Ceritinib-Induced Regression of an Insulin-Like Growth Factor-Driven Neuroepithelial Brain Tumor


 , , , add
Show full author list
, , , add
Show full author list
Abstract
:1. Introduction
2. Results
2.1. Clinical Description
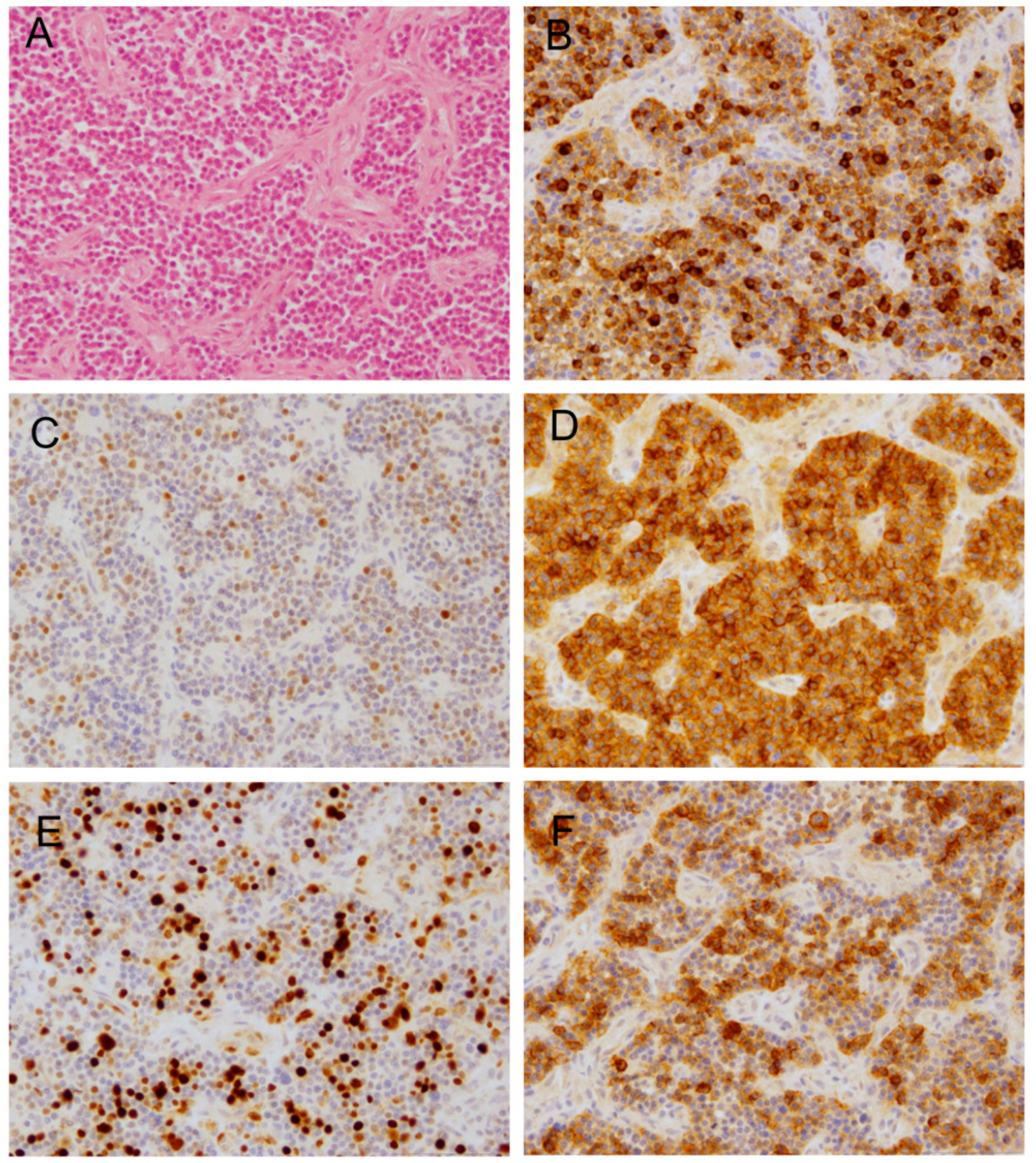
2.2. Histopathology
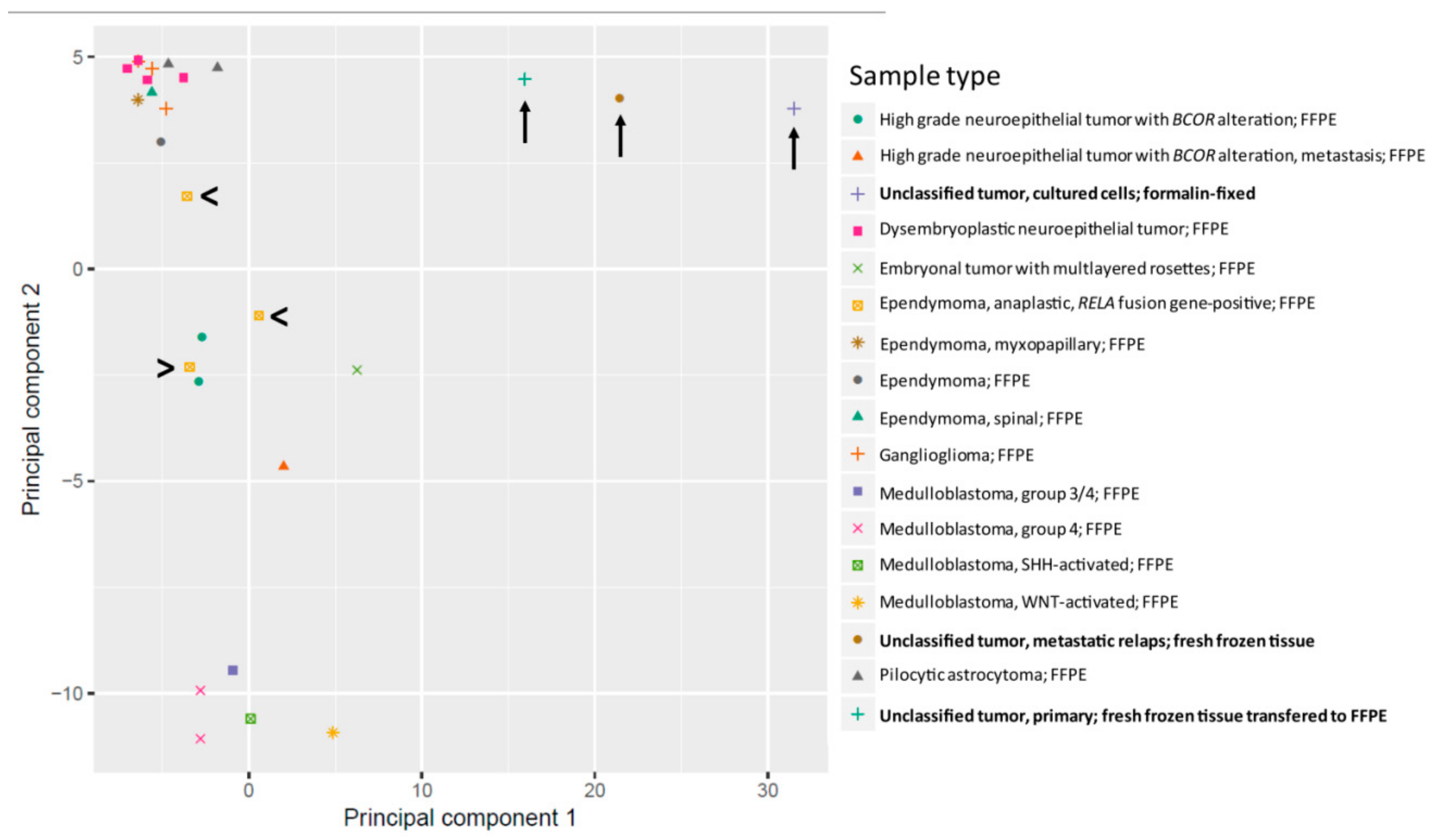
2.3. Methylation Analysis Reveals a not Classifiable Tumor Entity
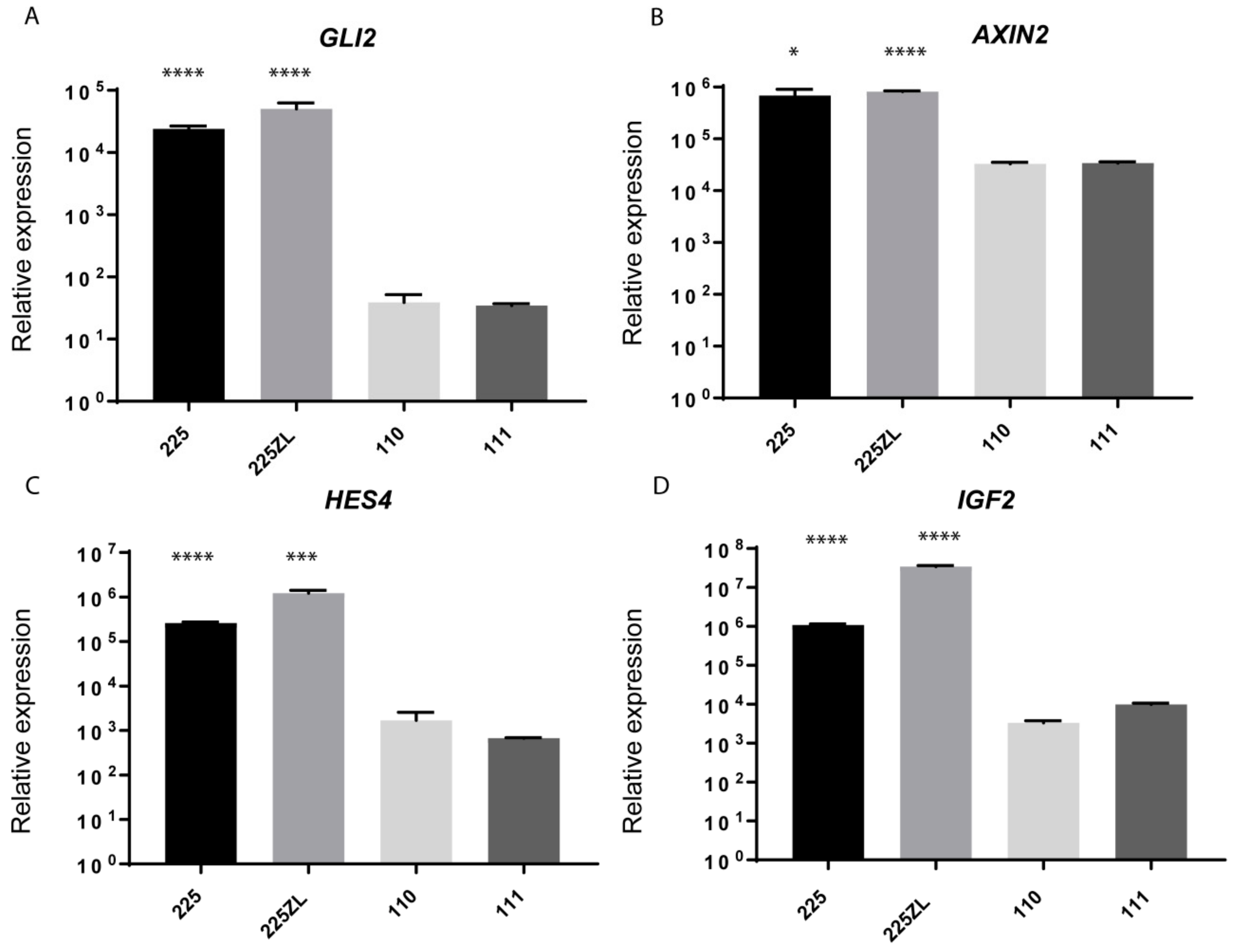
2.4. Whole-Transcriptome Sequencing Reveals the Activation of Several Embryonic Pathways
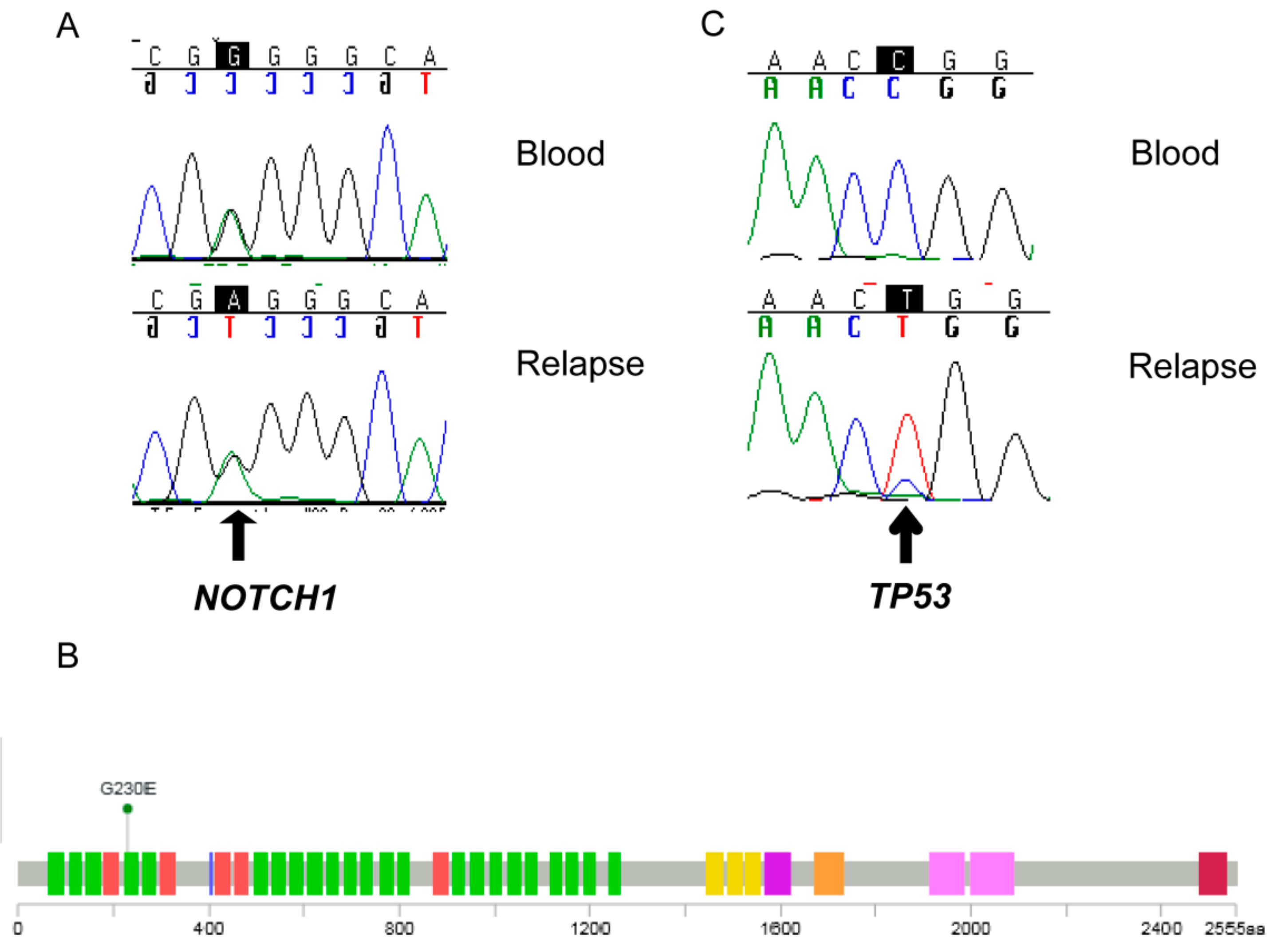
2.5. Detection of a NOTCH 1 Germline Mutation
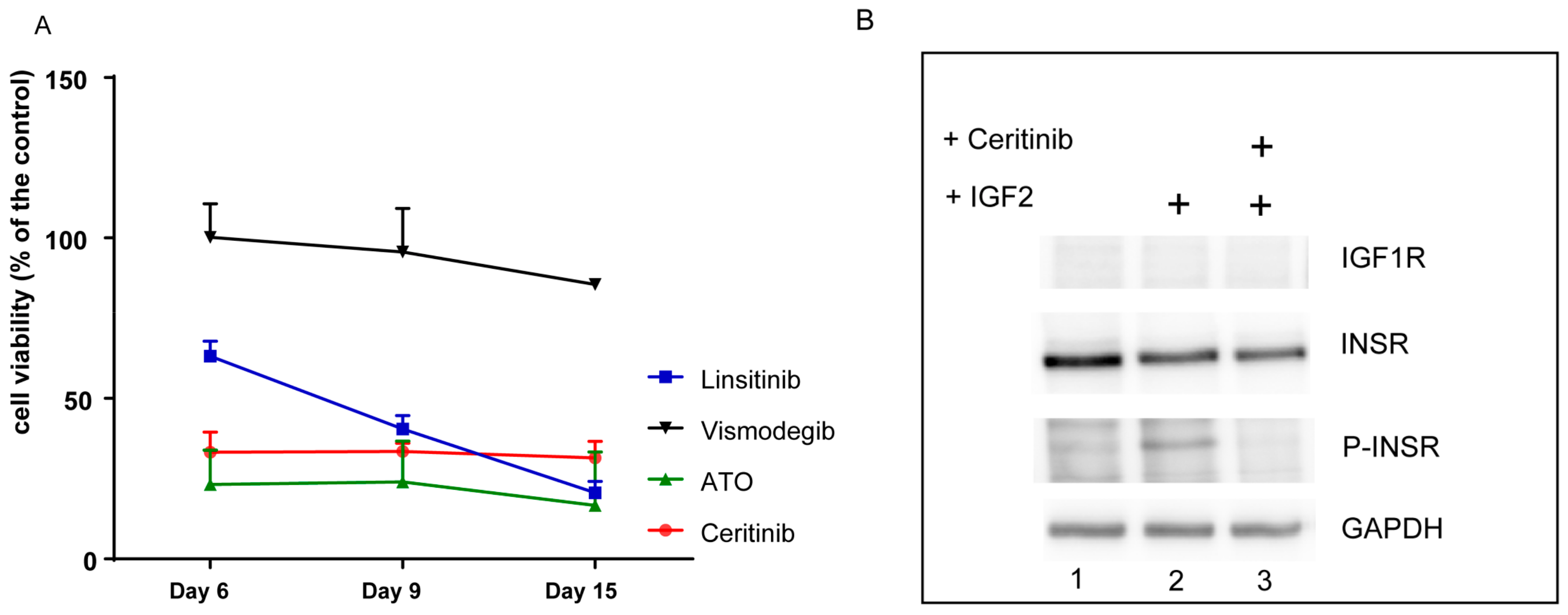
2.6. The Primary Tumor Cells are Sensitive to Inhibition with ATO and Ceritinib
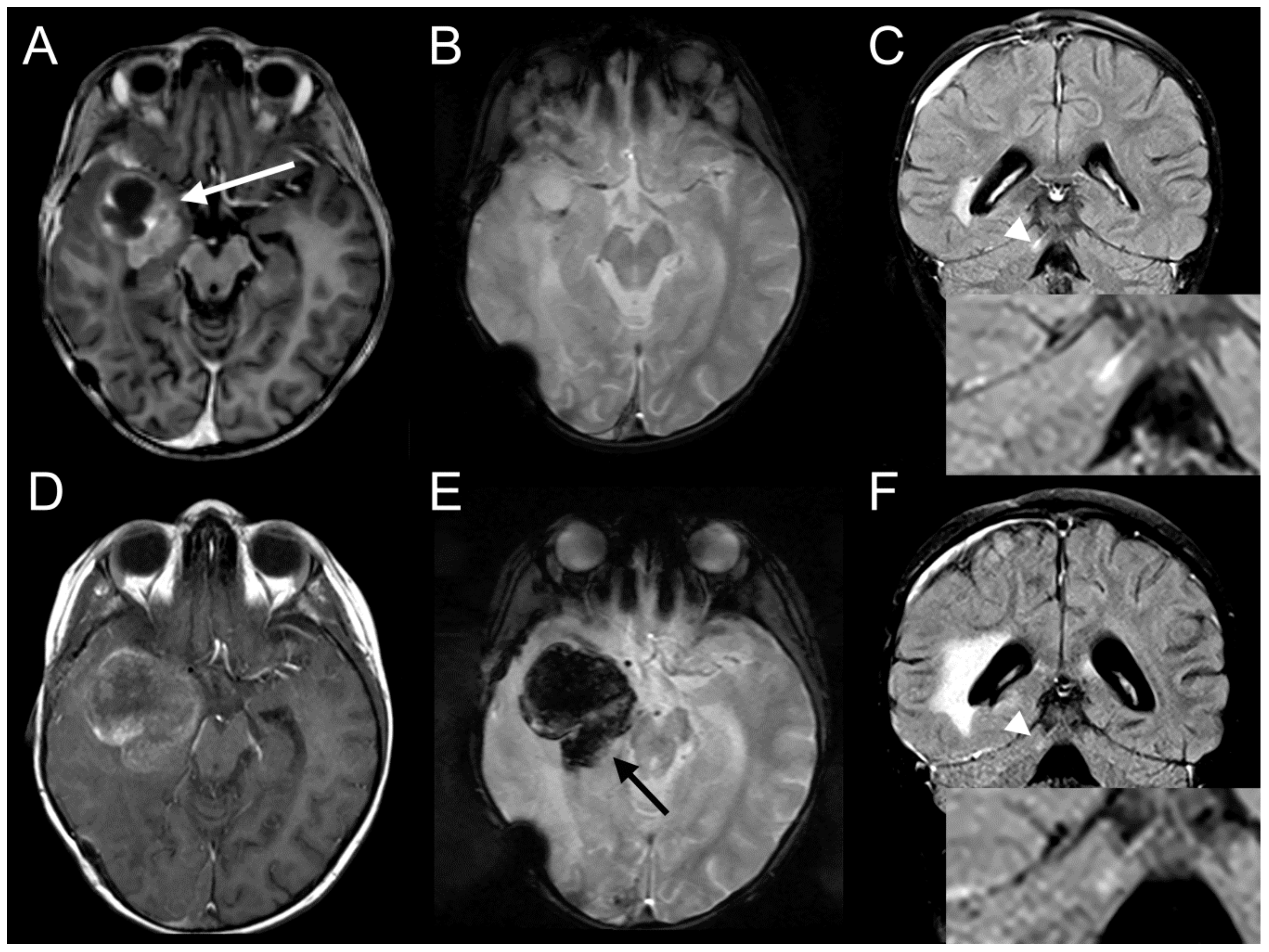
2.7. Ceritinib Induces Tumor Regression in Vivo
2.8. Ceritinib Penetrates the Human Brain
3. Discussion
4. Materials and Methods
4.1. Tissue Samples and Cells
4.2. Nucleic Acid Extraction
4.3. DNA Sequencing
4.4. qRT-PCR
4.5. RNA Sequencing
4.6. Methylation Analysis and Copy Number Variation Profiles
4.7. Cellular Proliferation Assays
4.8. Phosphorylation Assay
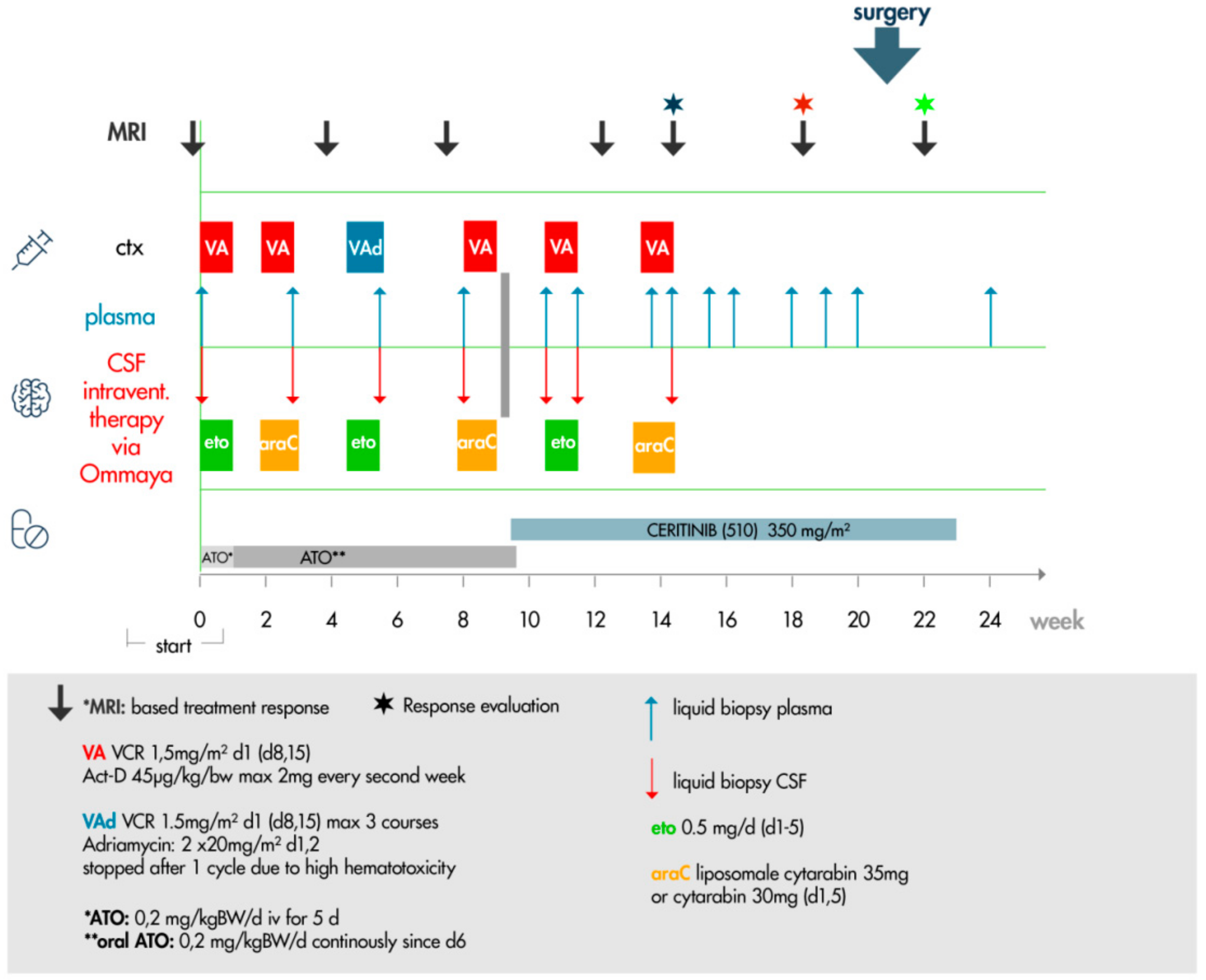
4.9. Targeted Therapy Protocol
4.10. Arsenic Concentration
4.11. Ceritinib Quantification
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Act-D | dactinomycin/ actinomycin-D |
| araC | cytarabine |
| ATO | arsenic trioxide |
| BBB | Blood-brain barrier |
| BCC | basal cell carcinoma |
| CSF | cerebrospinal fluid |
| Eto | etoposide |
| HGNET-BCOR | High grade neuroepithelial tumor with BCOR alteration |
| IGF | insulin-like growth factor |
| IPA | Ingenuity Pathway Analysis |
| iv | intravenous |
| MRI | magnetic resonance imaging |
| RNA seq | whole-transcriptome sequencing |
| ROS1 | proto-oncogene receptor tyrosine kinase |
| SHH | sonic hedgehog |
| TPM | transcripts per kilobase million |
| VCR | vincristine |
References
- Lodhia, K.A.; Tienchaiananda, P.; Haluska, P. Understanding the Key to Targeting the IGF Axis in Cancer: A Biomarker Assessment. Front. Oncol. 2015, 5, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondy, C.A.; Lee, W.H. Patterns of insulin-like growth factor and IGF receptor gene expression in the brain. Functional implications. Ann. N. Y. Acad. Sci. 1993, 692, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Del Valle, L.; Enam, S.; Lassak, A.; Wang, J.Y.; Croul, S.; Khalili, K.; Reiss, K. Insulin-like growth factor I receptor activity in human medulloblastomas. Clin. Cancer Res. 2002, 8, 1822–1830. [Google Scholar] [PubMed]
- Hartmann, W.; Koch, A.; Brune, H.; Waha, A.; Schuller, U.; Dani, I.; Denkhaus, D.; Langmann, W.; Bode, U.; Wiestler, O.D.; et al. Insulin-like growth factor II is involved in the proliferation control of medulloblastoma and its cerebellar precursor cells. Am. J. Pathol. 2005, 166, 1153–1162. [Google Scholar] [CrossRef]
- Gong, Y.; Ma, Y.; Sinyuk, M.; Loganathan, S.; Thompson, R.C.; Sarkaria, J.N.; Chen, W.; Lathia, J.D.; Mobley, B.C.; Clark, S.W.; et al. Insulin-mediated signaling promotes proliferation and survival of glioblastoma through Akt activation. Neuro Oncol. 2016, 18, 48–57. [Google Scholar] [CrossRef]
- Korshunov, A.; Neben, K.; Wrobel, G.; Tews, B.; Benner, A.; Hahn, M.; Golanov, A.; Lichter, P. Gene expression patterns in ependymomas correlate with tumor location, grade, and patient age. Am. J. Pathol. 2003, 163, 1721–1727. [Google Scholar] [CrossRef]
- Simpson, A.; Petnga, W.; Macaulay, V.M.; Weyer-Czernilofsky, U.; Bogenrieder, T. Insulin-Like Growth Factor (IGF) Pathway Targeting in Cancer: Role of the IGF Axis and Opportunities for Future Combination Studies. Target. Oncol. 2017, 12, 571–597. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Kim, D.W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; De Pas, T.; et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 1189–1197. [Google Scholar] [CrossRef]
- Marsilje, T.H.; Pei, W.; Chen, B.; Lu, W.; Uno, T.; Jin, Y.; Jiang, T.; Kim, S.; Li, N.; Warmuth, M.; et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulf onyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J. Med. Chem. 2013, 56, 5675–5690. [Google Scholar] [CrossRef]
- Van Erp, A.E.M.; Hillebrandt-Roeffen, M.H.S.; van Houdt, L.; Fleuren, E.D.G.; van der Graaf, W.T.A.; Versleijen-Jonkers, Y.M.H. Targeting Anaplastic Lymphoma Kinase (ALK) in Rhabdomyosarcoma (RMS) with the Second-Generation ALK Inhibitor Ceritinib. Target. Oncol. 2017, 12, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Vewinger, N.; Huprich, S.; Seidmann, L.; Russo, A.; Alt, F.; Bender, H.; Sommer, C.; Samuel, D.; Lehmann, N.; Backes, N.; et al. IGF1R Is a Potential New Therapeutic Target for HGNET-BCOR Brain Tumor Patients. Int. J. Mol. Sci. 2019, 20, 3027. [Google Scholar] [CrossRef] [PubMed]
- Gessi, M.; Giagnacovo, M.; Modena, P.; Elefante, G.; Gianno, F.; Buttarelli, F.R.; Arcella, A.; Donofrio, V.; Camassei, F.D.; Nozza, P.; et al. Role of Immunohistochemistry in the Identification of Supratentorial C11ORF95-RELA Fused Ependymoma in Routine Neuropathology. Am. J. Surg. Pathol. 2019, 43, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Andl, T.; Grachtchouk, V.; Wang, A.; Liu, J.; Syu, L.J.; Ferris, J.; Wang, T.S.; Glick, A.B.; Millar, S.E.; et al. Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical Wnt/beta3-catenin signaling. Nat. Genet. 2008, 40, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Hajicek, N.; Kozasa, T. Regulation and physiological functions of G12/13-mediated signaling pathways. Neurosignals 2009, 17, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, J.M.; Shelat, A.A.; Carcaboso, A.M.; Kranenburg, T.A.; Arnold, L.A.; Boulos, N.; Wright, K.; Johnson, R.A.; Poppleton, H.; Mohankumar, K.M.; et al. An integrated in vitro and in vivo high-throughput screen identifies treatment leads for ependymoma. Cancer Cell 2011, 20, 384–399. [Google Scholar] [CrossRef]
- Pietsch, T.; Wohlers, I.; Goschzik, T.; Dreschmann, V.; Denkhaus, D.; Dorner, E.; Rahmann, S.; Klein-Hitpass, L. Supratentorial ependymomas of childhood carry C11orf95-RELA fusions leading to pathological activation of the NF-kappaB signaling pathway. Acta Neuropathol. 2014, 127, 609–611. [Google Scholar] [CrossRef]
- Giovannone, B.; Lee, E.; Laviola, L.; Giorgino, F.; Cleveland, K.A.; Smith, R.J. Two novel proteins that are linked to insulin-like growth factor (IGF-I) receptors by the Grb10 adapter and modulate IGF-I signaling. J. Biol. Chem. 2003, 278, 31564–31573. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Regairaz, M.; Munier, F.; Sartelet, H.; Castaing, M.; Marty, V.; Renauleaud, C.; Doux, C.; Delbe, J.; Courty, J.; Fabre, M.; et al. Mutation-Independent Activation of the Anaplastic Lymphoma Kinase in Neuroblastoma. Am. J. Pathol. 2016, 186, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryeom, S.W. The cautionary tale of side effects of chronic Notch1 inhibition. J. Clin. Investig. 2011, 121, 508–509. [Google Scholar] [CrossRef] [Green Version]
- Fellner, C. Vismodegib (erivedge) for advanced Basal cell carcinoma. Pharm. Ther. 2012, 37, 670–682. [Google Scholar]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.C.; Peaceman, D.; Ozdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Investig. 2011, 121, 148–160. [Google Scholar] [CrossRef] [Green Version]
- Paret, C.; Russo, A.; Otto, H.; Mayer, A.; Zahnreich, S.; Wagner, W.; Samuel, D.; Scharnhorst, D.; Solomon, D.A.; Dhall, G.; et al. Personalized therapy: CNS HGNET-BCOR responsiveness to arsenic trioxide combined with radiotherapy. Oncotarget 2017, 8, 114210–114225. [Google Scholar] [CrossRef] [Green Version]
- Torka, P.; Al Ustwani, O.; Wetzler, M.; Wang, E.S.; Griffiths, E.A. Swallowing a bitter pill-oral arsenic trioxide for acute promyelocytic leukemia. Blood Rev. 2016, 30, 201–211. [Google Scholar] [CrossRef]
- Beckwith, H.; Yee, D. Minireview: Were the IGF Signaling Inhibitors All Bad? Mol. Endocrinol. 2015, 29, 1549–1557. [Google Scholar] [CrossRef] [Green Version]
- Mulvihill, M.J.; Cooke, A.; Rosenfeld-Franklin, M.; Buck, E.; Foreman, K.; Landfair, D.; O’Connor, M.; Pirritt, C.; Sun, Y.; Yao, Y.; et al. Discovery of OSI-906: A selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med. Chem. 2009, 1, 1153–1171. [Google Scholar] [CrossRef] [PubMed]
- Poondru, S.; Chaves, J.; Yuen, G.; Parker, B.; Conklin, E.; Singh, M.; Nagata, M.; Gill, S. Mass balance, pharmacokinetics, and metabolism of linsitinib in cancer patients. Cancer Chemother. Pharmacol. 2016, 77, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Shahi, M.H.; Rey, J.A.; Castresana, J.S. The sonic hedgehog-GLI1 signaling pathway in brain tumor development. Expert Opin. Ther. Targets 2012, 16, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Ng, J.M.; Curran, T. Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell 2008, 13, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Kaste, S.C.; Chemaitilly, W.; Bowers, D.C.; Laughton, S.; Smith, A.; Gottardo, N.G.; Partap, S.; Bendel, A.; Wright, K.D.; et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 2017, 8, 69295–69302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehme, K.A.; Nitsch, J.; Riester, R.; Handgretinger, R.; Schleicher, S.B.; Kluba, T.; Traub, F. Arsenic trioxide potentiates the effectiveness of etoposide in Ewing sarcomas. Int. J. Oncol. 2016, 49, 2135–2146. [Google Scholar] [CrossRef] [Green Version]
- Meister, M.T.; Boedicker, C.; Graab, U.; Hugle, M.; Hahn, H.; Klingebiel, T.; Fulda, S. Arsenic trioxide induces Noxa-dependent apoptosis in rhabdomyosarcoma cells and synergizes with antimicrotubule drugs. Cancer Lett. 2016, 381, 287–295. [Google Scholar] [CrossRef]
- Tzaridis, T.; Milde, T.; Pajtler, K.W.; Bender, S.; Jones, D.T.; Muller, S.; Wittmann, A.; Schlotter, M.; Kulozik, A.E.; Lichter, P.; et al. Low-dose Actinomycin-D treatment re-establishes the tumoursuppressive function of P53 in RELA-positive ependymoma. Oncotarget 2016, 7, 61860–61873. [Google Scholar] [CrossRef]
- Slavc, I.; Peyrl, A.; Chocholous, M.; Reisinger, D.; Mayr, L.; Azizi, A.; Dieckmann, K.; Haberler, C.; Czech, T. MBCL-27. RESPONSE OF RECURRENT MALIGNANT CHILDHOOD CNS TUMORS TO A MEMMAT BASED METRONOMIC ANTIANGIOGENIC COMBINATION THERAPY VARIES DEPENDENT ON TUMOR TYPE: EXPERIENCE IN 71 PATIENTS. Neuro Oncol. 2018, 20, i122. [Google Scholar] [CrossRef]
- Kumana, C.R.; Au, W.Y.; Lee, N.S.; Kou, M.; Mak, R.W.; Lam, C.W.; Kwong, Y.L. Systemic availability of arsenic from oral arsenic-trioxide used to treat patients with hematological malignancies. Eur. J. Clin. Pharmacol. 2002, 58, 521–526. [Google Scholar] [CrossRef]
- Kiguchi, T.; Yoshino, Y.; Yuan, B.; Yoshizawa, S.; Kitahara, T.; Akahane, D.; Gotoh, M.; Kaise, T.; Toyoda, H.; Ohyashiki, K. Speciation of arsenic trioxide penetrates into cerebrospinal fluid in patients with acute promyelocytic leukemia. Leuk. Res. 2010, 34, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, V.M.; Middleton, M.R.; Protheroe, A.S.; Tolcher, A.; Dieras, V.; Sessa, C.; Bahleda, R.; Blay, J.Y.; LoRusso, P.; Mery-Mignard, D.; et al. Phase I study of humanized monoclonal antibody AVE1642 directed against the type 1 insulin-like growth factor receptor (IGF-1R), administered in combination with anticancer therapies to patients with advanced solid tumors. Ann. Oncol. 2013, 24, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; De Pas, T.; Riely, G.J.; et al. Activity and safety of ceritinib in patients with ALK-rearranged non-small-cell lung cancer (ASCEND-1): Updated results from the multicentre, open-label, phase 1 trial. Lancet Oncol. 2016, 17, 452–463. [Google Scholar] [CrossRef]
- Aiken, R.; Axelson, M.; Harmenberg, J.; Klockare, M.; Larsson, O.; Wassberg, C. Phase I clinical trial of AXL1717 for treatment of relapsed malignant astrocytomas: Analysis of dose and response. Oncotarget 2017, 8, 81501–81510. [Google Scholar] [CrossRef] [PubMed]
- Ekman, S.; Frodin, J.E.; Harmenberg, J.; Bergman, A.; Hedlund, A.; Dahg, P.; Alvfors, C.; Stahl, B.; Bergstrom, S.; Bergqvist, M. Clinical Phase I study with an Insulin-like Growth Factor-1 receptor inhibitor: Experiences in patients with squamous non-small cell lung carcinoma. Acta Oncol. 2011, 50, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef]
- Southgate, L.; Sukalo, M.; Karountzos, A.S.V.; Taylor, E.J.; Collinson, C.S.; Ruddy, D.; Snape, K.M.; Dallapiccola, B.; Tolmie, J.L.; Joss, S.; et al. Haploinsufficiency of the NOTCH1 Receptor as a Cause of Adams-Oliver Syndrome With Variable Cardiac Anomalies. Circ. Cardiovasc. Genet. 2015, 8, 572–581. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef]
- Westhoff, B.; Colaluca, I.N.; D’Ario, G.; Donzelli, M.; Tosoni, D.; Volorio, S.; Pelosi, G.; Spaggiari, L.; Mazzarol, G.; Viale, G.; et al. Alterations of the Notch pathway in lung cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 22293–22298. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.J.; Sanborn, Z.; Arnett, K.L.; Bayston, L.J.; Liao, W.; Proby, C.M.; Leigh, I.M.; Collisson, E.A.; Gordon, P.B.; Jakkula, L.; et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17761–17766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modena, P.; Lualdi, E.; Facchinetti, F.; Veltman, J.; Reid, J.F.; Minardi, S.; Janssen, I.; Giangaspero, F.; Forni, M.; Finocchiaro, G.; et al. Identification of tumor-specific molecular signatures in intracranial ependymoma and association with clinical characteristics. J. Clin. Oncol. 2006, 24, 5223–5233. [Google Scholar] [CrossRef] [PubMed]
- Puget, S.; Grill, J.; Valent, A.; Bieche, I.; Dantas-Barbosa, C.; Kauffmann, A.; Dessen, P.; Lacroix, L.; Geoerger, B.; Job, B.; et al. Candidate genes on chromosome 9q33-34 involved in the progression of childhood ependymomas. J. Clin. Oncol. 2009, 27, 1884–1892. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Chao, W.S.; Katsube, K.; Yamaguchi, A. Distinct roles of EGF repeats for the Notch signaling system. Exp. Cell Res. 2005, 302, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Rodilla, V.; Villanueva, A.; Obrador-Hevia, A.; Robert-Moreno, A.; Fernandez-Majada, V.; Grilli, A.; Lopez-Bigas, N.; Bellora, N.; Alba, M.M.; Torres, F.; et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 6315–6320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, S.; Li, Y.; Miller, E.E.; Shih, D.J.; Taylor, M.D.; Stearns, T.M.; Bronson, R.T.; Ackerman, S.L.; Yoon, J.K.; Yun, K. Notch1-induced brain tumor models the sonic hedgehog subgroup of human medulloblastoma. Cancer Res. 2013, 73, 5381–5390. [Google Scholar] [CrossRef]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Zhu, Z.; Chai, Y. Crizotinib resistance overcome by ceritinib in an ALK-positive non-small cell lung cancer patient with brain metastases: A case report. Medicine 2017, 96, e8652. [Google Scholar] [CrossRef]
- Kort, A.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Brain accumulation of the EML4-ALK inhibitor ceritinib is restricted by P-glycoprotein (P-GP/ABCB1) and breast cancer resistance protein (BCRP/ABCG2). Pharmacol. Res. 2015, 102, 200–207. [Google Scholar] [CrossRef]
- Katayama, R.; Sakashita, T.; Yanagitani, N.; Ninomiya, H.; Horiike, A.; Friboulet, L.; Gainor, J.F.; Motoi, N.; Dobashi, A.; Sakata, S.; et al. P-glycoprotein Mediates Ceritinib Resistance in Anaplastic Lymphoma Kinase-rearranged Non-small Cell Lung Cancer. EBioMedicine 2016, 3, 54–66. [Google Scholar] [CrossRef]
- Fromm, M.F. Importance of P-glycoprotein at blood-tissue barriers. Trends Pharmacol. Sci. 2004, 25, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Phoenix, T.N.; Patmore, D.M.; Boop, S.; Boulos, N.; Jacus, M.O.; Patel, Y.T.; Roussel, M.F.; Finkelstein, D.; Goumnerova, L.; Perreault, S.; et al. Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell 2016, 29, 508–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santarpia, M.; Daffina, M.G.; D’Aveni, A.; Marabello, G.; Liguori, A.; Giovannetti, E.; Karachaliou, N.; Cao, M.G.; Rosell, R.; Altavilla, G. Spotlight on ceritinib in the treatment of ALK+ NSCLC: Design, development and place in therapy. Drug Des. Dev. Ther. 2017, 11, 2047–2063. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Salzberg, S.L. TopHat-Fusion: An algorithm for discovery of novel fusion transcripts. Genome Biol. 2011, 12, R72. [Google Scholar] [CrossRef]
- Assenov, Y.; Müller, F.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 2014, 11, 1138. [Google Scholar] [CrossRef]
- Paret, C.; Theruvath, J.; Russo, A.; Kron, B.; El Malki, K.; Lehmann, N.; Wingerter, A.; Neu, M.A.; Gerhold-Ay, A.; Wagner, W.; et al. Activation of the basal cell carcinoma pathway in a patient with CNS HGNET-BCOR diagnosis: Consequences for personalized targeted therapy. Oncotarget 2016, 7, 83378–83391. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Type | Sample ID | Methylation Classifier Result | Chromosomal Aberrations |
|---|---|---|---|
| Primary tumor | 176 | unclassified | Gain on chromosome 2 and 11q |
| First metastatic relapse | 225 | unclassified | Tendency to gain on chromosomes 1p, 2, 7q partially, 11q, 21q; loss of chromosomes 17p, 19p |
| in vitro culture of tumor cells | 225 ZL | unclassified | Gain on chromosomes 1p, 2, 7q partially, 11q, 21q; loss of chromosomes 17p, 18q, 19p |
| Ingenuity Canonical Pathways | -Log (p-value) |
|---|---|
| Notch Signaling | 3.62 |
| Basal Cell Carcinoma Signaling | 2.55 |
| Gα12/13 Signaling | 2.13 |
| Transcript 1 | Chr | Position | Transcript 2 | Chr | Position |
|---|---|---|---|---|---|
| NCOA1 intron7 | 2 | 24914529–24916118 | GIGYF2 exon8 | 2 | 233626104–233626145 |
| NCOA1 intron12 | 2 | 24933980–24949455 | GIGYF2 intron8 | 2 | 233626146–233651857 |
| NCOA1 intron12 | 2 | 24933980–24949455 | C11orf95 intron3 | 11 | 63532726–63533276 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, A.; Paret, C.; Alt, F.; Burhenne, J.; Fresnais, M.; Wagner, W.; Glaser, M.; Bender, H.; Huprich, S.; Harter, P.N.; et al. Ceritinib-Induced Regression of an Insulin-Like Growth Factor-Driven Neuroepithelial Brain Tumor. Int. J. Mol. Sci. 2019, 20, 4267. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174267
Russo A, Paret C, Alt F, Burhenne J, Fresnais M, Wagner W, Glaser M, Bender H, Huprich S, Harter PN, et al. Ceritinib-Induced Regression of an Insulin-Like Growth Factor-Driven Neuroepithelial Brain Tumor. International Journal of Molecular Sciences. 2019; 20(17):4267. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174267
Chicago/Turabian StyleRusso, Alexandra, Claudia Paret, Francesca Alt, Jürgen Burhenne, Margaux Fresnais, Wolfgang Wagner, Martin Glaser, Hannah Bender, Sabrina Huprich, Patrick N. Harter, and et al. 2019. "Ceritinib-Induced Regression of an Insulin-Like Growth Factor-Driven Neuroepithelial Brain Tumor" International Journal of Molecular Sciences 20, no. 17: 4267. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174267