.; Kanazawa, A.; Shimamoto, Y. Sequence variation of non-coding regions of chloroplast DNA of soybean and related wild species and its implications for the evolution of different chloroplast haplotypes. Theor. Appl. Genet. 2000, 101, 724–732. [Google Scholar] [CrossRef]
- Hoang, N.V.; Furtado, A.; McQualter, R.B.; Henry, R.J. Next generation sequencing of total DNA from sugarcane provides no evidence for chloroplast heteroplasmy. New Negat. Plant Sci. 2015, 1, 33–45. [Google Scholar] [CrossRef]
- Su, Y.; Huang, L.; Wang, Z.; Wang, T. Comparative chloroplast genomics between the invasive weed Mikania micrantha and its indigenous congener Mikania cordata: Structure variation, identification of highly divergent regions, divergence time estimation, and phylogenetic analysis. Mol. Phylogenet. Evol. 2018, 126, 181–195. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Li, W.H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef]
- Ingvarsson, P.K.; Ribstein, S.; Taylor, D.R. Molecular evolution of insertions and deletion in the chloroplast genome of Silene. Mol. Biol. Evol. 2003, 20, 1737–1740. [Google Scholar] [CrossRef]
- Martin, G.E.; Rousseau-Gueutin, M.; Cordonnier, S.; Lima, O.; Michon-Coudouel, S.; Naquin, D.; De Carvalho, J.F.; Ainouche, A.; Salmon, A.; Ainouche, A. The first complete chloroplast genome of the genistoid legume Lupinus Luteus: Evidence for a novel major lineage-specific rearrangement and new insights regarding plastome evolution in the legume family. Ann. Bot. 2014, 113, 1197–1210. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Ma, J.; Yang, B.X.; Li, R.Y.; Zhu, W.; Sun, L.L.; Tian, J.K.; Zhang, L. The complete chloroplast genome sequence of Taxus chinensis var. mairei (Taxaceae): Loss of an inverted repeat region and comparative analysis with related species. Gene 2014, 540, 201–209. [Google Scholar] [CrossRef]
- Group, C.P.W.; Hollingsworth, P.M.; Forrest, L.L.; Spouge, J.L.; Hajibabaei, M.; Ratnasingham, S.; Fazekas, A.J. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar] [CrossRef] [Green Version]
- Atherton, R.A.; McComish, B.J.; Shepherd, L.D.; Berry, L.A.; Albert, N.W.; Lockhart, P.J. Whole genome sequencing of enriched chloroplast DNA using the Illumina GAII platform. Plant Methods 2010, 6, 22. [Google Scholar] [CrossRef]
- Doorduin, L.; Gravendeel, B.; Lammers, Y.; Ariyurek, Y.; Chin-A-Woeng, T.; Vrieling, K. The complete chloroplast genome of 17 individuals of pest species Jacobaea vulgaris: SNPs, microsatellites and barcoding markers for population and phylogenetic studies. DNA Res. 2011, 18, 93–105. [Google Scholar] [CrossRef]
- Awad, M.; Fahmy, R.M.; Mosa, K.A.; Helmy, M.; El-Feky, F.A. Identification of effective DNA barcodes for Triticum plants through chloroplast genome-wide analysis. Comput. Biol. Chem. 2017, 71, 20–31. [Google Scholar] [CrossRef]
- Chen, X.L.; Zhou, J.G.; Cui, Y.X.; Wang, Y.; Duan, B.Z.; Yao, H. Identification of Ligularia herbs using the complete chloroplast genome as a super-barcode. Front. Pharmacol. 2018, 9, 695. [Google Scholar] [CrossRef]
- Yousefzadeh, H.; Khodadost, A.; Abdollahi, H.; Ali, S.S.; Kozlowski, G.; Bina, H. Biogeography and phylogenetic relationships of Hyrcanian wild apple using cpDNA and ITS noncoding sequences. Syst. Biodivers. 2019. [Google Scholar] [CrossRef]
- Niu, Y.T.; Jabbour, F.; Barrett, R.L.; Ye, J.F.; Zhang, Z.Z.; Lu, K.Q.; Lu, L.M.; Chen, Z.D. Combining complete chloroplast genome sequences with target loci data and morphology to resolve species limits in Triplostegia (Caprifoliaceae). Mol. Phylogenet. Evol. 2018, 129, 15–26. [Google Scholar] [CrossRef]
- D’Hont, A.; Paulet, F.; Glaszmann, J.C. Oligoclonal interspecific origin of ‘North Indian’ and ‘Chinese’ sugarcanes. Chromosome Res. 2002, 10, 253–262. [Google Scholar] [CrossRef]
- Zhang, J.S.; Zhang, Q.; Li, L.T.; Tang, H.B.; Zhang, Q.; Chen, Y.; Arrow, J.; Zhang, X.T.; Wang, A.Q.; Miao, C.Y.; et al. Recent polyploidization events in three Saccharum founding species. Plant Biotechnol. J. 2018, 17, 264–274. [Google Scholar] [CrossRef]
- D’Hont, A. Unraveling the genome structure of polyploids using FISH and GISH; examples of sugarcane and banana. Cytogenet. Genome Res. 2005, 109, 27–33. [Google Scholar] [CrossRef]
- Zhang, J.S.; Zhang, X.T.; Tang, H.B.; Zhang, Q.; Hua, X.T.; Ma, X.K.; Zhu, F.; Jones, T.; Zhu, X.G.; Bowers, J.; et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 2018, 50, 1565–1573. [Google Scholar] [CrossRef]
- Garsmeur, O.; Droc, G.; Rudie, A.; Grimwood, J.; Potier, B.; Aitken, K.S.; Jenkins, J.; Martin, G.; Charron, C.; Hervouet, C.; et al. A mosaic monoploid reference sequence for the highly complex genome of sugarcane. Nat. Commun. 2018, 9, 2638. [Google Scholar] [CrossRef]
- Irvine, J.E. Saccharum species as horticultural classes. Theor. Appl. Genet. 1999, 98, 186–194. [Google Scholar] [CrossRef]
- D’Hont, A.; Ison, D.; Alix, K.; Roux, C.; Glaszmann, J.C. Determination of basic chromosome numbers in the genus Saccharum by physical mapping of ribosomal RNA genes. Genome 1998, 41, 221–225. [Google Scholar] [CrossRef]
- Ha, S.; Moore, P.H.; Heinz, D.; Kato, S.; Ohmido, N.; Fukui, K. Quantitative chromosome map of the polyploid Saccharum spontaneum by multicolor fluorescence in situ hybridization and imaging methods. Plant Mol. Biol. 1999, 39, 1165–1173. [Google Scholar] [CrossRef]
- Asano, T.; Tsudzuki, T.; Takahashi, S.; Shimada, H.; Kadowaki, K. Complete nucleotide sequence of the sugarcane (Saccharum officinarum) chloroplast genome: A comparative analysis of four monocot chloroplast genomes. DNA Res. 2004, 11, 93–99. [Google Scholar] [CrossRef]
- Júnior, T.C.; Carraro, D.M.; Benatti, M.R.; Barbosa, A.C.; Kitajima, J.P.; Carrer, H. Structural features and transcript-editing analysis of sugarcane (Saccharum officinarum L.) chloroplast genome. Curr. Genet. 2004, 46, 366–373. [Google Scholar] [CrossRef]
- Vidigal, P.M.P.; Coelho, A.S.G.; Novaes, E.; Barbosa, M.H.P.; Peternelli, L.A. Complete chloroplast genome sequence and annotation of the Saccharum hybrid cultivar RB867515. Genome Announc. 2016, 4. [Google Scholar] [CrossRef]
- Evans, D.L.; Joshi, S.V. Complete chloroplast genomes of Saccharum spontaneum, Saccharum officinarum and Miscanthus floridulus (Panicoideae: Andropogoneae) reveal the plastid view on sugarcane origins. Syst. Biodivers. 2016, 14, 548–571. [Google Scholar] [CrossRef]
- Dutta, S.; Mohanty, S.; Tripathy, B.C. Role of temperature stress on chloroplast biogenesis and protein import in pea. Plant Physiol. 2009, 150, 1050–1061. [Google Scholar] [CrossRef]
- Taylor, N.L.; Tan, Y.F.; Jacoby, R.P.; Millar, A.H. Abiotic environmental stress induced changes in the Arabidopsis thaliana chloroplast, mitochondria and peroxisome proteomes. J. Proteom. 2009, 72, 367–378. [Google Scholar] [CrossRef]
- Ling, Q.H.; Jarvis, P. Regulation of chloroplast protein import by the ubiquitin E3 ligase SP1 is important for stress tolerance in plants. Curr. Biol. 2015, 25, 2527–2534. [Google Scholar] [CrossRef]
- Dinha, S.N.; Sai, T.Z.T.; Nawaz, G.; Leea, K.; Kanga, H. Abiotic stresses affect differently the intron splicing and expression of chloroplast genes in coffee plants (Coffea arabica) and rice (Oryza sativa). J. Plant Physiol. 2016, 201, 85–94. [Google Scholar] [CrossRef]
- Wang, S.; Bai, G.; Wang, S.; Yang, L.Y.; Yang, F.; Wang, Y.; Zhu, J.K.; Hua, J. Chloroplast RNA-binding protein RBD1 promotes chilling tolerance through 23S rRNA processing in Arabidopsis. PLoS Genet. 2016, 12, e1006027. [Google Scholar] [CrossRef]
- Nawaz, G.; Lee, K.; Park, S.J.; Kim, Y.O.; Kang, H. A chloroplast-targeted cabbage DEAD-box RNA helicase BrRH22 confers abiotic stress tolerance to transgenic Arabidopsis plants by affecting translation of chloroplast transcripts. Plant Physiol. Biochem. 2018, 127, 336–342. [Google Scholar] [CrossRef]
- Serrano, I.; Audran, C.; Rivas, S. Chloroplasts at work during plant innate immunity. J. Exp. Bot. 2016, 67, 3845–3854. [Google Scholar] [CrossRef]
- Browse, J.; Xin, Z. Temperature sensing and cold acclimation. Curr. Opin. Plant Biol. 2001, 4, 241–246. [Google Scholar] [CrossRef]
- Berry, J.; Bjorkman, O. Photosynthetic response and adaptation to temperature in higher plants. Annu. Rev. Plant Physiol. 1980, 31, 491–543. [Google Scholar] [CrossRef]
- Yang, Y.; Yu, Q.; Yang, Y.; Su, Y.; Ahmad, W.; Guo, J.; Gao, S.; Xu, L.; Que, Y. Identification of cold-related miRNAs in sugarcane by small RNA sequencing and functional analysis of a cold inducible ScmiR393 to cold stress. Environ. Exp. Bot. 2018, 155, 464–476. [Google Scholar] [CrossRef]
- Zhang, B.Q.; Yang, L.T.; Li, Y.R. Physiological and biochemical characteristics related to cold resistance in Sugarcane. Sugar Technol. 2015, 17, 49–58. [Google Scholar] [CrossRef]
- Nogueira, F.T.; De Rosa, V.E.; Menossi, M.; Ulian, E.C.; Arruda, P. RNA expression profiles and data mining of sugarcane response to low temperature. Plant Physiol. 2003, 132, 1811–1824. [Google Scholar] [CrossRef]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef]
- Wang, X.M.; Zhou, T.; Bai, G.Q.; Zhao, Y.M. Complete chloroplast genome sequence of Fagopyrum dibotrys: Genome features, comparative analysis and phylogenetic relationships. Sci. Rep. 2018, 8, 12379. [Google Scholar] [CrossRef]
- Dong, W.P.; Xu, C.; Li, C.H.; Sun, J.H.; Zuo, Y.J.; Shi, S.; Cheng, T.; Guo, J.J.; Zhou, S.L. Ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef]
- Wang, C.L.; Ding, M.Q.; Zou, C.Y.; Zhu, X.M.; Tang, Y.; Zhou, M.L.; Shao, J.R. Comparative analysis of four buckwheat species based on morphology and complete chloroplast genome sequences. Sci. Rep. 2017, 7, 6514. [Google Scholar] [CrossRef]
- Song, Y.; Dong, W.P.; Liu, B.; Xu, C.; Yao, X.; Gao, J.; Corlett, R.T. Comparative analysis of complete chloroplast genome sequences of two tropical trees Machilus yunnanensis and Machilus balansae in the family Lauraceae. Front. Plant Sci. 2015, 6, 662. [Google Scholar] [CrossRef]
- Chung, H.J.; Jung, J.D.; Park, H.W.; Kim, J.H.; Cha, H.W.; Min, S.R.; Jeong, W.J.; Liu, J.R. The complete chloroplast genome sequences of Solanum tuberosum, and comparative analysis with Solanaceae species identified the presence of a 241-bp deletion in cultivated potato chloroplast DNA sequence. Plant Cell Rep. 2006, 25, 1369–1379. [Google Scholar] [CrossRef]
- Xue, Z.Q.; Xue, J.H.; Victorovna, K.M.; Ma, K.P. The complete chloroplast DNA sequence of Trapa maximowiczii Korsh. (Trapaceae), and comparative analysis with other Myrtales species. Aquat. Bot. 2017, 143, 54–62. [Google Scholar] [CrossRef]
- Saski, C.; Lee, S.B.; Fjellheim, S.; Guda, C.; Jansen, R.K.; Lou, H.; Tomkins, J.; Rognli, O.A.; Daniell, H.; Clarke, J.L. Compete chloroplast genome sequences of Hordeum vulgare, Sorghum bicolor and Agrostis stolonifera, and comparative analysis with other grass genomes. Theo. Appl. Genet. 2007, 115, 571–590. [Google Scholar] [CrossRef]
- Park, I.; Yang, S.Y.; Kim, W.J.; Noh, P.; Lee, H.O.; Moon, B.C. The complete chloroplast genomes of six Ipomoea species and Indel marker development for the discrimination of authentic pharbitidis semen (seeds of I. nil or I. purpurea). Front. Plant Sci. 2018, 9, 965. [Google Scholar] [CrossRef]
- Nguyen, V.B.; Giang, V.N.L.; Waminal, N.E.; Park, H.S.; Kim, N.H.; Jang, W.; Lee, J.; Yang, T.J. Comprehensive comparative analysis of chloroplast genomes from seven Panax species and development of an authentication system based on species-unique single nucleotide polymorphism markers. J. Ginseng Res. 2018. [Google Scholar] [CrossRef]
- Li, Y.T.; Zhang, J.; Li, L.F.; Gao, L.J.; Xu, J.T.; Yang, M.S. Structural and comparative analysis of the complete chloroplast genome of Pyrus hopeiensis—“wild plants with a tiny population”—and three other Pyrus species. Int. J. Mol. Sci. 2018, 19, 3262. [Google Scholar] [CrossRef]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Holaday, A.S.; Martindale, W.; Alred, R.; Brooks, A.L.; Leegood, R.C. Changes in activities of enzymes of carbon metabolism in leaves during exposure of plants to low temperature. Plant Physiol. 1992, 98, 1105–1114. [Google Scholar] [CrossRef]
- Mohanty, S.; Grimm, B.; Tripathy, B.C. Light and dark modulation of chlorophyll biosynthetic genes in response to temperature. Planta 2006, 224, 692–699. [Google Scholar] [CrossRef]
- Tewari, A.K.; Tripathy, B.C. Acclimation of chlorophyll biosynthetic reactions to temperature stress in cucumber (Cucumis sativus L.). Planta 1999, 208, 431–437. [Google Scholar] [CrossRef]
- Allen, D.J.; Ort, D.R. Impact of chilling temperatures on photosynthesis in warm-climate plants. Trends Plant Sci. 2001, 6, 36–42. [Google Scholar] [CrossRef]
- Govindacharya, S.; Bukhov, N.G.; Joly, D.; Carpentier, R. Photosystem II inhibition by moderate light under low temperature in intact leaves of chilling-sensitive and tolerant plants. Physiol. Plant 2004, 121, 322–333. [Google Scholar] [CrossRef]
- Huner, N.P.A.; Öquist, G.; Hurry, V.M.; Krol, M.; Falk, S.; Griffith, M. Photosynthesis, photoinhibition and low temperature acclimation in cold tolerant plants. Photosynth. Res. 1993, 37, 19–39. [Google Scholar] [CrossRef]
- Wise, R.R.; Olson, A.J.; Schrader, S.M.; Sharkey, T.D. Electron transport is the functional limitation of photosynthesis in field-grown Pima cotton plants at high temperature. Plant Cell Environ. 2004, 27, 717–724. [Google Scholar] [CrossRef]
- Yu, J.Q.; Zhou, Y.H.; Huang, L.F.; Allen, D. Chill-induced inhibition of photosynthesis: Genotypic variation within Cucumis sativus. Plant Cell Physiol. 2002, 43, 1182–1188. [Google Scholar] [CrossRef]
- Zhou, Y.H.; Yu, J.Q.; Mao, W.H.; Huang, L.F.; Song, X.S.; Nogue’s, S. Genotypic variation of Rubisco expression, photosynthetic electron flow and antioxidant metabolism in the chloroplasts of chill-exposed cucumber plants. Plant Cell Physiol. 2006, 47, 192–199. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, S.; Jiang, Y.; Lin, Z.; Luo, J.; Li, M.; Guo, J.; Su, Y.; Xu, L.; Que, Y. The physiological and agronomic responses to nitrogen dosage in different sugarcane varieties. Front. Plant Sci. 2019, 10, 406. [Google Scholar] [CrossRef]
- Doyle, J.J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, 6–11. [Google Scholar] [CrossRef]
- Kimura, M.A. simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-seriesmethods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Melotto-Passarin, D.M.; Tambarussi, E.V.; Dressano, K.; De Martin, V.F.; Carrer, H. Characterization of chloroplast DNA microsatellites from Saccharum spp and related species. Genet. Mol. Res. 2011, 10, 2024–2033. [Google Scholar] [CrossRef]
- Dinh, T.H.; Watanabe, K.; Takaragawa, H.; Nakabaru, M.; Kawamitsu, Y. Photosynthetic response and nitrogen use efficiency of sugarcane under drought stress conditions with different nitrogen application levels. Plant Prod. Sci. 2017, 20, 412–422. [Google Scholar] [CrossRef] [Green Version]
- Shu, S.; Tang, Y.Y.; Yuan, Y.H.; Sun, J.; Zhong, M.; Guo, S.R. The role of 24-epibrassinolide in the regulation of photosynthetic characteristics and nitrogen metabolism of tomato seedlings under a combined low temperature and weak light stress. Plant Physiol. Biochem. 2016, 107, 344–353. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functions | Family Genes | List of Genes |
|---|---|---|
| Photosynthesis genes | Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ, ycf4 |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Subunits of Cytochrome b6/f/complex | petA, petB, petD, petG, petL, petN | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF 1, atpH, atpI | |
| Subunits of Rubisco | rbcL | |
| Subunits of NADH-dehydrogenase | ndhA1, ndhB1, *, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Self-replicating genes | Large subunit ribosomal proteins | rpl21, *, rpl14, rpl16, rpl20, rpl22, rpl23 *, rpl32, rpl33, rpl36 |
| Small subunit ribosomal proteins | rps2, rps3, rps4, rps7, rps8, rps11, rps12#, rps14, rps15, rps16 1, rps18, rps19 | |
| DNA dependent RNA polymerase | rpoA, B, C1, C2 | |
| Ribosomal RNA genes | rrn23*, rrn4.5*, rrn 5*, rrn16* | |
| Transfer RNA genes | trnL-UUU1, trnG-UUG, trnS-GCU, trnS-UGA, trnG-GCC, trnM-CAU, trnS-CGA1, trnT-GGU, trnG-UUC, trnT-GUA, trnA-GUC, trnC-GCA, trnA-UCU, trnS-GGA, trnT-UGU, trnL-UAA1, trnP-GAA, trnV-UAC1, trnM-CAU, trnT-CCA, trnP-UGG, trnH-GUG, trnM-CAU *, trnL-CAA *, trnV-GAC, trnT-CGU1,*, trnA-UGC1,*, trnA-ACG, trnA-GUU *, trnL-UAG, trnA-ACG, trnV-GAC, trnH-GUG | |
| Other genes | Envelop membrane protein | cemA |
| Translational initiation factor | infA | |
| c-type cytochrome synthesis gene | ccsA | |
| ATP-dependent/Protease | clpP | |
| Maturase | matK | |
| Unknown function protein-coding gene | ycf1*, ycf2 *, ycf 32 |
| Gene Order in cp Genome | Gene Name | Position | Code of Base in Reference | Code in S. spontaneum | Code of Amino Acid in Reference | Amino Acids in S. spontaneum |
|---|---|---|---|---|---|---|
| 13 | rpoC1 | 2059 | ATA | GTA | I | V |
| 14 | rpoC2 | 3228 | GGC | GGT | G | G |
| 19 | atpA | 572 | GGT | GAT | G | D |
| 23 | ycf3 | 300 | GCA | GCG | A | A |
| 29 | atpB | 313 | ACT | GCT | T | A |
| 29 | atpB | 940 | GCC | ACC | A | T |
| 38 | psbE | 57 | TAC | TAT | Y | Y |
| 52 | rpoA | 202 | GGG | TGG | G | W |
| 57 | rpl14 | 335 | TTT | TAT | F | Y |
| 59 | rps3 | 267 | CCC | CCA | P | P |
| 65 | ndhB | 776 | GAC | GGA | D | G |
| 65 | ndhB | 777 | GAC | GGA | D | G |
| 70 | ndhF | 868 | ATA | CTA | I | L |
| 70 | ndhF | 1804 | ATC | CTC | I | L |
| 72 | ccsA | 537 | TTT | TTA | F | L |
| 72 | ccsA | 538 | CTT | AGA | L | R |
| 72 | ccsA | 539 | CTT | AGA | L | R |
| 72 | ccsA | 540 | CTT | AGA | L | R |
| 72 | ccsA | 939 | TAT | TAC | Y | Y |
| 73 | ndhD | 1240 | TTA | GTA | L | V |
| 78 | ndhA | 858 | TCT | TCC | S | S |
| 79 | ndhH | 793 | ATC | GTC | I | V |
| 84 | ndhB | 776 | GAC | GGA | D | G |
| 84 | ndhB | 777 | GAC | GGA | D | G |
| Amino Acid | Symbol | Codon | Saccharum officinarum | S. spontaneum | ||
|---|---|---|---|---|---|---|
| Number | RSCU | Number | RSCU | |||
| * | Ter | UAA | 40 | 1.579 | 39 | 1.520 |
| * | Ter | UAG | 19 | 0.750 | 19 | 0.740 |
| * | Ter | UGA | 17 | 0.671 | 19 | 0.740 |
| A | Ala | GCA | 368 | 1.198 | 373 | 1.192 |
| A | Ala | GCC | 189 | 0.615 | 194 | 0.62 |
| A | Ala | GCG | 134 | 0.436 | 137 | 0.438 |
| A | Ala | GCU | 538 | 1.751 | 548 | 1.751 |
| C | Cys | UGC | 55 | 0.498 | 55 | 0.491 |
| C | Cys | UGU | 166 | 1.502 | 169 | 1.509 |
| D | Asp | GAC | 155 | 0.437 | 155 | 0.433 |
| D | Asp | GAU | 555 | 1.563 | 561 | 1.567 |
| E | Glu | GAA | 794 | 1.497 | 803 | 1.484 |
| E | Glu | GAG | 267 | 0.503 | 279 | 0.516 |
| F | Phe | UUC | 403 | 0.718 | 422 | 0.732 |
| F | Phe | UUU | 720 | 1.282 | 731 | 1.268 |
| G | Gly | GGA | 586 | 1.570 | 596 | 1.567 |
| G | Gly | GGC | 151 | 0.404 | 150 | 0.394 |
| G | Gly | GGG | 275 | 0.737 | 284 | 0.747 |
| G | Gly | GGU | 481 | 1.289 | 491 | 1.291 |
| H | His | CAC | 120 | 0.512 | 122 | 0.501 |
| H | His | CAU | 349 | 1.488 | 365 | 1.499 |
| I | Ile | AUA | 522 | 0.956 | 540 | 0.970 |
| I | Ile | AUC | 307 | 0.562 | 308 | 0.553 |
| I | Ile | AUU | 810 | 1.483 | 823 | 1.478 |
| K | Lys | AAA | 743 | 1.450 | 751 | 1.437 |
| K | Lys | AAG | 282 | 0.550 | 294 | 0.563 |
| L | Leu | CUA | 324 | 0.894 | 338 | 0.910 |
| L | Leu | CUC | 138 | 0.380 | 137 | 0.369 |
| L | Leu | CUG | 116 | 0.320 | 116 | 0.313 |
| L | Leu | CUU | 476 | 1.313 | 500 | 1.346 |
| L | Leu | UUA | 722 | 1.992 | 730 | 1.966 |
| L | Leu | UUG | 399 | 1.100 | 407 | 1.096 |
| M | Met | AUA | 2 | 0.013 | 2 | 0.013 |
| M | Met | AUG | 456 | 2.961 | 458 | 2.961 |
| M | Met | GUG | 4 | 0.026 | 4 | 0.026 |
| N | Asn | AAC | 208 | 0.515 | 218 | 0.527 |
| N | Asn | AAU | 599 | 1.485 | 609 | 1.473 |
| P | Pro | CCA | 216 | 1.013 | 222 | 1.026 |
| P | Pro | CCC | 208 | 0.975 | 212 | 0.979 |
| P | Pro | CCG | 93 | 0.436 | 94 | 0.434 |
| P | Pro | CCU | 336 | 1.576 | 338 | 1.561 |
| Q | Gln | CAA | 524 | 1.555 | 532 | 1.549 |
| Q | Gln | CAG | 150 | 0.445 | 155 | 0.451 |
| R | Arg | AGA | 361 | 1.758 | 376 | 1.764 |
| R | Arg | AGG | 110 | 0.536 | 114 | 0.535 |
| R | Arg | CGA | 273 | 1.330 | 285 | 1.337 |
| R | Arg | CGC | 106 | 0.516 | 110 | 0.516 |
| R | Arg | CGG | 94 | 0.458 | 100 | 0.469 |
| R | Arg | CGU | 288 | 1.403 | 294 | 1.379 |
| S | Ser | AGC | 104 | 0.434 | 106 | 0.428 |
| S | Ser | AGU | 297 | 1.241 | 303 | 1.225 |
| S | Ser | UCA | 235 | 0.982 | 237 | 0.958 |
| S | Ser | UCC | 286 | 1.195 | 302 | 1.221 |
| S | Ser | UCG | 122 | 0.510 | 130 | 0.526 |
| S | Ser | UCU | 392 | 1.638 | 406 | 1.642 |
| T | Thr | ACA | 300 | 1.109 | 304 | 1.094 |
| T | Thr | ACC | 203 | 0.750 | 211 | 0.759 |
| T | Thr | ACG | 133 | 0.492 | 137 | 0.493 |
| T | Thr | ACU | 446 | 1.649 | 460 | 1.655 |
| V | Val | GUA | 431 | 1.504 | 438 | 1.516 |
| V | Val | GUC | 134 | 0.468 | 137 | 0.474 |
| V | Val | GUG | 150 | 0.524 | 152 | 0.526 |
| V | Val | GUU | 431 | 1.504 | 429 | 1.484 |
| W | Trp | UGG | 347 | 1.000 | 353 | 1.000 |
| Y | Tyr | UAC | 151 | 0.411 | 154 | 0.410 |
| Y | Tyr | UAU | 583 | 1.589 | 598 | 1.590 |
| S. officinarum | S. spontaneum | ||||||
|---|---|---|---|---|---|---|---|
| SSR | Size/bp | Start Position | End Position | SSR | Size/bp | Start Position | End Position |
| (A)13 | 13 | 3754 | 3766 | (A)15 | 15 | 3753 | 3767 |
| (A)11 | 11 | 4114 | 4124 | (A)10 | 10 | 4115 | 4124 |
| (T)10 | 10 | 6446 | 6455 | (T)11 | 11 | 6112 | 6122 |
| (A)11 | 11 | 7777 | 7787 | (T)14 | 14 | 6446 | 6459 |
| (T)10 | 10 | 9056 | 9065 | (A)11 | 11 | 7781 | 7791 |
| (G)10 | 10 | 11,003 | 11,012 | (T)10 | 10 | 9060 | 9069 |
| (T)11 | 11 | 13,372 | 13,382 | (G)10 | 10 | 11,007 | 11,016 |
| (T)13 | 13 | 16,556 | 16,568 | (T)11 | 11 | 13,375 | 13,385 |
| (A)10 | 10 | 18,659 | 18,668 | (A)10 | 10 | 15,948 | 15,957 |
| (A)12 | 12 | 19,205 | 19,216 | (T)14 | 14 | 16,554 | 16,567 |
| (T)12 | 12 | 21,069 | 21,080 | (A)12 | 12 | 19,197 | 19,208 |
| (A)10 | 10 | 31,914 | 31,923 | (T)10 | 10 | 21,061 | 21,070 |
| (T)11 | 11 | 34,093 | 34,103 | (A)10 | 10 | 31,904 | 31,913 |
| (T)10 | 10 | 34,862 | 34,871 | (T)11 | 11 | 34,083 | 34,093 |
| (T)10 | 10 | 35,824 | 35,833 | (T)10 | 10 | 34,852 | 34,861 |
| (T)10 | 10 | 38,697 | 38,706 | (T)10 | 10 | 35,814 | 35,823 |
| (T)10 | 10 | 44,257 | 44,266 | (T)11 | 11 | 38,687 | 38,697 |
| (T)14 | 14 | 52,367 | 52,380 | (T)11 | 11 | 38,836 | 38,846 |
| (T)10 | 10 | 56,710 | 56,719 | (T)14 | 14 | 52,356 | 52,369 |
| (T)10 | 10 | 56,892 | 56,901 | (T)10 | 10 | 56,700 | 56,709 |
| (T)10 | 10 | 57,363 | 57,372 | (T)10 | 10 | 57,351 | 57,360 |
| (A)11 | 11 | 63,551 | 63,561 | (T)11 | 11 | 60,122 | 60,132 |
| (A)11 | 11 | 65,513 | 65,523 | (T)10 | 10 | 66,448 | 66,457 |
| (T)10 | 10 | 67,486 | 67,495 | (T)10 | 10 | 67,465 | 67,474 |
| (T)11 | 11 | 68,139 | 68,149 | (T)12 | 12 | 68,133 | 68,144 |
| (A)10 | 10 | 73,938 | 73,947 | C2 | 65 | 73,929 | 73,993 |
| (T)12 | 12 | 78,482 | 78,493 | (T)12 | 12 | 78,474 | 78,485 |
| C1 | 28 | 79,044 | 79,071 | C3 | 28 | 79,031 | 79,058 |
| (T)14 | 14 | 81,260 | 81,273 | (T)13 | 13 | 81,243 | 81,255 |
| ? | ? | ? | ? | (A)10 | 10 | 108,278 | 108,287 |
| Investigate Time | SPAD | p Value | Fv/Fm | ||
|---|---|---|---|---|---|
| S. officinarum | S. spontaneum | S. officinarum | S. spontaneum | ||
| Before cold stress | 29.25 ± 1.50A | 45.50 ± 1.40C | 0.000008 | - | - |
| Culture at 4 °C for 3 days | 28.02 ± 1.10A | 41.42 ± 1.88D | 0.000013 | - | - |
| Culture at 4 °C for 7 days | 27.73 ± 1.16A | 40.42 ± 1.25D | 0.001401 | 0.194 ± 0.096A | 0.364 ± 0.152B |
| Removing to field for recovery 10 days | 23.37 ± 2.64B | 39.48 ± 3.34D | 0.000100 | 0 (5/6 plants) | 0 (5/6 plants) |
| Species | S. officinarum | S. spontaneum | S. officinarum Hybrids | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Accession | Badila | IJ76-514 | Yunnan 83-184 | IJ76-287 | SES234B | RB867515 | Q155 | SP80-3280 | NCo310 | RB72454 | Q165 |
| Length/bp | 141,187 | 141,176 | 141,181 | 141,348 | 141,185 | 141,181 | 141,181 | 141,182 | 141,182 | 141,151 | 141,181 |
| LSC/bp | 83,065 | 83,042 | 83,047 | 83,226 | 83,063 | 83,047 | 83,047 | 83,048 | 83,048 | 83,017 | 83,047 |
| SSC/bp | 12,544 | 12,544 | 12,544 | 12,544 | 12,544 | 12,544 | 12,544 | 12,546 | 12,544 | 12,544 | 12,544 |
| IRa/bp | 22,789 | 22,795 | 22,795 | 22,789 | 22,789 | 22,795 | 22,795 | 22,794 | 22,795 | 22,795 | 22,795 |
| IRb/bp | 22,789 | 22,795 | 22,795 | 22,789 | 22,789 | 22,795 | 22,795 | 22,794 | 22,795 | 22,795 | 22,795 |
| Accession numbers in NCBI | Obtained in this study | NC_035224.1 | Obtained in this study | LS975131.1 | LN849912.1 | KX507245.1 | NC_029221.1 | NC_005878.2 | NC_006084.1 | LN849914.1 | LN896359.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, F.; He, L.; Gao, S.; Su, Y.; Li, F.; Xu, L. Comparative Analysis of two Sugarcane Ancestors Saccharum officinarum and S. spontaneum based on Complete Chloroplast Genome Sequences and Photosynthetic Ability in Cold Stress. Int. J. Mol. Sci. 2019, 20, 3828. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153828
Xu F, He L, Gao S, Su Y, Li F, Xu L. Comparative Analysis of two Sugarcane Ancestors Saccharum officinarum and S. spontaneum based on Complete Chloroplast Genome Sequences and Photosynthetic Ability in Cold Stress. International Journal of Molecular Sciences. 2019; 20(15):3828. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153828
Chicago/Turabian StyleXu, Fu, Lilian He, Shiwu Gao, Yachun Su, Fusheng Li, and Liping Xu. 2019. "Comparative Analysis of two Sugarcane Ancestors Saccharum officinarum and S. spontaneum based on Complete Chloroplast Genome Sequences and Photosynthetic Ability in Cold Stress" International Journal of Molecular Sciences 20, no. 15: 3828. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20153828