Osteostatin Inhibits Collagen-Induced Arthritis by Regulation of Immune Activation, Pro-Inflammatory Cytokines, and Osteoclastogenesis


Abstract
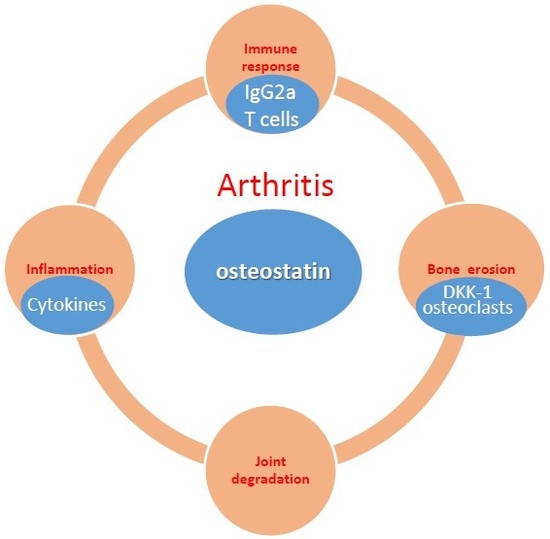
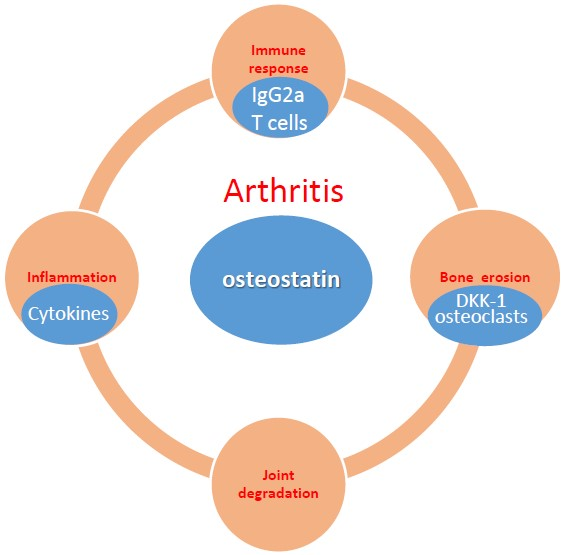
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
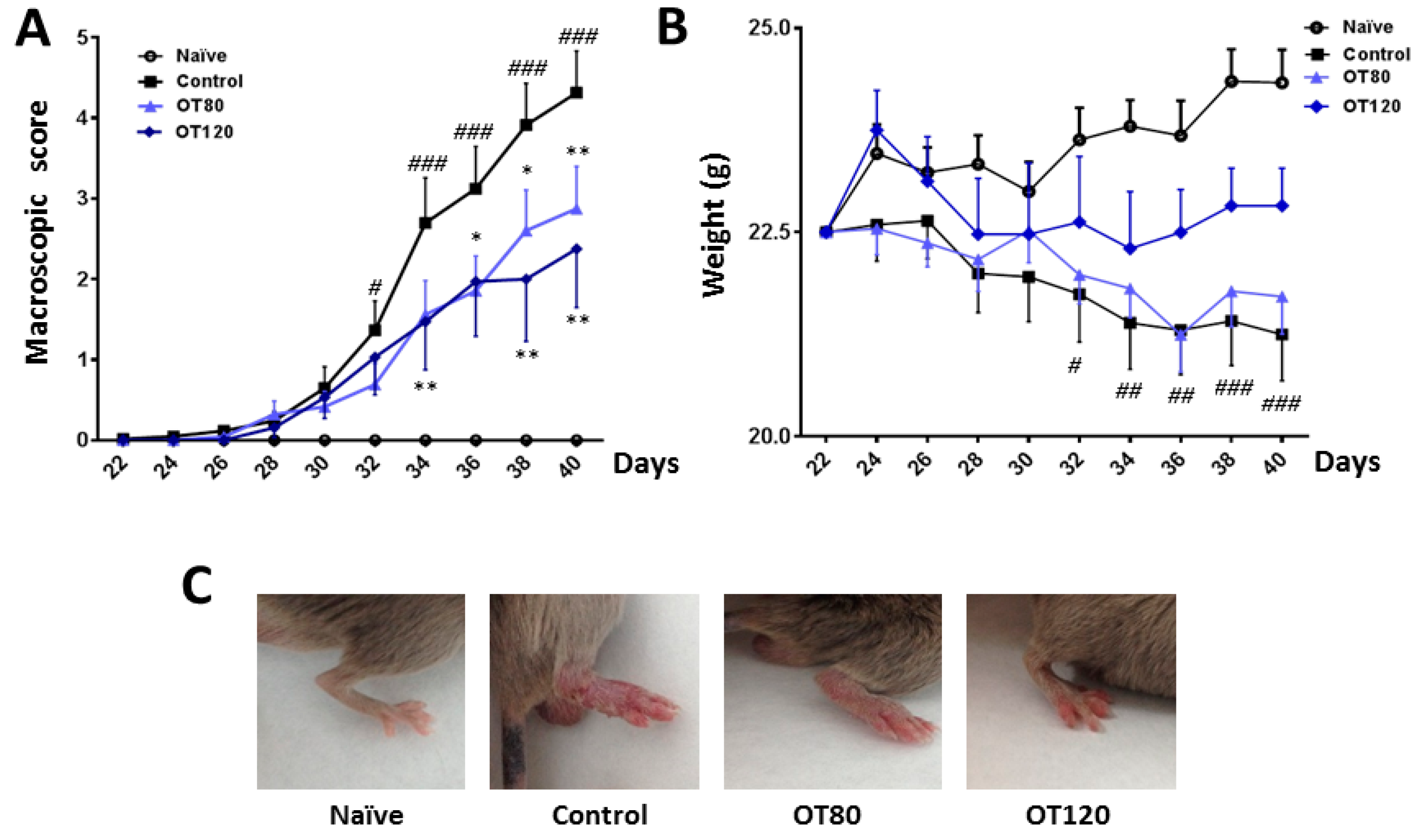
2.1. Effects of Osteostatin on the Progression of Arthritis
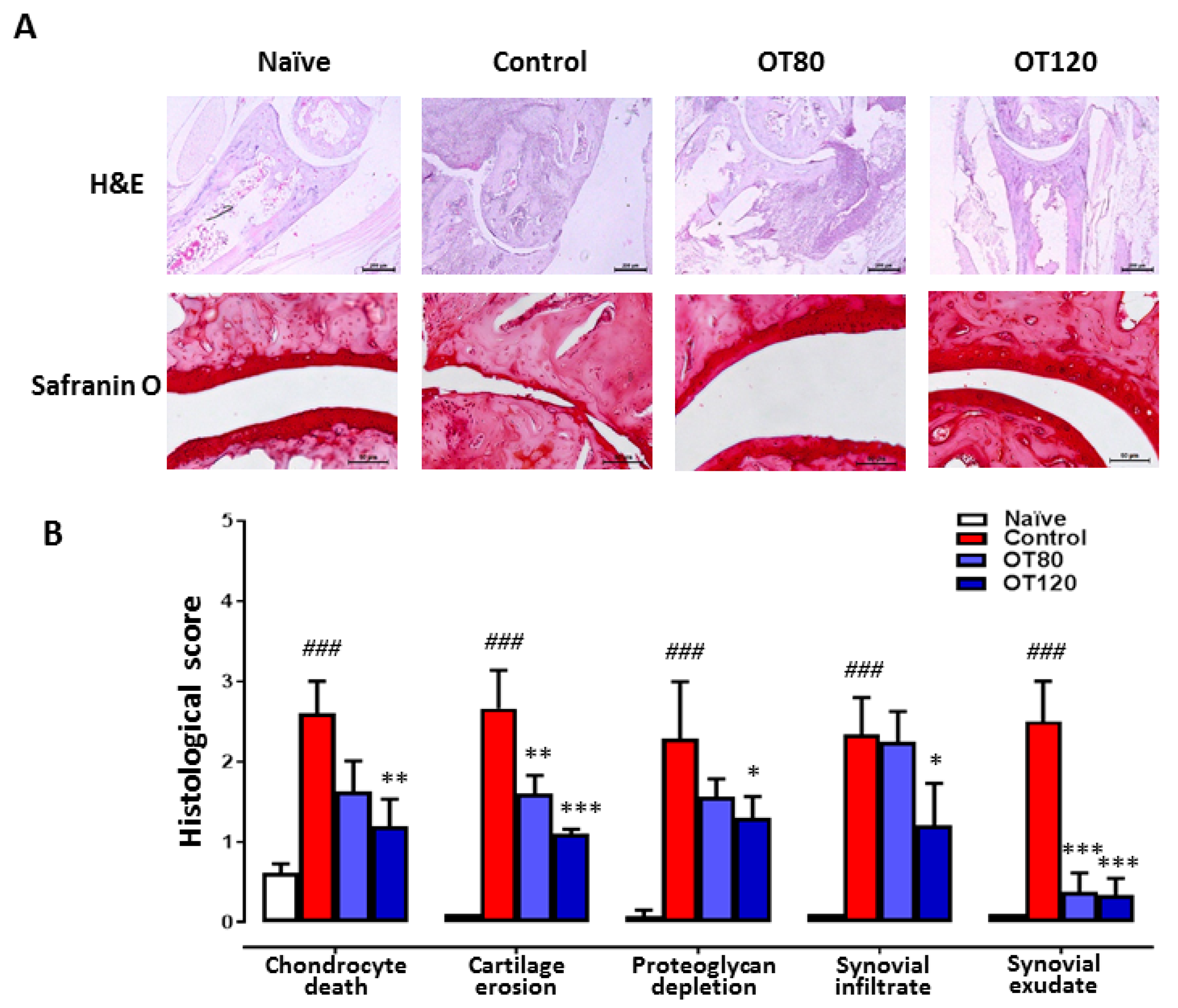
2.2. Joint Histological Analysis
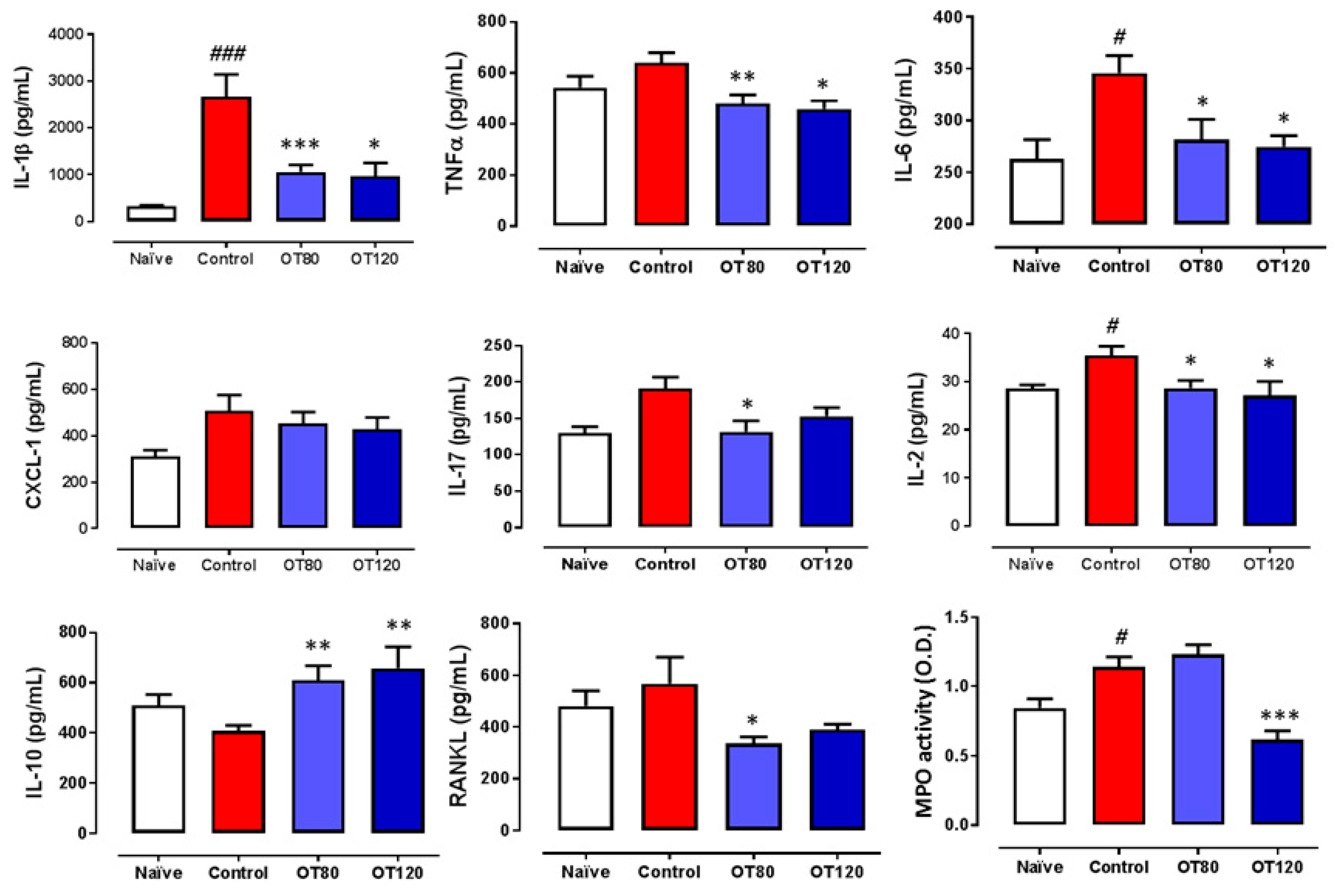
2.3. Effects on Local Cytokine Levels and Myeloperoxidase Activity
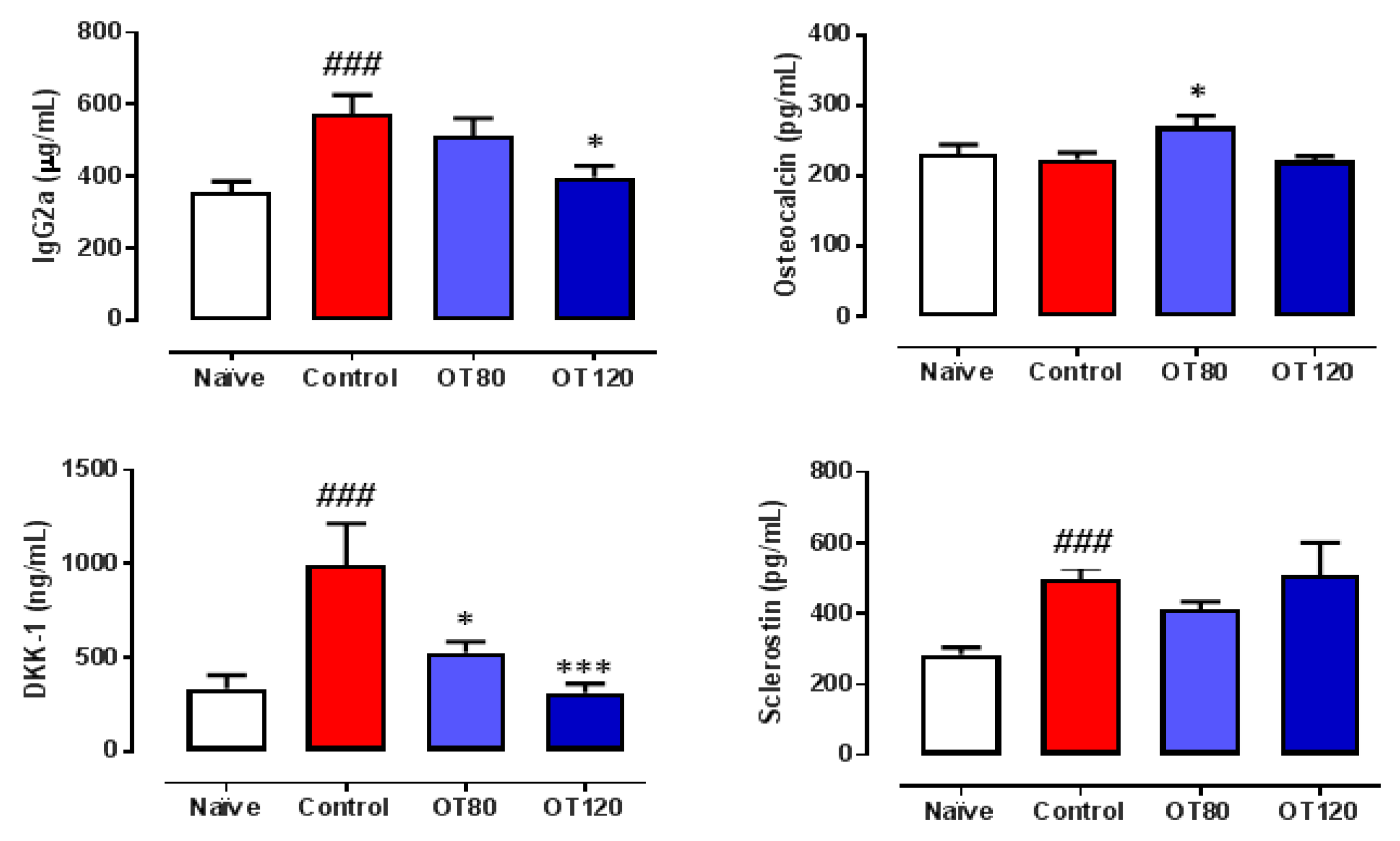
2.4. Effects on Serum IgG2a and Bone Metabolism Biomarkers
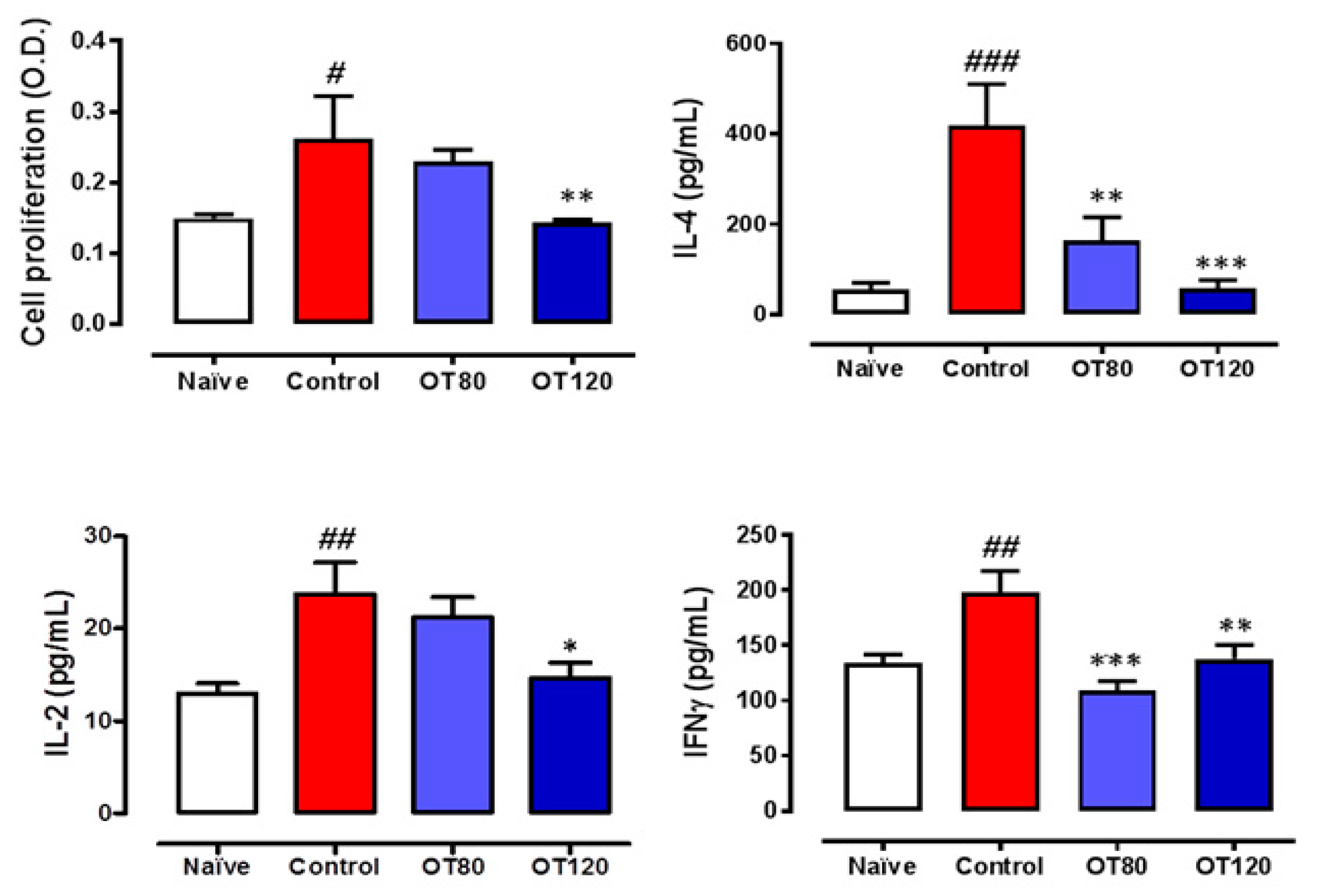
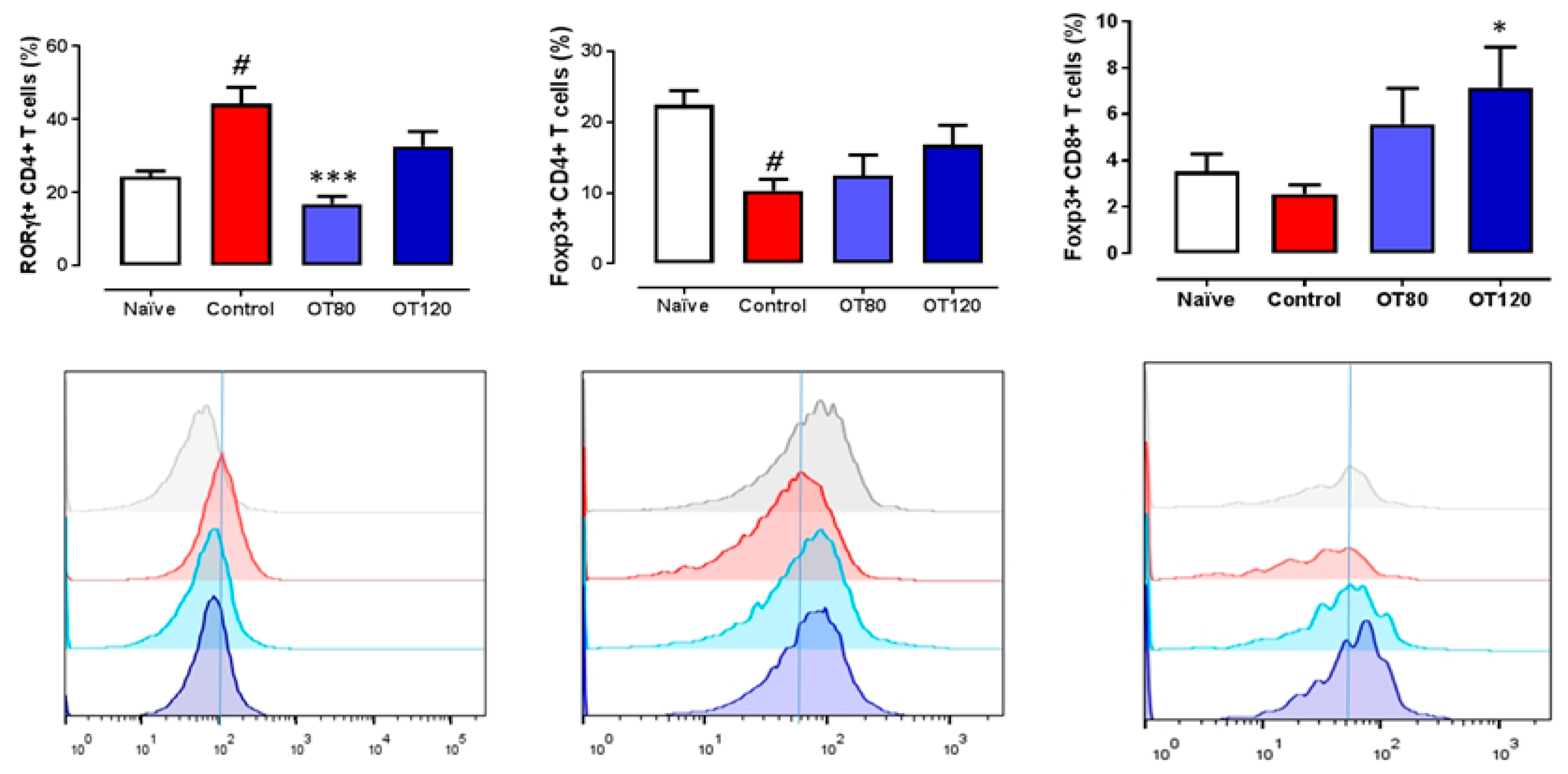
2.5. Lymph Node T Cell Proliferation and Release of Cytokines
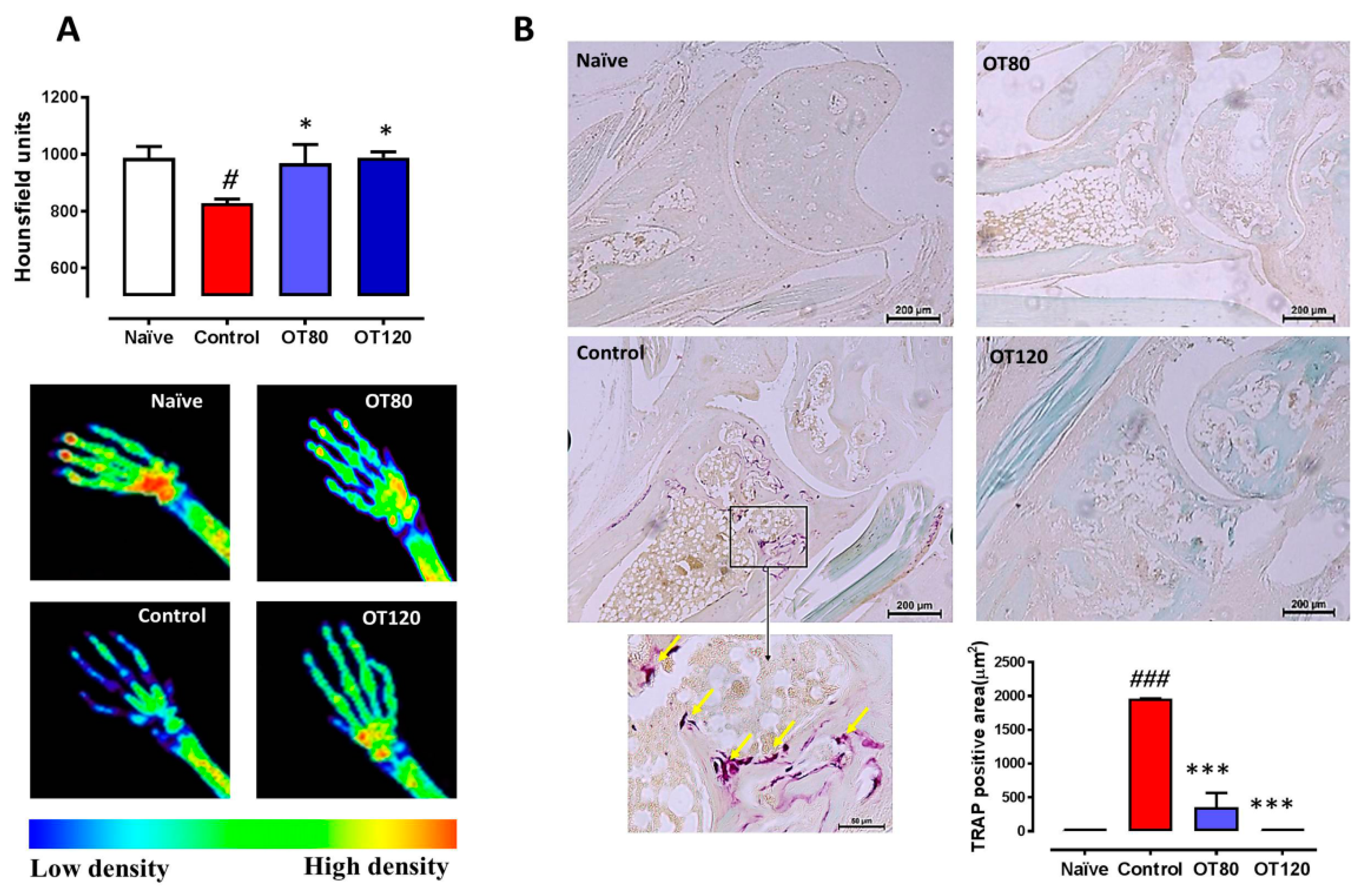
2.6. Bone Degradation
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Induction of Arthritis
4.3. Experimental Groups and Treatment
4.4. Arthritis Score
4.5. Histological Analysis
4.6. Determination of Mediators in Paw Homogenates
4.7. Serum Determinations
4.8. Lymph Node Cells Isolation, Proliferation Assay and Cytokine Determination
4.9. Flow Cytometry
4.10. X-ray Analysis
4.11. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- de Castro, L.F.; Lozano, D.; Portal-Nuñez, S.; Maycas, M.; De la Fuente, M.; Caeiro, J.R.; Esbrit, P. Comparison of the skeletal effects induced by daily administration of PTHrP (1–36) and PTHrP (107–139) to ovariectomized mice. J. Cell. Physiol. 2012, 227, 1752–1760. [Google Scholar] [CrossRef] [PubMed]
- de Gortazar, A.R.; Alonso, V.; Alvarez-Arroyo, M.V.; Esbrit, P. Transient exposure to PTHrP (107–139) exerts anabolic effects through vascular endothelial growth factor receptor 2 in human osteoblastic cells in vitro. Calcif. Tissue Int. 2006, 79, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Trejo, C.G.; Lozano, D.; Manzano, M.; Doadrio, J.C.; Salinas, A.J.; Dapia, S.; Gomez-Barrena, E.; Vallet-Regi, M.; Garcia-Honduvilla, N.; Bujan, J.; et al. The osteoinductive properties of mesoporous silicate coated with osteostatin in a rabbit femur cavity defect model. Biomaterials 2010, 31, 8564–8573. [Google Scholar] [CrossRef] [PubMed]
- Fenton, A.J.; Martin, T.J.; Nicholson, G.C. Carboxyl-terminal parathyroid hormone-related protein inhibits bone resorption by isolated chicken osteoclasts. J. Bone Miner. Res. 1994, 9, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of Rheumatoid Arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Scholtysek, C.; Kronke, G.; Schett, G. Inflammation-associated changes in bone homeostasis. Inflamm. Allergy Drug Targets 2012, 11, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Szentpetery, A.; Horvath, A.; Gulyas, K.; Petho, Z.; Bhattoa, H.P.; Szanto, S.; Szucs, G.; FitzGerald, O.; Schett, G.; Szekanecz, Z. Effects of targeted therapies on the bone in arthritides. Autoimmun. Rev. 2017, 36, 313–320. [Google Scholar] [CrossRef]
- Kohno, H.; Shigeno, C.; Kasai, R.; Akiyama, H.; Iida, H.; Tsuboyama, T.; Sato, K.; Konishi, J.; Nakamura, T. Synovial fluids from patients with osteoarthritis and rheumatoid arthritis contain high levels of parathyroid hormone-related peptide. J. Bone Miner. Res. 1997, 12, 847–854. [Google Scholar] [CrossRef]
- Fischer, J.; Dickhut, A.; Rickert, M.; Richter, W. Human articular chondrocytes secrete parathyroid hormone-related protein and inhibit hypertrophy of mesenchymal stem cells in coculture during chondrogenesis. Arthritis Rheum. 2010, 62, 2696–2706. [Google Scholar] [CrossRef]
- Chen, X.; Macica, C.M.; Nasiri, A.; Broadus, A.E. Regulation of articular chondrocyte proliferation and differentiation by indian hedgehog and parathyroid hormone-related protein in mice. Arthritis Rheum. 2008, 58, 3788–3797. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, T.; Yoshida, T.; Koshihara, Y.; Sakamoto, H.; Kanai, H.; Yamamoto, S.; Ito, H. The increase of parathyroid hormone-related peptide and cytokine levels in synovial fluid of elderly rheumatoid arthritis and osteoarthritis. Endocr. J. 1999, 46, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Platas, J.; Guillen, M.I.; Gomar, F.; Castejon, M.A.; Esbrit, P.; Alcaraz, M.J. Anti-senescence and Anti-inflammatory Effects of the C-terminal Moiety of PTHrP Peptides in OA Osteoblasts. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 172, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Myers, L.K.; Rosloniec, E.F.; Cremer, M.A.; Kang, A.H. Collagen-induced arthritis, an animal model of autoimmunity. Life Sci. 1997, 61, 1861–1878. [Google Scholar] [CrossRef]
- Williams, P.J.; Jones, R.H.; Rademacher, T.W. Correlation between IgG anti-type II collagen levels and arthritic severity in murine arthritis. Autoimmunity 1998, 27, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Watson, W.C.; Townes, A.S. Genetic susceptibility to murine collagen II autoimmune arthritis. Proposed relationship to the IgG2 autoantibody subclass response, complement C5, major histocompatibility complex (MHC) and non-MHC loci. J. Exp. Med. 1985, 162, 1878–1891. [Google Scholar] [CrossRef] [PubMed]
- Lories, R.J.; Corr, M.; Lane, N.E. To Wnt or not to Wnt: The bone and joint health dilemma. Nat. Rev. Rheumatol. 2013, 9, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Teitelbaum, S.L. Osteoclasts and Arthritis. J. Bone Miner. Res. 2009, 24, 1142–1146. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.O. Collagen-induced arthritis in mice: A major role for tumor necrosis factor-alpha. Methods Mol. Biol. 2007, 361, 265–284. [Google Scholar] [PubMed]
- Cush, J.J.; Splawski, J.B.; Thomas, R.; McFarlin, J.E.; Schulze-Koops, H.; Davis, L.S.; Fujita, K.; Lipsky, P.E. Elevated interleukin-10 levels in patients with rheumatoid arthritis. Arthritis Rheum. 1995, 38, 96–104. [Google Scholar] [CrossRef]
- Isomaki, P.; Luukkainen, R.; Saario, R.; Toivanen, P.; Punnonen, J. Interleukin-10 functions as an antiinflammatory cytokine in rheumatoid synovium. Arthritis Rheum. 1996, 39, 386–395. [Google Scholar] [CrossRef]
- Finnegan, A.; Kaplan, C.D.; Cao, Y.; Eibel, H.; Glant, T.T.; Zhang, J. Collagen-induced arthritis is exacerbated in IL-10-deficient mice. Arthritis Res. Ther. 2003, 5, R18–R24. [Google Scholar] [CrossRef] [PubMed]
- Ronnelid, J.; Lysholm, J.; Engstrom-Laurent, A.; Klareskog, L.; Heyman, B. Local anti-type II collagen antibody production in rheumatoid arthritis synovial fluid. Evidence for an HLA-DR4-restricted IgG response. Arthritis Rheum. 1994, 37, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Croxford, A.M.; Whittingham, S.; McNaughton, D.; Nandakumar, K.S.; Holmdahl, R.; Rowley, M.J. Type II collagen-specific antibodies induce cartilage damage in mice independent of inflammation. Arthritis Rheum. 2013, 65, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Plows, D.; Kontogeorgos, G.; Kollias, G. Mice lacking mature T and B lymphocytes develop arthritic lesions after immunization with type II collagen. J. Immunol. 1999, 162, 1018–1023. [Google Scholar] [PubMed]
- Nandakumar, K.S.; Backlund, J.; Vestberg, M.; Holmdahl, R. Collagen type II (CII)-specific antibodies induce arthritis in the absence of T or B cells but the arthritis progression is enhanced by CII-reactive T cells. Arthritis Res. Ther. 2004, 6, R544–R550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Paul, W.E. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol. Rev. 2010, 238, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Lubberts, E. Th17 cytokines and arthritis. Semin. Immunopathol. 2010, 32, 43–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Usui, T.; Kobayashi, S.; Iguchi-Hashimoto, M.; Ito, H.; Yoshitomi, H.; Nakamura, T.; Shimizu, M.; Kawabata, D.; Yukawa, N.; et al. Gamma/delta T cells are the predominant source of interleukin-17 in affected joints in collagen-induced arthritis, but not in rheumatoid arthritis. Arthritis Rheum. 2009, 60, 2294–2303. [Google Scholar] [CrossRef]
- Lubberts, E.; Koenders, M.I.; Oppers-Walgreen, B.; Van Den, B.L.; Coenen-De Roo, C.J.; Joosten, L.A.; van den Berg, W.B. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004, 50, 650–659. [Google Scholar] [CrossRef]
- Koenders, M.I.; Marijnissen, R.J.; Devesa, I.; Lubberts, E.; Joosten, L.A.; Roth, J.; van Lent, P.L.; van de Loo, F.A.; van den Berg, W.B. Tumor necrosis factor-interleukin-17 interplay induces S100A8, interleukin-1beta, and matrix metalloproteinases, and drives irreversible cartilage destruction in murine arthritis: Rationale for combination treatment during arthritis. Arthritis Rheum. 2011, 63, 2329–2339. [Google Scholar] [CrossRef]
- Lubberts, E.; Van Den, B.L.; Oppers-Walgreen, B.; Schwarzenberger, P.; Coenen-De Roo, C.J.; Kolls, J.K.; Joosten, L.A.; van den Berg, W.B. IL-17 promotes bone erosion in murine collagen-induced arthritis through loss of the receptor activator of NF-kappa B ligand/osteoprotegerin balance. J. Immunol. 2003, 170, 2655–2662. [Google Scholar] [CrossRef] [PubMed]
- Cools, N.; Ponsaerts, P.; Van Tendeloo, V.F.; Berneman, Z.N. Regulatory T cells and human disease. Clin. Dev. Immunol. 2007, 2007, 89195. [Google Scholar] [CrossRef] [PubMed]
- Kelchtermans, H.; Geboes, L.; Mitera, T.; Huskens, D.; Leclercq, G.; Matthys, P. Activated CD4+CD25+ regulatory T cells inhibit osteoclastogenesis and collagen-induced arthritis. Ann. Rheum. Dis. 2009, 68, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Notley, C.A.; McCann, F.E.; Inglis, J.J.; Williams, R.O. anti-CD3 therapy expands the numbers of CD4+ and CD8+ Treg cells and induces sustained amelioration of collagen-induced arthritis. Arthritis Rheum. 2010, 62, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, M.M.; Frey, B.; Hess, A.; Zwerina, J.; Luther, J.; Nimmerjahn, F.; Engelke, K.; Kollias, G.; Hunig, T.; Schett, G.; et al. Regulatory T cells protect from local and systemic bone destruction in arthritis. J. Immunol. 2010, 184, 7238–7246. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Ma, X.; Gong, R.; Zhu, J.; Wei, L.; Yao, J. Recent advances in CD8(+) regulatory T cell research. Oncol. Lett. 2018, 15, 8187–8194. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Tsuruoka, M.; Ogura, H.; Okuyama, Y.; Arima, Y.; Hirano, T.; Murakami, M. IL-6 positively regulates Foxp3+CD8+ T cells in vivo. Int. Immunol. 2010, 22, 129–139. [Google Scholar] [CrossRef]
- Filaci, G.; Fenoglio, D.; Indiveri, F. CD81 T regulatory/suppressor cells and their relationships with autoreactivity and autoimmunity. Autoimmunity 2011, 44, 51–57. [Google Scholar] [CrossRef]
- Sun, J.; Yang, Y.; Huo, X.; Zhu, B.; Li, Z.; Jiang, X.; Xie, R.; Gao, L.; Sun, Y.; Fan, H.; et al. Efficient therapeutic function and mechanisms of human polyclonal CD8+CD103+Foxp3+ regulatory T cells on collagen-induced arthritis in mice. J. Immunol. Res. 2019, 2019, 8575407. [Google Scholar] [CrossRef]
- Stolina, M.; Adamu, S.; Ominsky, M.; Dwyer, D.; Asuncion, F.; Geng, Z.; Middleton, S.; Brown, H.; Pretorius, J.; Schett, G.; et al. RANKL is a marker and mediator of local and systemic bone loss in two rat models of inflammatory arthritis. J. Bone Miner. Res. 2005, 20, 1756–1765. [Google Scholar] [CrossRef]
- Nanes, M.S. Tumor necrosis factor-alpha: Molecular and cellular mechanisms in skeletal pathology. Gene 2003, 321, 1–15. [Google Scholar] [CrossRef]
- Diarra, D.; Stolina, M.; Polzer, K.; Zwerina, J.; Ominsky, M.S.; Dwyer, D.; Korb, A.; Smolen, J.; Hoffmann, M.; Scheinecker, C.; et al. Dickkopf-1 is a master regulator of joint remodeling. Nat. Med. 2007, 13, 156–163. [Google Scholar] [CrossRef]
- Nakashima, T.; Kobayashi, Y.; Yamasaki, S.; Kawakami, A.; Eguchi, K.; Sasaki, H.; Sakai, H. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappaB ligand: Modulation of the expression by osteotropic factors and cytokines. Biochem. Biophys. Res. Commun. 2000, 275, 768–775. [Google Scholar] [CrossRef]
- Kobayashi, K.; Takahashi, N.; Jimi, E.; Udagawa, N.; Takami, M.; Kotake, S.; Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J. Exp. Med. 2000, 191, 275–286. [Google Scholar] [CrossRef]
- Sen, M. Wnt signalling in rheumatoid arthritis. Rheumatology 2005, 44, 708–713. [Google Scholar] [CrossRef]
- Walsh, N.C. and Gravallese, E.M. Bone remodeling in rheumatic disease: A question of balance. Immunol. Rev. 2010, 233, 301–312. [Google Scholar] [CrossRef]
- Braun, T.; Zwerina, J. Positive regulators of osteoclastogenesis and bone resorption in rheumatoid arthritis. Arthritis Res. Ther. 2011, 13, 235. [Google Scholar] [CrossRef]
- Seror, R.; Boudaoud, S.; Pavy, S.; Nocturne, G.; Schaeverbeke, T.; Saraux, A.; Chanson, P.; Gottenberg, J.E.; Devauchelle-Pensec, V.; Tobon, G.J.; et al. Increased Dickkopf-1 in Recent-onset Rheumatoid Arthritis is a New Biomarker of Structural Severity. Data from the ESPOIR Cohort. Sci. Rep. 2016, 6, 18421. [Google Scholar] [CrossRef]
- Kerschan-Schindl, K.; Ebenbichler, G.; Foeger-Samwald, U.; Leiss, H.; Gesslbauer, C.; Herceg, M.; Stummvoll, G.; Marculescu, R.; Crevenna, R.; Pietschmann, P. Rheumatoid arthritis in remission: Decreased myostatin and increased serum levels of periostin. Wien. Klin. Wochenschr. 2019, 131, 1–7. [Google Scholar] [CrossRef]
- Marenzana, M.; Vugler, A.; Moore, A.; Robinson, M. Effect of sclerostin-neutralising antibody on periarticular and systemic bone in a murine model of rheumatoid arthritis: A microCT study. Arthritis Res. Ther. 2013, 15, R125. [Google Scholar] [CrossRef]
- Wehmeyer, C.; Frank, S.; Beckmann, D.; Bottcher, M.; Cromme, C.; Konig, U.; Fennen, M.; Held, A.; Paruzel, P.; Hartmann, C.; et al. Sclerostin inhibition promotes TNF-dependent inflammatory joint destruction. Sci. Transl. Med. 2016, 8, 330ra35. [Google Scholar] [CrossRef]
- Ji, Y.; Qiao, H.; He, J.; Li, W.; Chen, R.; Wang, J.; Wu, L.; Hu, R.; Duan, J.; Chen, Z. Functional oligopeptide as a novel strategy for drug delivery. J. Drug. Target. 2017, 25, 597–607. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS. Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Maicas, N.; Ibañez, L.; Alcaraz, M.J.; Ubeda, A.; Ferrandiz, M.L. Prostaglandin D2 regulates joint inflammation and destruction in murine collagen-induced arthritis. Arthritis Rheum. 2012, 64, 130–140. [Google Scholar] [CrossRef]
- Paya, M.; Terencio, M.C.; Ferrandiz, M.L.; Alcaraz, M.J. Involvement of secretory phospholipase A2 activity in the zymosan rat air pouch model of inflammation. Br. J. Pharmacol. 1996, 117, 1773–1779. [Google Scholar] [CrossRef] [Green Version]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nácher-Juan, J.; Terencio, M.C.; Alcaraz, M.J.; Ferrándiz, M.L. Osteostatin Inhibits Collagen-Induced Arthritis by Regulation of Immune Activation, Pro-Inflammatory Cytokines, and Osteoclastogenesis. Int. J. Mol. Sci. 2019, 20, 3845. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20163845
Nácher-Juan J, Terencio MC, Alcaraz MJ, Ferrándiz ML. Osteostatin Inhibits Collagen-Induced Arthritis by Regulation of Immune Activation, Pro-Inflammatory Cytokines, and Osteoclastogenesis. International Journal of Molecular Sciences. 2019; 20(16):3845. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20163845
Chicago/Turabian StyleNácher-Juan, Josep, María Carmen Terencio, María José Alcaraz, and María Luisa Ferrándiz. 2019. "Osteostatin Inhibits Collagen-Induced Arthritis by Regulation of Immune Activation, Pro-Inflammatory Cytokines, and Osteoclastogenesis" International Journal of Molecular Sciences 20, no. 16: 3845. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20163845