Genetic Ablation and Guanylyl Cyclase/Natriuretic Peptide Receptor-A: Impact on the Pathophysiology of Cardiovascular Dysfunction

Abstract
:1. Introduction
2. Historical Perspectives and Background
3. Ablation of Nppa and Nppb Triggers Hypertension and Cardiovascular Dysfunction
3.1. Gene Ablation of Nppa Increases Blood Pressure
3.2. Role of Nppa/Nppb in Cardiac Remodeling and Dysfunction
4. Genetic Disruption of Npr1 and Pathophysiology of Hypertension and Cardiovascular Events
4.1. Effect of Npr1 Gene Ablation on Hypertension
4.2. Effect of Npr1 Gene Disruption on the Pathophysiology of Cardiac Dysfunction
4.3. Effect of Npr1 Ablation on the Renin-Angiotensin System and Cardiovascular Disorders
5. Consequences of Genetic Duplication of Npr1
6. Genetic Disruption of Npr1 and Cardiac Hypertrophy, Fibrosis, and Inflammation
7. Association of Nppa, Nppb, and Npr1 Polymorphisms with Cardiovascular Dysfunction
8. Therapeutic Use of Natriuretic Peptides
9. Current and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- De Bold, A.J.; Borenstein, H.B.; Veress, A.T.; Sonnenberg, H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci. 1981, 28, 89–94. [Google Scholar] [CrossRef]
- de Bold, A.J. Atrial natriuretic factor: A hormone produced by the heart. Science 1985, 230, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Drewett, J.G.; Garbers, D.L. The family of guanylyl cyclase receptors and their ligands. Endocr. Rev. 1994, 15, 135–162. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.M.; Ballermann, B.J.; Gunning, M.E.; Zeidel, M.L. Diverse biological actions of atrial natriuretic peptide. Physiol. Rev. 1990, 70, 665–699. [Google Scholar] [CrossRef] [PubMed]
- McGrath, M.F.; de Bold, M.L.; de Bold, A.J. The endocrine function of the heart. Trends Endocrinol. Metab. 2005, 16, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.N. Biology of natriuretic peptides and their receptors. Peptides 2005, 26, 901–932. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.N. Emerging Roles of Natriuretic Peptides and their Receptors in Pathophysiology of Hypertension and Cardiovascular Regulation. J. Am. Soc. Hypertens 2008, 2, 210–226. [Google Scholar] [CrossRef] [PubMed]
- de Bold, A.J.; Ma, K.K.; Zhang, Y.; de Bold, M.L.; Bensimon, M.; Khoshbaten, A. The physiological and pathophysiological modulation of the endocrine function of the heart. Can. J. Physiol. Pharmacol. 2001, 79, 705–714. [Google Scholar] [CrossRef]
- Pandey, K.N. Molecular and genetic aspects of guanylyl cyclase natriuretic peptide receptor-A in regulation of blood pressure and renal function. Physiol. Genom. 2018, 50, 913–928. [Google Scholar] [CrossRef]
- Pandey, K.N. The functional genomics of guanylyl cyclase/natriuretic peptide receptor-A: Perspectives and paradigms. FEBS J. 2011, 278, 1792–1807. [Google Scholar] [CrossRef] [Green Version]
- LaPointe, M.C. Molecular regulation of the brain natriuretic peptide gene. Peptides 2005, 26, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Schulz, S. C-type natriuretic peptide and guanylyl cyclase B receptor. Peptides 2005, 26, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, A.; Seidman, C.E. Atrial natriuretic factor and related peptide hormones. Annu. Rev. Biochem. 1991, 60, 229–255. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Minamino, N.; Kangawa, K.; Matsuo, H. Cloning and sequence analysis of cDNA encoding a precursor for rat brain natriuretic peptide. Biochem. Biophys. Res. Commun. 1989, 159, 1420–1426. [Google Scholar] [CrossRef]
- Suga, S.; Nakao, K.; Hosoda, K.; Mukoyama, M.; Ogawa, Y.; Shirakami, G.; Arai, H.; Saito, Y.; Kambayashi, Y.; Inouye, K. Phenotype-related alteration in expression of natriuretic peptide receptors in aortic smooth muscle cells. Circ. Res. 1992, 71, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Koller, K.J.; de Sauvage, F.J.; Lowe, D.G.; Goeddel, D.V. Conservation of the kinaselike regulatory domain is essential for activation of the natriuretic peptide receptor guanylyl cyclases. Mol. Cell. Biol. 1992, 12, 2581–2590. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R.; Gardner, D.G.; Samson, W.K. Natriuretic peptides. NEJM 1998, 339, 321–328. [Google Scholar] [PubMed]
- Shi, S.J.; Nguyen, H.T.; Sharma, G.D.; Navar, L.G.; Pandey, K.N. Genetic disruption of atrial natriuretic peptide receptor-A alters renin and angiotensin II levels. Am. J. Physiol. Ren. Physiol. 2001, 281, F665–F673. [Google Scholar] [CrossRef]
- Zhao, D.; Vellaichamy, E.; Somanna, N.K.; Pandey, K.N. Guanylyl cyclase/natriuretic peptide receptor-A gene disruption causes increased adrenal angiotensin II and aldosterone levels. Am. J. Physiol. Ren. Physiol. 2007, 293, F121–F127. [Google Scholar] [CrossRef]
- Garbers, D.L.; Chrisman, T.D.; Wiegn, P.; Katafuchi, T.; Albanesi, J.P.; Bielinski, V.; Barylko, B.; Redfield, M.M.; Burnett, J.C., Jr. Membrane guanylyl cyclase receptors: An update. Trends Endocrinol. Metab. 2006, 17, 251–258. [Google Scholar] [CrossRef]
- Gardner, D.G. Natriuretic peptides: Markers or modulators of cardiac hypertrophy? Trends Endocrinol. Metab. 2003, 14, 411–416. [Google Scholar] [CrossRef]
- Richards, A.M. Natriuretic peptides: Update on Peptide release, bioactivity, and clinical use. Hypertension 2007, 50, 25–30. [Google Scholar] [CrossRef]
- Vollmer, A.M. The role of atrial natriuretic peptide in the immune system. Peptides 2005, 26, 1087–1094. [Google Scholar] [CrossRef]
- Vasan, R.S.; Benjamin, E.J.; Larson, M.G.; Leip, E.P.; Wang, T.J.; Wilson, P.W.; Levy, D. Plasma natriuretic peptides for community screening for left ventricular hypertrophy and systolic dysfunction: The Framingham heart study. JAMA 2002, 288, 1252–1259. [Google Scholar] [CrossRef]
- Pandey, K.N.; Singh, S. Molecular cloning and expression of murine guanylate cyclase/atrial natriuretic factor receptor cDNA. J. Biol. Chem. 1990, 265, 12342–12348. [Google Scholar]
- Schulz, S.; Singh, S.; Bellet, R.A.; Singh, G.; Tubb, D.J.; Chin, H.; Garbers, D.L. The primary structure of a plasma membrane guanylate cyclase demonstrates diversity within this new receptor family. Cell 1989, 58, 1155–1162. [Google Scholar] [CrossRef]
- Lucas, K.A.; Pitari, G.M.; Kazerounian, S.; Ruiz-Stewart, I.; Park, J.; Schulz, S.; Chepenik, K.P.; Waldman, S.A. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol. Rev. 2000, 52, 375–414. [Google Scholar]
- Tremblay, J.; Desjardins, R.; Hum, D.; Gutkowska, J.; Hamet, P. Biochemistry and physiology of the natriuretic peptide receptor guanylyl cyclases. Mol. Cell. Biochem. 2002, 230, 31–47. [Google Scholar] [CrossRef]
- Koller, K.J.; Goddel, D.V. Molecular biology of the natriuretic peptides and their receptors. Circulation 1992, 86, 1081–1088. [Google Scholar] [CrossRef]
- Garbers, D.L. Guanylyl cyclase receptors and their endocrine, paracrine, and autocrine ligands. Cell 1992, 71, 1–4. [Google Scholar] [CrossRef]
- Fuller, F.; Porter, J.G.; Arfsten, A.E.; Miller, J.; Schilling, J.W.; Scarborough, R.M.; Lewicki, J.A.; Schenk, D.B. Atrial natriuretic peptide clearance receptor. Complete sequence and functional expression of cDNA clones. J. Biol. Chem. 1988, 263, 9395–9401. [Google Scholar]
- Mani, I.; Garg, R.; Pandey, K.N. Role of FQQI motif in the internalization, trafficking, and signaling of guanylyl-cyclase/natriuretic peptide receptor-A in cultured murine mesangial cells. Am. J. Physiol. Ren. Physiol. 2016, 310, F68–F84. [Google Scholar] [CrossRef] [Green Version]
- Mani, I.; Pandey, K.N. Emerging concepts of receptor endocytosis and concurrent intracellular signaling: Mechanisms of guanylyl cyclase/natriuretic peptide receptor-A activation and trafficking. Cell Signal. 2019, 60, 17–30. [Google Scholar] [CrossRef]
- Pandey, K.N. Stoichiometric analysis of internalization, recycling, and redistribution of photoaffinity-labeled guanylate cyclase/atrial natriuretic factor receptors in cultured murine Leydig tumor cells. J. Biol. Chem. 1993, 268, 4382–4390. [Google Scholar]
- Brackmann, M.; Schuchmann, S.; Anand, R.; Braunewell, K.H. Neuronal Ca2+ sensor protein VILIP-1 affects cGMP signalling of guanylyl cyclase B by regulating clathrin-dependent receptor recycling in hippocampal neurons. J. Cell Sci. 2005, 118, 2495–2505. [Google Scholar] [CrossRef] [Green Version]
- Koh, G.Y.; Nussenzveig, D.R.; Okolicany, J.; Price, D.A.; Maack, T. Dynamics of atrial natriuretic factor-guanylate cyclase receptors and receptor-ligand complexes in cultured glomerular mesangial and renomedullary interstitial cells. J. Biol. Chem. 1992, 267, 11987–11994. [Google Scholar]
- Kuhn, M. Cardiac and intestinal natriuretic peptides: Insights from genetically modified mice. Peptides 2005, 26, 1078–1085. [Google Scholar] [CrossRef]
- Kishimoto, I.; Tokudome, T.; Nakao, K.; Kangawa, K. Natriuretic peptide system: An overview of studies using genetically engineered animal models. FEBS J. 2011, 278, 1830–1841. [Google Scholar] [CrossRef]
- Misono, K.S.; Philo, J.S.; Arakawa, T.; Ogata, C.M.; Qiu, Y.; Ogawa, H.; Young, H.S. Structure, signaling mechanism and regulation of the natriuretic peptide receptor guanylate cyclase. FEBS J. 2011, 278, 1818–1829. [Google Scholar] [CrossRef] [Green Version]
- Potter, L.R. Natriuretic peptide metabolism, clearance and degradation. FEBS J. 2011, 278, 1808–1817. [Google Scholar] [CrossRef] [Green Version]
- Goetz, K.L. Evidence that atriopeptin is not a physiological regulator of sodium excretion. Hypertension 1990, 15, 9–19. [Google Scholar] [CrossRef]
- Goetz, K.L. Renal natriuretic peptide (urodilatin?) and atriopeptin: Evolving concepts. Am. J. Physiol. 1991, 261, F921–F932. [Google Scholar] [CrossRef]
- Espiner, E.A. Physiology of natriuretic peptides. J. Intern. Med. 1994, 235, 527–541. [Google Scholar] [CrossRef]
- Sagnella, G.A. Measurement and significance of circulating natriuretic peptides in cardiovascular disease. Clin. Sci. (Lond.) 1998, 95, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Blaine, E. Atrial natriuretic factor plays a significant role in blody fluid hemostasis. Hypertension 1990, 15, 2–8. [Google Scholar] [CrossRef]
- McGrath, M.F.; de Bold, A.J. Determinants of natriuretic peptide gene expression. Peptides 2005, 26, 933–943. [Google Scholar] [CrossRef]
- Holmes, S.J.; Espiner, E.A.; Richards, A.M.; Yandle, T.G.; Frampton, C. Renal, endocrine, and hemodynamic effects of human brain natriuretic peptide in normal man. J. Clin. Endocrinol. Metab. 1993, 76, 91–96. [Google Scholar]
- Lang, C.C.; McAlpine, H.M.; Choy, A.M.; Pringle, T.H.; Coutie, W.J.; Struthers, A.D. Effect of pericardiocentesis on plasma levels of brain natriuretic peptide in cardiac tamponade. Am. J. Cardiol. 1992, 70, 1628–1629. [Google Scholar] [CrossRef]
- Lang, C.C.; Choy, A.M.; Struthers, A.D. Atrial and brain natriuretic peptides: A dual natriuretic peptide system potentially involved in circulatory homeostasis. Clin. Sci. (Lond.) 1992, 83, 519–527. [Google Scholar] [CrossRef]
- Yoshimura, M.; Yasue, H.; Morita, E.; Sakaino, N.; Jougasaki, M.; Kurose, M.; Mukoyama, M.; Saito, Y.; Nakao, K.; Imura, H. Hemodynamic, renal, and hormonal responses to brain natriuretic peptide infusion in patients with congestive heart failure. Circulation 1991, 84, 1581–1588. [Google Scholar] [CrossRef]
- Darbar, D.; Davidson, N.C.; Gillespie, N.; Choy, A.M.; Lang, C.C.; Shyr, Y.; McNeill, G.P.; Pringle, T.H.; Struthers, A.D. Diagnostic value of B-type natriuretic peptide concentrations in patients with acute myocardial infarction. Am. J. Cardiol. 1996, 78, 284–287. [Google Scholar] [CrossRef]
- Anand-Srivastava, M.B.; Trachte, G.J. Atrial natriuretic factor receptors and signal transduction mechanisms. Pharmacol. Rev. 1993, 45, 455–497. [Google Scholar]
- Lisy, O.; Huntley, B.K.; McCormick, D.J.; Kurlansky, P.A.; Burnett, J.C., Jr. Design synthesis, and actions of a novel chimeric natriuretic peptide: CD-NP. J. Am. Coll. Cardiol. 2008, 52, 60–68. [Google Scholar] [CrossRef]
- Lee, C.Y.; Lieu, H.; Burnett, J.C., Jr. Designer natriuretic peptides. J. Investig. Med. 2009, 57, 18–21. [Google Scholar] [CrossRef]
- Kuhn, M. Molecular Physiology of Membrane Guanylyl Cyclase Receptors. Physiol. Rev. 2016, 96, 751–804. [Google Scholar] [CrossRef]
- Schwab, T.R.; Edwards, B.S.; Heublein, D.M.; Burnett, J.C., Jr. Role of atrial natriuretic peptide in volume-expansion natriuresis. Am. J. Physiol. 1986, 251, R310–R313. [Google Scholar] [CrossRef]
- Delporte, C.; Poloczek, P.; Tastenoy, M.; Winand, J.; Christophe, J. Atrial natriuretic peptide binds to ANP-R1 receptors in neuroblastoma cells or is degraded extracellularly at the Ser-Phe bond. Eur. J. Pharmacol. 1992, 227, 247–256. [Google Scholar] [CrossRef]
- Delporte, C.; Winand, J.; Poloczek, P.; von Geldern, T.; Christophe, J. Discovery of a potent atrial natriuretic peptide antagonist for ANPA receptors in the human neuroblastoma NB-OK-1 cell line. Eur. J. Pharmacol. 1992, 224, 183–188. [Google Scholar] [CrossRef]
- Ohyama, Y.; Miyamoto, K.; Morishita, Y.; Matsuda, Y.; Saito, Y.; Minamino, N.; Kangawa, K.; Matsuo, H. Stable expression of natriuretic peptide receptors: Effects of HS-142–1, a non-peptide ANP antagonist. Biochem. Biophys. Res. Commun. 1992, 189, 336–342. [Google Scholar] [CrossRef]
- Khurana, M.L.; Pandey, K.N. Receptor-mediated stimulatory effect of atrial natriuretic factor, brain natriuretic peptide, and C-type natriuretic peptide on testosterone production in purified mouse Leydig cells: Activation of cholesterol side-chain cleavage enzyme. Endocrinology 1993, 133, 2141–2149. [Google Scholar] [CrossRef]
- Smithies, O.; Maeda, N. Gene targeting approaches to complex genetic diseases: Atherosclerosis and essential hypertension. Proc. Natl. Acad. Sci. USA 1995, 92, 5266–5272. [Google Scholar] [CrossRef]
- Matsukawa, N.; Grzesik, W.J.; Takahashi, N.; Pandey, K.N.; Pang, S.; Yamauchi, M.; Smithies, O. The natriuretic peptide clearance receptor locally modulates the physiological effects of the natriuretic peptide system. Pro. Natl. Acad. Sci. USA 1999, 96, 7403–7408. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Krege, J.H.; Kluckman, K.D.; Hagaman, J.R.; Hodgin, J.B.; Best, C.F.; Jennette, J.C.; Coffman, T.M.; Maeda, N.; Smithies, O. Genetic control of blood pressure and the angiotensinogen locus. Proc. Natl. Acad. Sci. USA 1995, 92, 2735–2739. [Google Scholar] [CrossRef]
- Oliver, P.M.; Fox, J.E.; Kim, R.; Rockman, H.A.; Kim, H.S.; Reddick, R.L.; Pandey, K.N.; Milgram, S.L.; Smithies, O.; Maeda, N. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc. Natl. Acad. Sci. USA 1997, 94, 14730–14735. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.J.; Vellaichamy, E.; Chin, S.Y.; Smithies, O.; Navar, L.G.; Pandey, K.N. Natriuretic peptide receptor A mediates renal sodium excretory responses to blood volume expansion. Am. J. Physiol. Ren. Physiol. 2003, 285, F694–F702. [Google Scholar] [CrossRef] [Green Version]
- Vellaichamy, E.; Khurana, M.L.; Fink, J.; Pandey, K.N. Involvement of the NF-kappa B/matrix metalloproteinase pathway in cardiac fibrosis of mice lacking guanylyl cyclase/natriuretic peptide receptor A. J. Biol. Chem. 2005, 280, 19230–19242. [Google Scholar] [CrossRef]
- Vellaichamy, E.; Zhao, D.; Somanna, N.; Pandey, K.N. Genetic disruption of guanylyl cyclase/natriuretic peptide receptor-A upregulates ACE and AT1 receptor gene expression and signaling: Role in cardiac hypertrophy. Physiol. Genom. 2007, 31, 193–202. [Google Scholar] [CrossRef]
- Das, S.; Au, E.; Krazit, S.T.; Pandey, K.N. Targeted disruption of guanylyl cyclase-A/natriuretic peptide receptor-A gene provokes renal fibrosis and remodeling in null mutant mice: Role of proinflammatory cytokines. Endocrinology 2010, 151, 5841–5850. [Google Scholar] [CrossRef]
- Das, S.; Periyasamy, R.; Pandey, K.N. Activation of IKK/NF-kappaB provokes renal inflammatory responses in guanylyl cyclase/natriuretic peptide receptor-A gene-knockout mice. Physiol. Genom. 2012, 44, 430–442. [Google Scholar] [CrossRef]
- Kumar, P.; Periyasamy, R.; Das, S.; Neerukonda, S.; Mani, I.; Pandey, K.N. All-trans retinoic acid and sodium butyrate enhance natriuretic peptide receptor a gene transcription: Role of histone modification. Mol. Pharmacol. 2014, 85, 946–957. [Google Scholar] [CrossRef]
- Oliver, P.M.; John, S.W.; Purdy, K.E.; Kim, R.; Maeda, N.; Goy, M.F.; Smithies, O. Natriuretic peptide receptor 1 expression influences blood pressures of mice in a dose-dependent manner. Proc. Natl. Acad. Sci. USA 1998, 95, 2547–2551. [Google Scholar] [CrossRef] [Green Version]
- Pandey, K.N.; Oliver, P.M.; Maeda, N.; Smithies, O. Hypertension associated with decreased testosterone levels in natriuretic peptide receptor-A gene-knockout and gene-duplicated mutant mouse models. Endocrinology 1999, 140, 5112–5119. [Google Scholar] [CrossRef]
- Vellaichamy, E.; Das, S.; Subramanian, U.; Maeda, N.; Pandey, K.N. Genetically altered mutant mouse models of guanylyl cyclase/natriuretic peptide receptor-A exhibit the cardiac expression of proinflammatory mediators in a gene-dose-dependent manner. Endocrinology 2014, 155, 1045–1056. [Google Scholar] [CrossRef]
- Kumar, P.; Gogulamudi, V.R.; Periasamy, R.; Raghavaraju, G.; Subramanian, U.; Pandey, K.N. Inhibition of HDAC enhances STAT acetylation, blocks NF-kappaB, and suppresses the renal inflammation and fibrosis in Npr1 haplotype male mice. Am. J. Physiol. Ren. Physiol. 2017, 313, F781–F795. [Google Scholar] [CrossRef]
- Gogulamudi, V.R.; Mani, I.; Subramanian, U.; Pandey, K.N. Genetic disruption of Npr1 depletes regulatory T cells and provokes high levels of proinflammatory cytokines and fibrosis in the kidneys of female mutant mice. Am. J. Physiol. Ren. Physiol. 2019, 316, F1254–F1272. [Google Scholar] [CrossRef]
- Periyasamy, R.; Das, S.; Pandey, K.N. Genetic disruption of guanylyl cyclase/natriuretic peptide receptor-A upregulates renal (pro) renin receptor expression in Npr1 null mutant mice. Peptides 2019, 114, 17–28. [Google Scholar] [CrossRef]
- Subramanian, U.; Kumar, P.; Mani, I.; Chen, D.; Kessler, I.; Periyasamy, R.; Raghavaraju, G.; Pandey, K.N. Retinoic acid and sodium butyrate suppress the cardiac expression of hypertrophic markers and proinflammatory mediators in Npr1 gene-disrupted haplotype mice. Physiol. Genom. 2016, 48, 477–490. [Google Scholar] [CrossRef]
- Kim, H.S.; Lu, G.; John, S.W.M.; Maeda, N.; Smithies, O. Molecular phenotyping for analyzing subtle genetic effects in mice application to an angiotensinogen gene titration. Proc. Natl. Acad. Sci. USA 2002, 99, 4602–4607. [Google Scholar] [CrossRef]
- Takahashi, N.; Smithies, O. Gene targeting approaches to analyzing hypertension. J. Am. Soc. Nephrol. 1999, 10, 1598–1605. [Google Scholar]
- John, S.W.; Krege, J.H.; Oliver, P.M.; Hagaman, J.R.; Hodgin, J.B.; Pang, S.C.; Flynn, T.G.; Smithies, O. Genetic decreases in atrial natriuretic peptide and salt-sensitive hypertension. Science 1995, 267, 679–681. [Google Scholar] [CrossRef]
- Tamura, N.; Ogawa, Y.; Chusho, H.; Nakamura, K.; Nakao, K.; Suda, M.; Kasahara, M.; Hashimoto, R.; Katsuura, G.; Mukoyama, M.; et al. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc. Natl. Acad. Sci. USA 2000, 97, 4239–4244. [Google Scholar] [CrossRef] [Green Version]
- Melo, L.G.; Veress, A.T.; Ackermann, U.; Steinhelper, M.E.; Pang, S.C.; Tse, Y.; Sonnenberg, H. Chronic regulation of arterial blood pressure in ANP transgenic and knockout mice: Role of cardiovascular sympathetic tone. Cardiovasc. Res. 1999, 43, 437–444. [Google Scholar] [CrossRef]
- Steinhelper, M.E.; Cochrane, K.L.; Field, L.J. Hypotension in transgenic mice expressing atrial natriuretic factor fusion genes. Hypertension 1990, 16, 301–307. [Google Scholar] [CrossRef]
- Lin, K.F.; Chao, J.; Chao, L. Human atrial natriuretic peptide gene delivery reduces blood pressure in hypertensive rats. Hypertension 1995, 26, 847–853. [Google Scholar] [CrossRef]
- Schillinger, K.J.; Tsai, S.Y.; Taffet, G.E.; Reddy, A.K.; Marian, A.J.; Entman, M.L.; Oka, K.; Chan, L.; O’Malley, B.W. Regulatable atrial natriuretic peptide gene therapy for hypertension. Proc. Natl. Acad. Sci. USA 2005, 102, 13789–13794. [Google Scholar] [CrossRef] [Green Version]
- Klinger, J.R.; Cutaia, M. The natriuretic peptides. Clinical applications in patients with COPD. Chest 1996, 110, 1136–1138. [Google Scholar] [CrossRef]
- Naruse, M.; Takeyama, Y.; Tanabe, A.; Hiroshige, J.; Naruse, K.; Yoshimoto, T.; Tanaka, M.; Katagiri, T.; Demura, H. Atrial and brain natriuretic peptides in cardiovascular diseases. Hypertension 1994, 23, I231–I234. [Google Scholar] [CrossRef]
- Klinger, J.R.; Petit, R.D.; Curtin, L.A.; Warburton, R.R.; Wrenn, D.S.; Steinhelper, M.E.; Field, L.J.; Hill, N.S. Cardiopulmonary responses to chronic hypoxia in transgenic mice that overexpress ANP. J. Appl. Physiol. 1993, 75, 198–205. [Google Scholar] [CrossRef]
- Hohne, C.; Drzimalla, M.; Krebs, M.O.; Boemke, W.; Kaczmarczyk, G. Atrial natriuretic peptide ameliorates hypoxic pulmonary vasoconstriction without influencing systemic circulation. J. Physiol. Pharmacol. 2003, 54, 497–510. [Google Scholar]
- Klinger, J.R.; Warburton, R.R.; Pietras, L.; Oliver, P.; Fox, J.; Smithies, O.; Hill, N.S. Targeted disruption of the gene for natriuretic peptide receptor-A worsens hypoxia-induced cardiac hypertrophy. Am. J. Physiol. 2002, 282, H58–H65. [Google Scholar] [CrossRef]
- Klinger, J.R.; Warburton, R.R.; Pietras, L.A.; Smithies, O.; Swift, R.; Hill, N.S. Genetic disruption of atrial natriuretic peptide causes pulmonary hypertension in normoxic and hypoxic mice. Am. J. Physiol. 1999, 276, L868–L874. [Google Scholar] [CrossRef]
- Sun, J.Z.; Chen, S.J.; Li, G.; Chen, Y.F. Hypoxia reduces atrial natriuretic peptide clearance receptor gene expression in ANP knockout mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L511–L519. [Google Scholar] [CrossRef]
- Melo, L.G.; Sonnenberg, H. Requirement for prostaglandin synthesis in secretion of atrial natriuretic factor from isolated rat heart. Regul. Pept. 1995, 60, 79–87. [Google Scholar] [CrossRef]
- Melo, L.G.; Veress, A.T.; Chong, C.K.; Pang, S.C.; Flynn, T.G.; Sonnenberg, H. Salt-sensitive hypertension in ANP knockout mice: Potential role of abnormal plasma renin activity. Am. J. Physiol. 1998, 274, R255–R261. [Google Scholar] [CrossRef]
- Itoh, H.; Nakao, K.; Mukoyama, M.; Yamada, T.; Hosoda, K.; Shirakami, G.; Morii, N.; Sugawara, A.; Saito, Y.; Shiono, S.; et al. Chronic blockade of endogenous atrial natriuretic polypeptide (ANP) by monoclonal antibody against ANP accelerates the development of hypertension in spontaneously hypertensive and deoxycorticosterone acetate-salt-hypertensive rats. J. Clin. Investig. 1989, 84, 145–154. [Google Scholar] [CrossRef]
- Felker, G.M.; Petersen, J.W.; Mark, D.B. Natriuretic peptides in the diagnosis and management of heart failure. CMAJ 2006, 175, 611–617. [Google Scholar] [CrossRef] [Green Version]
- Reinhart, K.; Meisner, M.; Brunkhorst, F.M. Markers for sepsis diagnosis: What is useful? Crit. Care Clin. 2006, 22, 503–519. [Google Scholar] [CrossRef]
- See, R.; de Lemos, J.A. Current status of risk stratification methods in acute coronary syndromes. Curr. Cardiol. Rep. 2006, 8, 282–288. [Google Scholar] [CrossRef]
- Jaffe, A.S.; Babuin, L.; Apple, F.S. Biomarkers in acute cardiac disease: The present and the future. J. Am. Coll. Cardiol. 2006, 48, 1–11. [Google Scholar] [CrossRef]
- Ellmers, L.J.; Scott, N.J.; Piuhola, J.; Maeda, N.; Smithies, O.; Frampton, C.M.; Richards, A.M.; Cameron, V.A. Npr1-regulated gene pathways contributing to cardiac hypertrophy and fibrosis. J. Mol. Endocrinol. 2007, 38, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Das, S.; Pandey, K.N. Interactive roles of Npr1 gene-dosage and salt diets on cardiac angiotensin II, aldosterone and pro-inflammatory cytokines levels in mutant mice. J. Hypertens. 2013, 31, 134–144. [Google Scholar] [CrossRef]
- Chen, H.H.; Burnett, J.C., Jr. The natriuretic peptides in heart failure: Diagnostic and therapeutic potentials. Proc. Assoc. Am. Physicians 1999, 111, 406–416. [Google Scholar] [CrossRef]
- Nakao, K.; Itoh, H.; Saito, Y.; Mukoyama, M.; Ogawa, Y. The natriuretic peptide family. Curr. Opin. Nephrol. Hypertens. 1996, 5, 4–11. [Google Scholar] [CrossRef]
- Tsutamoto, T.; Kanamori, T.; Morigami, N.; Sugimoto, Y.; Yamaoka, O.; Kinoshita, M. Possibility of downregulation of atrial natriuretic peptide receptor coupled to guanylate cyclase in peripheral vascular beds of patients with chronic severe heart failure. Circulation 1993, 87, 70–75. [Google Scholar] [CrossRef]
- Mukoyama, M.; Nakao, K.; Hosoda, K.; Suga, S.; Saito, Y.; Ogawa, Y.; Shirakami, G.; Jougasaki, M.; Obata, K.; Yasue, H.; et al. Brain natriuretic peptide as a novel cardiac hormone in humans. Evidence for an exquisite dual natriuretic peptide system, atrial natriuretic peptide and brain natriuretic peptide. J. Clin. Investig. 1991, 87, 1402–1412. [Google Scholar] [CrossRef]
- Knowles, J.W.; Esposito, G.; Mao, L.; Hagaman, J.R.; Fox, J.E.; Smithies, O.; Rockman, H.A.; Maeda, N. Pressure-independent enhancement of cardiac hypertrophy in natriuretic peptide receptor A-deficient mice. J. Clin. Investig. 2001, 107, 975–984. [Google Scholar] [CrossRef]
- Xue, H.; Wang, S.; Wang, H.; Sun, K.; Song, X.; Zhang, W.; Fu, C.; Han, Y.; Hui, R. Atrial natriuretic peptide gene promoter polymorphism is associated with left ventricular hypertrophy in hypertension. Clin. Sci. (Lond.) 2008, 114, 131–137. [Google Scholar] [CrossRef]
- Doust, J.A.; Pietrzak, E.; Dobson, A.; Glasziou, P. How well does B-type natriuretic peptide predict death and cardiac events in patients with heart failure: Systematic review. BWJ 2005, 330, 625. [Google Scholar] [CrossRef]
- Khan, I.A.; Fink, J.; Nass, C.; Chen, H.; Christenson, R.; de Filippi, C.R. N-terminal pro-B-type natriuretic peptide and B-type natriuretic peptide for identifying coronary artery disease and left ventricular hypertrophy in ambulatory chronic kidney disease patients. Am. J. Cardiol. 2006, 97, 1530–1534. [Google Scholar] [CrossRef]
- Girsen, A.; Ala-Kopsala, M.; Makikallio, K.; Vuolteenaho, O.; Rasanen, J. Cardiovascular hemodynamics and umbilical artery N-terminal peptide of proB-type natriuretic peptide in human fetuses with growth restriction. UOG: ISUOG 2007, 29, 296–303. [Google Scholar] [CrossRef]
- Kocylowski, R.D.; Dubiel, M.; Gudmundsson, S.; Sieg, I.; Fritzer, E.; Alkasi, O.; Breborowicz, G.H.; von Kaisenberg, C.S. Biochemical tissue-specific injury markers of the heart and brain in postpartum cord blood. Am. J. Obstet. Gynecol. 2009, 200, 273 e1–273 e25. [Google Scholar] [CrossRef]
- Zahabi, A.; Picard, S.; Fortin, N.; Reudelhuber, T.L.; Deschepper, C.F. Expression of constitutively active guanylate cyclase in cardiomyocytes inhibits the hypertrophic effects of isoproterenol and aortic constriction on mouse hearts. J. Biol. Chem. 2003, 278, 47694–47699. [Google Scholar] [CrossRef]
- Deschepper, C.F.; Masciotra, S.; Zahabi, A.; Boutin-Ganache, I.; Picard, S.; Reudelhuber, T.L. Function alterations of the Nppa promoter are linked to cardiac ventricular hypertrophy in WKY/WKHA rat crosses. Circ. Res. 2001, 88, 223–228. [Google Scholar] [CrossRef]
- Lattion, A.L.; Michel, J.B.; Arnauld, E.; Corvol, P.; Soubrier, F. Myocardial recruitment during ANF mRNA increase with volume overload in the rat. Am. J. Physiol. 1986, 251, H890–H896. [Google Scholar] [CrossRef]
- Hunter, J.J.; Chien, K.R. Signaling pathways for cardiac hypertrophy and failure. NEJM 1999, 341, 1276–1283. [Google Scholar] [CrossRef]
- Hasegawa, K.; Fujiwara, H.; Doyama, K.; Miyamae, M.; Fujiwara, T.; Suga, S.; Mukoyama, M.; Nakao, K.; Imura, H.; Sasayama, S. Ventricular expression of brain natriuretic peptide in hypertrophic cardiomyopathy. Circulation 1993, 88, 372–380. [Google Scholar] [CrossRef]
- Hasegawa, K.; Fujiwara, H.; Doyama, K.; Mukoyama, M.; Nakao, K.; Fujiwara, T.; Imura, H.; Kawai, C. Ventricular expression of atrial and brain natriuretic peptides in dilated cardiomyopathy. An immunohistocytochemical study of the endomyocardial biopsy specimens using specific monoclonal antibodies. Am. J. Pathol. 1993, 142, 107–116. [Google Scholar]
- Knowlton, K.U.; Rockman, H.A.; Itani, M.; Vovan, A.; Seidman, C.E.; Chien, K.R. Divergent pathways mediate the induction of ANF transgenes in neonatal and hypertrophic ventricular myocardium. J. Clin. Investig. 1995, 96, 1311–1318. [Google Scholar] [CrossRef]
- Calderone, A.; Thaik, C.M.; Takahashi, N.; Chang, D.L.; Colucci, W.S. Nitric oxide, atrial natriuretic peptide, and cyclic GMP inhibit the growth-promoting effects of norepinephrine in cardiac myocytes and fibroblasts. J. Clin. Investig. 1998, 101, 812–818. [Google Scholar] [CrossRef]
- Horio, T.; Nishikimi, T.; Yoshihara, F.; Matsuo, H.; Takishita, S.; Kangawa, K. Inhibitory regulation of hypertrophy by endogenous atrial natriuretic peptide in cultured cardiac myocytes. Hypertension 2000, 35, 19–24. [Google Scholar] [CrossRef]
- Wu, C.F.; Bishopric, N.H.; Pratt, R.E. Atrial natriuretic peptide induces apoptosis in neonatal rat cardiac myocytes. J. Biol. Chem. 1997, 272, 14860–14866. [Google Scholar] [CrossRef]
- Silberbach, M.; Gorenc, T.; Hershberger, R.E.; Stork, P.J.; Steyger, P.S.; Roberts, C.T., Jr. Extracellular signal-regulated protein kinase activation is required for the anti-hypertrophic effect of atrial natriuretic factor in neonatal rat ventricular myocytes. J. Biol. Chem. 1999, 274, 24858–24864. [Google Scholar] [CrossRef]
- Kishimoto, I.; Tokudome, T.; Horio, T.; Garbers, D.L.; Nakao, K.; Kangawa, K. Natriuretic Peptide Signaling via Guanylyl Cyclase (GC)-A: An Endogenous Protective Mechanism of the Heart. Curr. Cardiol. Rev. 2009, 5, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, Y.; Itoh, H.; Tamura, N.; Suga, S.; Yoshimasa, T.; Uehira, M.; Matsuda, S.; Shiono, S.; Nishimoto, H.; Nakao, K. Molecular cloning of the complementary DNA and gene that encode mouse brain natriuretic peptide and generation of transgenic mice that overexpress the brain natriuretic peptide gene. J. Clin. Investig. 1994, 93, 1911–1921. [Google Scholar] [CrossRef]
- Chusho, H.; Tamura, N.; Ogawa, Y.; Yasoda, A.; Suda, M.; Miyazawa, T.; Nakamura, K.; Nakao, K.; Kurihara, T.; Komatsu, Y.; et al. Dwarfism and early death in mice lacking C-type natriuretic peptide. Proc. Natl. Acad. Sci. USA 2001, 98, 4016–4021. [Google Scholar] [CrossRef] [Green Version]
- Yasoda, A.; Komatsu, Y.; Chusho, H.; Miyazawa, T.; Ozasa, A.; Miura, M.; Kurihara, T.; Rogi, T.; Tanaka, S.; Suda, M.; et al. Overexpression of CNP in chondrocytes rescues achondroplasia through a MAPK-dependent pathway. Nat. Med. 2004, 10, 80–86. [Google Scholar] [CrossRef]
- Wang, Y.; de Waard, M.C.; Sterner-Kock, A.; Stepan, H.; Schultheiss, H.P.; Duncker, D.J.; Walther, T. Cardiomyocyte-restricted over-expression of C-type natriuretic peptide prevents cardiac hypertrophy induced by myocardial infarction in mice. Eur. J. Heart Fail. 2007, 9, 548–557. [Google Scholar] [CrossRef]
- Lopez, M.J.; Wong, S.K.; Kishimoto, I.; Dubois, S.; Mach, V.; Friesen, J.; Garbers, D.L.; Beuve, A. Salt-resistant hypertension in mice lacking the guanylyl cyclase-A receptor for atrial natriuretic peptide. Nature 1995, 378, 65–68. [Google Scholar] [CrossRef]
- Tamura, N.; Doolittle, L.K.; Hammer, R.E.; Shelton, J.M.; Richardson, J.A.; Garbers, D.L. Critical roles of the guanylyl cyclase B receptor in endochondral ossification and development of female reproductive organs. Proc. Natl. Acad. Sci. USA 2004, 101, 17300–17305. [Google Scholar] [CrossRef] [Green Version]
- Langenickel, T.H.; Buttgereit, J.; Pagel-Langenickel, I.; Lindner, M.; Monti, J.; Beuerlein, K.; Al-Saadi, N.; Plehm, R.; Popova, E.; Tank, J.; et al. Cardiac hypertrophy in transgenic rats expressing a dominant-negative mutant of the natriuretic peptide receptor B. Proc. Natl. Acad. Sci. USA 2006, 103, 4735–4740. [Google Scholar] [CrossRef] [Green Version]
- Kishimoto, I.; Dubois, S.K.; Garbers, D.L. The heart communicates with the kidney exclusively through the guanylyl cyclase-A receptor: Acute handling of sodium and water in response to volume expansion. Proc. Natl. Acad. Sci. USA 1996, 93, 6215–6219. [Google Scholar] [CrossRef]
- Kishimoto, I.; Garbers, D.L. Physiological regulation of blood pressure and kidney function by guanylyl cyclase isoforms. Curr. Opin. Nephrol. Hypertens. 1997, 6, 58–63. [Google Scholar] [CrossRef]
- Nakayama, T.; Soma, M.; Takahashi, Y.; Rehemudula, D.; Kanmatsuse, K.; Furuya, K. Functional deletion mutation of the 5’-flanking region of type A human natriuretic peptide receptor gene and its association with essential hypertension and left ventricular hypertrophy in the Japanese. Circ. Res. 2000, 86, 841–845. [Google Scholar] [CrossRef]
- Pandey, K.N. Ligand-mediated endocytosis and intracellular sequestration of guanylyl cyclase/natriuretic peptide receptors: Role of GDAY motif. Mol. Cell. Biochem. 2010, 334, 81–98. [Google Scholar] [CrossRef]
- Nakanishi, M.; Saito, Y.; Kishimoto, I.; Harada, M.; Kuwahara, K.; Takahashi, N.; Kawakami, R.; Nakagawa, Y.; Tanimoto, K.; Yasuno, S.; et al. Role of natriuretic peptide receptor guanylyl cyclase-A in myocardial infarction evaluated using genetically engineered mice. Hypertension 2005, 46, 441–447. [Google Scholar] [CrossRef]
- Ellmers, L.J.; Knowles, J.W.; Kim, H.S.; Smithies, O.; Maeda, N.; Cameron, V.A. Ventricular expression of natriuretic peptides in Npr1(-/-) mice with cardiac hypertrophy and fibrosis. Am. J. Physiol. 2002, 283, H707–H714. [Google Scholar]
- Scott, N.J.; Ellmers, L.J.; Lainchbury, J.G.; Maeda, N.; Smithies, O.; Richards, A.M.; Cameron, V.A. Influence of natriuretic peptide receptor-1 on survival and cardiac hypertrophy during development. Biochim. Biophys. Acta 2009, 1792, 1175–1184. [Google Scholar] [CrossRef] [Green Version]
- Pandey, K.N.; Vellaichamy, E. Regulation of cardiac angiotensin-converting enzyme and angiotensin AT1 receptor gene expression in Npr1 gene-disrupted mice. Clin. Exp. Pharmacol. Physiol. 2010, 37, e70–e77. [Google Scholar] [CrossRef]
- Parthasarathy, A.; Gopi, V.; Umadevi, S.; Simna, A.; Sheik, M.J.; Divya, H.; Vellaichamy, E. Suppression of atrial natriuretic peptide/natriuretic peptide receptor-A-mediated signaling upregulates angiotensin-II-induced collagen synthesis in adult cardiac fibroblasts. Mol. Cell. Biochem. 2013, 378, 217–228. [Google Scholar] [CrossRef]
- Li, Y.; Kishimoto, I.; Saito, Y.; Harada, M.; Kuwahara, K.; Izumi, T.; Takahashi, N.; Kawakami, R.; Tanimoto, K.; Nakagawa, Y.; et al. Guanylyl cyclase-A inhibits angiotensin II type 1A receptor-mediated cardiac remodeling, an endogenous protective mechanism in the heart. Circulation 2002, 106, 1722–1728. [Google Scholar] [CrossRef]
- Kilic, A.; Bubikat, A.; Gassner, B.; Baba, H.A.; Kuhn, M. Local actions of atrial natriuretic peptide counteract angiotensin II stimulated cardiac remodeling. Endocrinology 2007, 148, 4162–4169. [Google Scholar] [CrossRef]
- Holtwick, R.; van Eickels, M.; Skryabin, B.V.; Baba, H.A.; Bubikat, A.; Begrow, F.; Schneider, M.D.; Garbers, D.L.; Kuhn, M. Pressure-independent cardiac hypertrophy in mice with cardiomyocyte-restricted inactivation of the atrial natriuretic peptide receptor guanylyl cyclase-A. J. Clin. Investig. 2003, 111, 1399–1407. [Google Scholar] [CrossRef] [Green Version]
- Morita, E.; Yasue, H.; Yoshimura, M.; Ogawa, H.; Jougasaki, M.; Matsumura, T.; Mukoyama, M.; Nakao, K. Increased plasma levels of brain natriuretic peptide in patients with acute myocardial infarction. Circulation 1993, 88, 82–91. [Google Scholar] [CrossRef]
- Phillips, P.A.; Sasadeus, J.; Hodsman, G.P.; Horowitz, J.; Saltups, A.; Johnston, C.I. Plasma atrial natriuretic peptide in patients with acute myocardial infarction: Effects of streptokinase. Br. Heart J. 1989, 61, 139–143. [Google Scholar] [CrossRef]
- Tomoda, H. Atrial natriuretic peptide in acute myocardial infarction. Am. J. Cardiol. 1988, 62, 1122–1123. [Google Scholar] [CrossRef]
- Newton-Cheh, C.; Larson, M.G.; Vasan, R.S.; Levy, D.; Bloch, K.D.; Surti, A.; Guiducci, C.; Kathiresan, S.; Benjamin, E.J.; Struck, J.; et al. Association of common variants in NPPA and NPPB with circulating natriuretic peptides and blood pressure. Nat. Genet. 2009, 41, 348–353. [Google Scholar] [CrossRef] [Green Version]
- Rubattu, S.; Bigatti, G.; Evangelista, A.; Lanzani, C.; Stanzione, R.; Zagato, L.; Manunta, P.; Marchitti, S.; Venturelli, V.; Bianchi, G.; et al. Association of atrial natriuretic peptide and type a natriuretic peptide receptor gene polymorphisms with left ventricular mass in human essential hypertension. J. Am. Coll. Cardiol. 2006, 48, 499–505. [Google Scholar] [CrossRef]
- Webber, M.A.; Marder, S.R. Better pharmacotherapy for schizophrenia: What does the future hold? Curr. Psychiatry Rep. 2008, 10, 352–358. [Google Scholar] [CrossRef]
- Pitzalis, M.V.; Sarzani, R.; Dessi-Fulgheri, P.; Iacoviello, M.; Forleo, C.; Lucarelli, K.; Pietrucci, F.; Salvi, F.; Sorrentino, S.; Romito, R.; et al. Allelic variants of natriuretic peptide receptor genes are associated with family history of hypertension and cardiovascular phenotype. J. Hypertens. 2003, 21, 1491–1496. [Google Scholar] [CrossRef]
- Usami, S.; Kishimoto, I.; Saito, Y.; Harada, M.; Kuwahara, K.; Nakagawa, Y.; Nakanishi, M.; Yasuno, S.; Kangawa, K.; Nakao, K. Association of CT dinucleotide repeat polymorphism in the 5’-flanking region of the guanylyl cyclase (GC)-A gene with essential hypertension in the Japanese. Hypertens. Res. 2008, 31, 89–96. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z.; Yu, C. Association of Polymorphisms in the Atrial Natriuretic Factor Gene with the Risk of Essential Hypertension: A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2016, 13, 458. [Google Scholar] [CrossRef]
- Ji, W.; Foo, J.N.; O’Roak, B.J.; Zhao, H.; Larson, M.G.; Simon, D.B.; Newton-Cheh, C.; State, M.W.; Levy, D.; Lifton, R.P. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 2008, 40, 592–599. [Google Scholar] [CrossRef] [Green Version]
- Lifton, R.P.; Gharavi, A.G.; Geller, D.S. Molecular mechanisms of human hypertension. Cell 2001, 104, 545–556. [Google Scholar] [CrossRef]
- Cannone, V.; Cefalu, A.B.; Noto, D.; Scott, C.G.; Bailey, K.R.; Cavera, G.; Pagano, M.; Sapienza, M.; Averna, M.R.; Burnett, J.C., Jr. The atrial natriuretic peptide genetic variant rs5068 is associated with a favorable cardiometabolic phenotype in a Mediterranean population. Diabetes Care 2013, 36, 2850–2856. [Google Scholar] [CrossRef]
- Cannone, V.; Scott, C.G.; Decker, P.A.; Larson, N.B.; Palmas, W.; Taylor, K.D.; Wang, T.J.; Gupta, D.K.; Bielinski, S.J.; Burnett, J.C., Jr. A favorable cardiometabolic profile is associated with the G allele of the genetic variant rs5068 in African Americans: The Multi-Ethnic Study of Atherosclerosis (MESA). PLoS ONE 2017, 12, e0189858. [Google Scholar] [CrossRef]
- Knowles, J.W.; Erickson, L.M.; Guy, V.K.; Sigel, C.S.; Wilder, J.C.; Maeda, N. Common variations in noncoding regions of the human natriuretic peptide receptor A gene have quantitative effects. Hum. Genet. 2003, 112, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.; DeStefano, A.L.; Larson, M.G.; O’Donnell, C.J.; Lifton, R.P.; Gavras, H.; Cupples, L.A.; Myers, R.H. Evidence for a gene influencing blood pressure on chromosome 17. Genome scan linkage results for longitudinal blood pressure phenotypes in subjects from the framingham heart study. Hypertension 2000, 36, 477–483. [Google Scholar] [CrossRef]
- Suwa, M.; Seino, Y.; Nomachi, Y.; Matsuki, S.; Funahashi, K. Multicenter prospective investigation on efficacy and safety of carperitide for acute heart failure in the ‘real world’ of therapy. Circ. J. 2005, 69, 283–290. [Google Scholar] [CrossRef]
- Park, K.; Itoh, H.; Yamahara, K.; Sone, M.; Miyashita, K.; Oyamada, N.; Sawada, N.; Taura, D.; Inuzuka, M.; Sonoyama, T.; et al. Therapeutic potential of atrial natriuretic peptide administration on peripheral arterial diseases. Endocrinology 2008, 149, 483–491. [Google Scholar] [CrossRef]
- Colucci, W.S.; Elkayam, U.; Horton, D.P.; Abraham, W.T.; Bourge, R.C.; Johnson, A.D.; Wagoner, L.E.; Givertz, M.M.; Liang, C.S.; Neibaur, M.; et al. Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. Nesiritide Study Group. NEJM 2000, 343, 246–253. [Google Scholar] [CrossRef]
- Nishikimi, T.; Kuwahara, K.; Nakagawa, Y.; Kangawa, K.; Minamino, N.; Nakao, K. Complexity of molecular forms of B-type natriuretic peptide in heart failure. Heart 2013, 99, 677–679. [Google Scholar] [CrossRef]
- Tokudome, T.; Kishimoto, I.; Yamahara, K.; Osaki, T.; Minamino, N.; Horio, T.; Sawai, K.; Kawano, Y.; Miyazato, M.; Sata, M.; et al. Impaired recovery of blood flow after hind-limb ischemia in mice lacking guanylyl cyclase-A, a receptor for atrial and brain natriuretic peptides. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1516–1521. [Google Scholar] [CrossRef]
- Kuhn, M.; Volker, K.; Schwarz, K.; Carbajo-Lozoya, J.; Flogel, U.; Jacoby, C.; Stypmann, J.; van Eickels, M.; Gambaryan, S.; Hartmann, M.; et al. The natriuretic peptide/guanylyl cyclase—A system functions as a stress-responsive regulator of angiogenesis in mice. J. Clin. Investig. 2009, 119, 2019–2030. [Google Scholar] [CrossRef]
- Dohi, K.; Ito, M. Novel diuretic strategies for the treatment of heart failure in Japan. Circ. J. 2014, 78, 1816–1823. [Google Scholar] [CrossRef]
- Hayek, S.; Nemer, M. Cardiac natriuretic peptides: From basic discovery to clinical practice. Cardiovasc. Ther. 2011, 29, 362–376. [Google Scholar] [CrossRef]
- Saito, Y. Roles of atrial natriuretic peptide and its therapeutic use. J. Cardiol. 2010, 56, 262–270. [Google Scholar] [CrossRef] [Green Version]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Investigators, Committees, Angiotensin-neprilysin inhibition versus enalapril in heart failure. NEJM 2014, 371, 993–1004. [Google Scholar] [CrossRef]


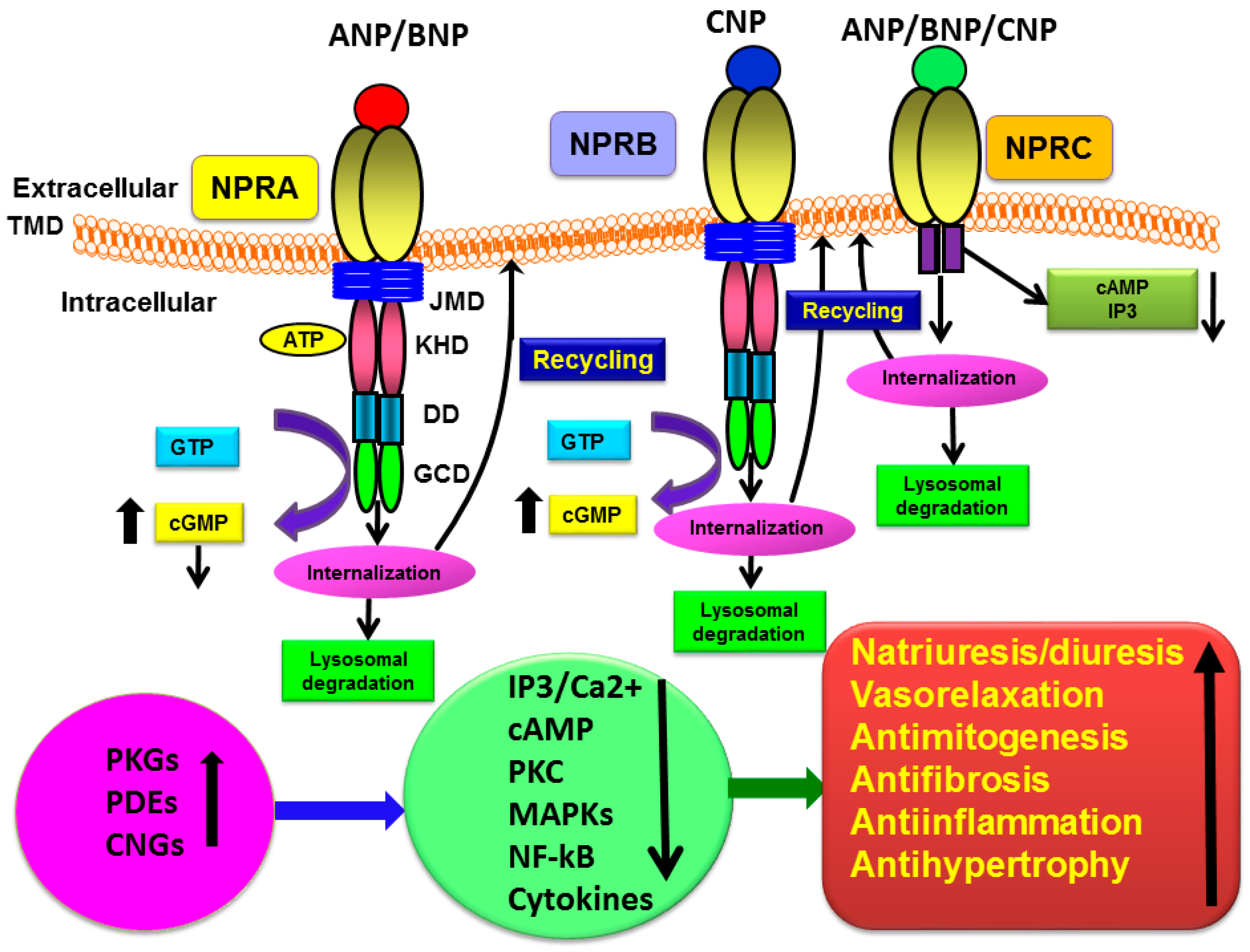{kind=link}
| Gene | Peptide/Protein | Gene-Knockout |
|---|---|---|
| Nomenclature | Nomenclature | Phenotype in Mouse |
| Nppa | ANP | Volume overload, high blood pressure and hypertension, salt sensitivity, fibrosis, and cardiac disorders [80,82,83,84] |
| Nppb | BNP | Hypertension, sodium excretion, vascular complication, and fibrosis [81,124] |
| Nppc | CNP | Dwarfism, reduced bone growth, and impaired endochondral ossification [125,126,127] |
| Npr1 | NPRA | High blood pressure and hypertension, salt sensitivity, volume overload, and cardiac hypertrophy, fibrosis, and inflammation [64,65,66,73,77,97,100,128] |
| Npr2 | NPRB | Dwarfism, seizures, female sterility, and decreased adiposity [129,130] |
| Npr3 | NPRC | Bone deformation and long bone overgrowth [62] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, K.N. Genetic Ablation and Guanylyl Cyclase/Natriuretic Peptide Receptor-A: Impact on the Pathophysiology of Cardiovascular Dysfunction. Int. J. Mol. Sci. 2019, 20, 3946. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20163946
Pandey KN. Genetic Ablation and Guanylyl Cyclase/Natriuretic Peptide Receptor-A: Impact on the Pathophysiology of Cardiovascular Dysfunction. International Journal of Molecular Sciences. 2019; 20(16):3946. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20163946
Chicago/Turabian StylePandey, Kailash N. 2019. "Genetic Ablation and Guanylyl Cyclase/Natriuretic Peptide Receptor-A: Impact on the Pathophysiology of Cardiovascular Dysfunction" International Journal of Molecular Sciences 20, no. 16: 3946. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20163946