1. Introduction
Ovarian cancer (OC) is one of the most frequent gynecological malignancy among women. Despite recent progress that has been made in the treatment of patients, OC is still characterized by a high mortality rate. It is the fifth leading cause of cancer-induced death in females in the world; the overall five-year survival rate is still only 15%–30% according to recent reports [
1,
2,
3]. The high mortality is at least partially due to the difficulties of early detection of the tumor. The routine diagnostic procedures (pelvic examination, transvaginal ultrasonography, CA125 antigen measurement) carried out in clinics are not suitable for early diagnosis [
4,
5,
6,
7]. During recent years several groups investigated the possibility of using circulating miRNAs as candidate biomarkers for diagnosis in several different human cancers, among them in OC. In addition to their diagnostic potential, the involvement of circulating miRNAs in tumor formation might be an equally important topic to study [
8,
9,
10].
MiRNAs are endogenously expressed, short (~22 nucleotides) non-coding single-stranded regulatory RNA molecules, known to interfere with the translation of the mRNA coded by the target genes. MiRNAs are negative regulators of gene expression, upon their sequence-specific binding to the 3′-UTR region of mRNAs, they can repress the translation of target mRNA or facilitate its cleavage and elimination [
11]. MiRNAs are promiscuous post-transcriptional regulators, due to their short “seed” sequence, a miRNA can interact with a large number of mRNAs, that way miRNAs are involved in the regulation of almost all important cellular, developmental, and pathological processes [
12]. It is well established that miRNAs are present and can be reliably detected in blood plasma since these circulating miRNAs are very stable, which is the result of their packaging into vesicles or interaction with proteins that protect miRNAs from RNase digestion. These features could make them not only ideal candidates for non-invasive plasma-based biomarkers but also regulatory factors contributing to tumor progression [
7,
13].
Circulating miRNAs could be released from cells through an active, regulated secretion process packaged into exosomes or microvesicles [
14]. Exosomes were shown to be involved in intercellular communication since they carry various bioactive molecules, among them miRNAs [
7,
15]. Exosomes can interact, fuse with the membrane of cells, and deliver their cargo to recipient target cells and thus modify their gene expression pattern [
16,
17,
18,
19,
20,
21]. Exosomes are considered as part of the tumor environment playing important roles in pre-metastatic environment formation, tumor progression, epithelial-to-mesenchymal transition, and the modulation of immune regulation [
14]. MiRNAs could be secreted from cells via binding to protein containing complexes like AGO2 or HDL. Although the functional role of circulating miRNAs is still largely unknown, it is known that the dysregulation of miRNA expression and the presence of circulating miRNAs have been linked to the formation of cancer among them OC, as well [
7,
10,
22]. It was shown that miRNAs could be oncomiRs or tumor suppressors, however this categorization is not straightforward, due to their extensive palette of target genes the same miRNA could play opposing roles in different processes [
23].
Several differentially expressed miRNAs were found in the plasma of OC patients, like members of the miR-200 family, miR-141, miR-125b, miR-222-3p or Let-7 among others [
14,
24,
25]. Despite the large number of reports no consensus regarding the circulating miRNA signature has been suggested so far, which would unambiguously distinguish OC patients from healthy individuals and an explanation of their potential biological significance in OC is still not clarified completely. The fact that ovarian cancer cells are rarely disseminated through the vasculature makes the interpretation of the pathophysiological role of circulating miRNAs difficult [
1,
15,
26].
The aim of this study was to analyze the circulating miRNA expression profile in serous epithelial OC patients and compare it to that of healthy individuals in order to contribute to the growing body of data available to establish a useful miRNA set for OC diagnosis obtained by the non-invasive liquid biopsy. We attempted to identify the miRNA expression profile, which is specific for tumor samples. We have also aimed to investigate the possible involvement of circulating miRNAs in OC development by analyzing the biological importance of miRNA targets and by the functional enrichment profile of their target gene set.
3. Discussion
In recent years several groups examined the biological importance of cell free miRNAs present in body fluids. It was suggested that circulating miRNAs have the potential to become non-invasive biomarkers for the early diagnosis of cancer [
9,
10,
24]. It is equally interesting however, to study the pathophysiological role of these miRNAs, since circulating miRNAs released from cells are known to be involved in intercellular communication and dysregulation of miRNAs in tissues is known to be associated with several cancers [
7,
9,
10,
14].
We have compared the expression profiles of circulating miRNA in blood plasma samples of six healthy females and 18 OC patients. Patients were divided into FIGO I, FIGO III, and FIGO IV stages, having six patients in each group. The nCounter Human v3 miRNA Panel of the NanoString System was used to measure the miRNA levels. MiRNA counts were low for most of the miRNAs, especially in the control samples. This might be due to the detection method, the NanoString method does not require an amplification step, so it is clearly different from those methods that use PCR for the miRNA measurement. It might suggest that the NanoString method is less sensitive for low abundance miRNAs.
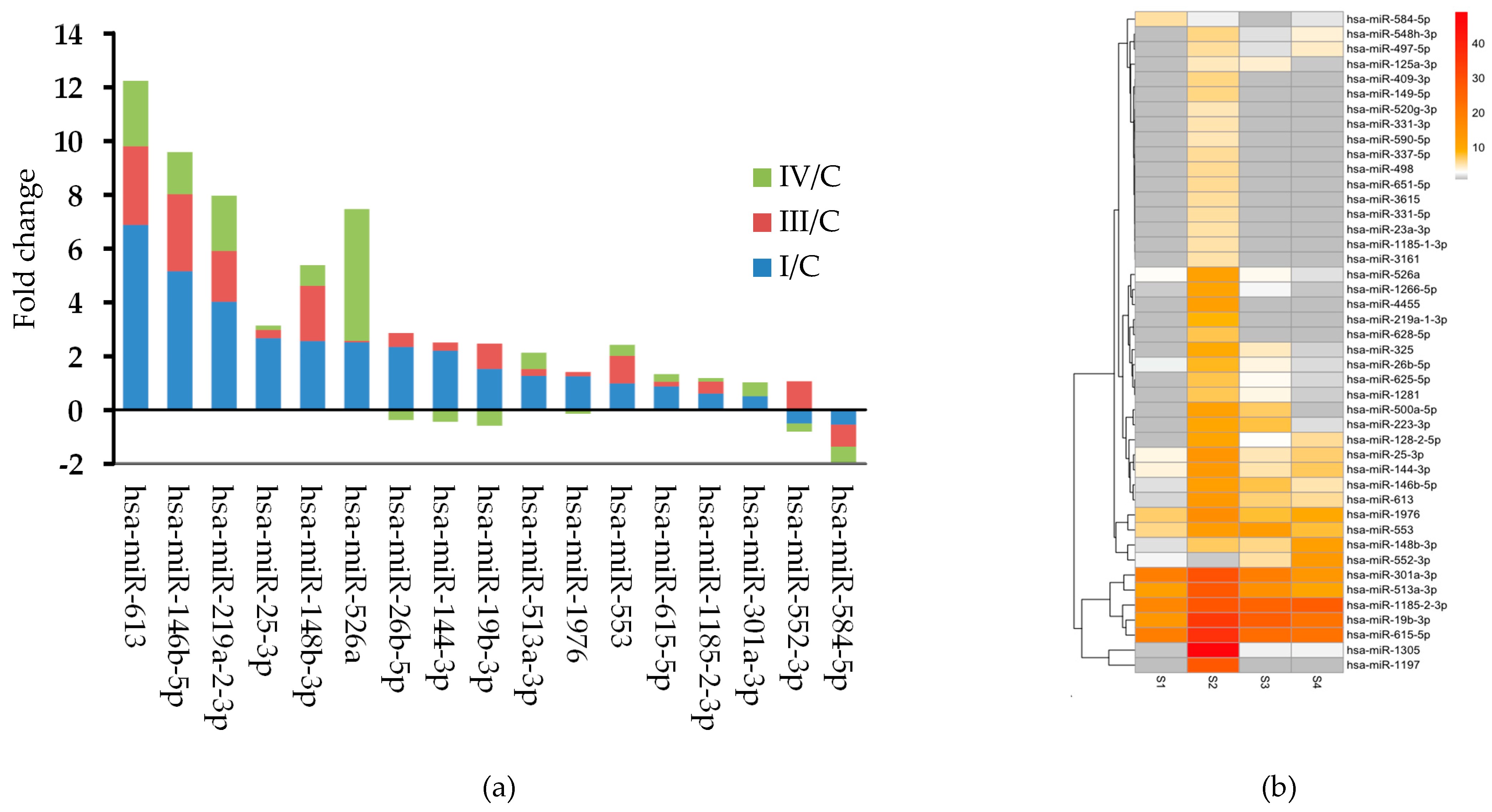
Comparing miRNA expression profiles in the control and OC patient samples we have identified 45 miRNAs showing different expression between controls and patients. Our data showed that 17 miRNAs out of 45 were present both in the control and patient plasma (Group 1 miRNAs), however their expression levels differed significantly between the four groups. With the exception of the tumor suppressor has-miR-584-5p all Group 1 miRNAs were upregulated. 19 miRNAs were found only in samples of FIGO I patients (Group 2 miRNA) and nine miRNAs were detected in all three patient groups but were absent in control samples (Group 3 miRNAs). The finding of miRNAs, which are present only in patient samples might be important from a diagnostic point of view, as it shows that circulating miRNAs have the potential to be used as non-invasive biomarkers. Our sample number, however, is too low to draw any firm conclusions.
The large majority of our differentially expressed miRNAs have been previously reported to play a role in different cancer types. A few of our differentially expressed miRNAs, however, were found to be associated with OC in previous reports (i.e., hsa-miR-144-3p, hsa-miR-337-5p, hsa-miR-500a-5p, hsa-miR-26b-5p, hsa-miR-125a-3p, hsa-miR-19b-3p) [
7,
14,
24,
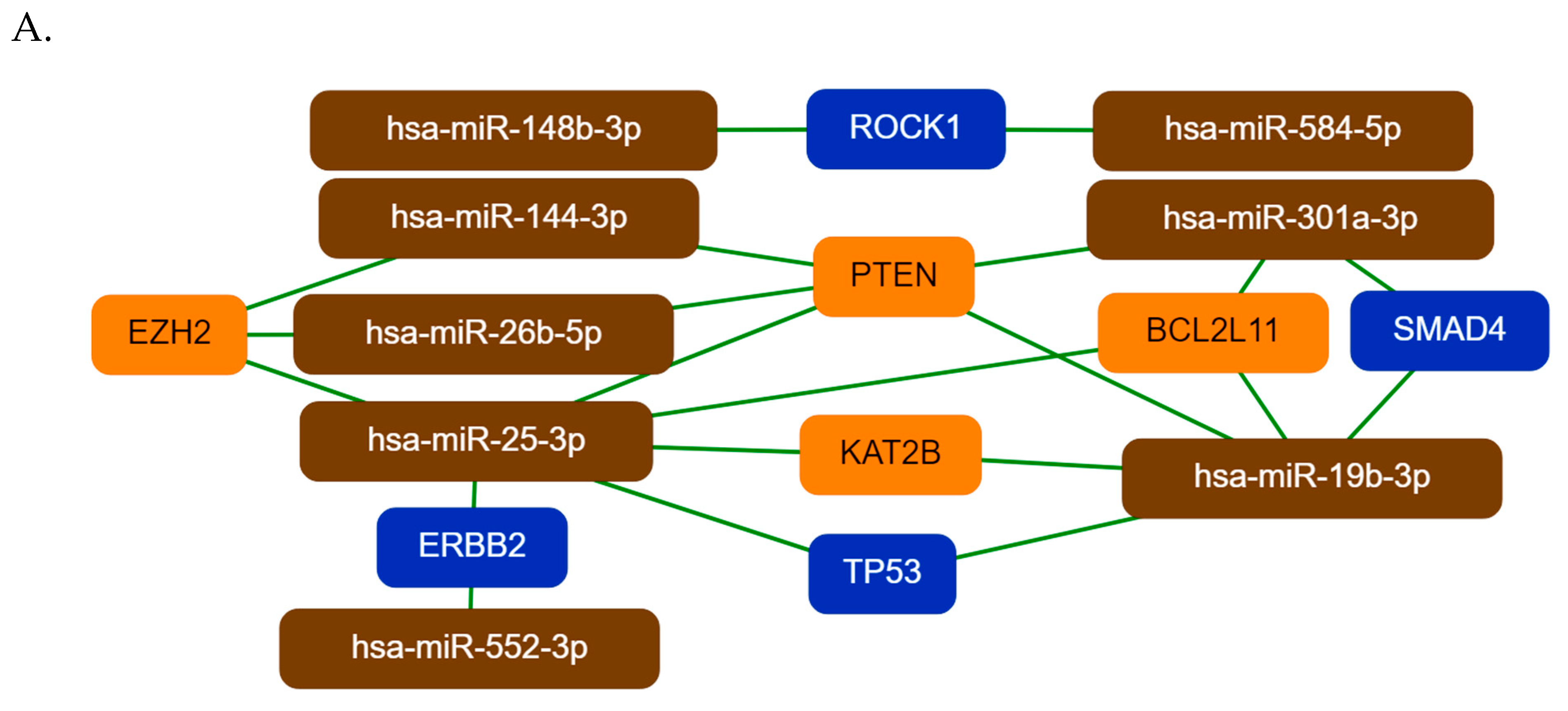
26]. Using the miRNet tool and a network-based approach we have constructed a miRNA–target interaction network for the miRNA groups. MiRNAs were ranked based on their degree-centrality value in the network, which reflects their biological importance. Hsa-mir-26b-5p, hsa-mir-19b-3p, and hsa-mir-25-3p were the top three ranked miRNAs for Group 1, hsa-mir-331-3p, hsa-mir-520g-3p, and hsa-mir-149-5p for Group 2 and hsa-mir-497-5p, hsa-mir-125a-3p, and hsa-mir-223-3p were the top three miRNAs for Group 3. The key miRNA–target interactions were visualized by the miTargetLink tool. For the Group 1 miRNAs, PTEN, EZH2, KAT2B, BCL2L11, TP53, SMAD4, and ERBB2 are the main targets, all known to be involved in tumorogenesis. PTEN and TP53 are known tumor suppressor proteins—impairing of KAT2B activity may contribute to genome instability; both oncogenic and tumor suppressive effects of EZH2 have been demonstrated in different cancer types and its expression is known to be regulated by miRNAs [
33,
34]. BCL2L11 is a tumor suppressor, it is an important regulator of apoptosis; loss of the SMAD4 activity may disrupt DNA damage response and repair mechanisms and enhance genomic instability [
35,
36]. The downregulation of these genes is in agreement with tumor formation, however the receptor tyrosine kinase ERBB2 is a proto-oncogene, the role of its downregulation by has-miR-25-3p and hsa-miR-552-3p is not known [
37].
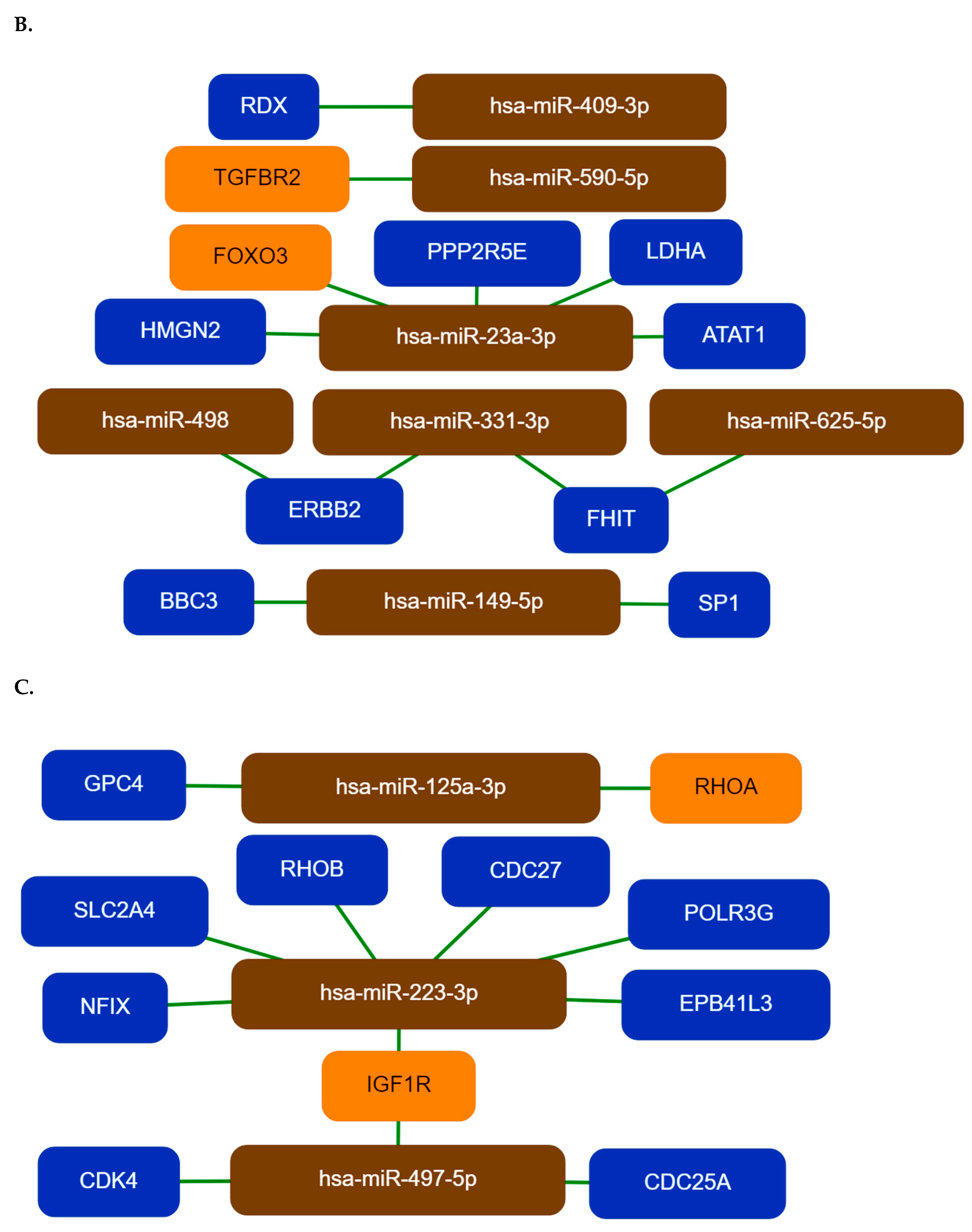
HMGN2, FOXO3, PPP2R5E, LDHA, ATAT1, FHIT, and the ERBB2 genes are the main targets for top Group 2 miRNAs, while the IGF1R, CDK4, CDC25A, SLC2A4/GLUT4, RHOB, CDC27, and POLR3G are the major targets for miRNAs of the third group. HMGN2 is an anti-tumor effector molecule of CD8
+T cells, FOXO3 is a core tumor suppressor in breast cancer; downregulation of PPP2R5E is a common event in acute myeloid leukemia [
38,
39,
40]. However, the ATAT1 activity is required for microtubule organization, it is specifically upregulated in colon cancer tissue and LDHA has an aberrantly high expression in multiple cancers [
41,
42].
Overexpression of CDC25A is known to be associated with malignancy and poor prognosis in cancer patients [
30]. Hsa-miR-223-3p targets the EPB41L3, a potential tumor suppressor gene, the NFIX gene that regulates both cell proliferation and migration, the SLC2A4/GLUT4 is a glucose transporter and a biomarker for many types of malignant tumors [
43,
44,
45]. A dualistic role of RHOB was reported, it could be a proto-oncogene or a tumor suppressor depending on the context of cancer development and progression. CDC27 is a tumor suppressor, its downregulation inhibits the proliferation of cancer cells [
46], POLR3G is required for proliferation, its depletion triggers proliferative arrest and differentiation of prostate cancer cells [
47].
Considering the negative regulatory role of miRNAs, it is noteworthy to find that not only tumor suppressor genes but proto-oncogenes are also present among the interacting targets. However, it is known, that miRNAs could have dualistic effect, a given miRNA can be an oncomiR or tumor suppressor depending on the cellular context [
23]. The miRTarBase and TargetLink databases were also used to predict experimentally validated targets of the differentially expressed miRNAs, several common targets were identified for three miRNA groups, the MET, SMAD7, EZH2, TERT, and IL6 genes for example are targeted by at least one member of each different miRNA group. The tyrosine–protein kinase MET is a proto-oncogene, its role in cell migration and in epithelial–mesenchymal transition (EMT) is well known [
48]. SMAD7 has a tumor suppressing role through blocking the TGF-β-stimulated cancer progression by increasing angiogenesis and inducing EMT [
49]. The telomerase reverse transcriptase TERT gene plays a crucial step in tumorogenesis, it is required to maintain the telomere length and telomerase activity to gain immortality [
50]. IL6 is an inflammatory cytokine which promotes metastasis in OC [
51].
The miRNA group specific target lists were used in a functional annotation analysis based on the enrichment of miRNA targets in the KEGG pathways and gene ontology biological processes terms. This analysis revealed that miRNA targets are enriched in known cancer pathways, signaling pathway which are crucial pathways in tumorogenesis. To name a few, FoxO signaling pathway, p53 signaling pathway, PI3-AKT pathway AMPK pathway, TGFβ signaling pathway, focal adhesion, proteoglycans in cancer, Hippo signaling pathway, ERBB signaling pathway, JAK-STAT signaling pathway, Estrogen signaling pathway, and MAPK signaling pathway were among the most significant ones. In GO-BP terms the positive regulation of gene expression, positive regulation of cell proliferation, negative regulation of cell proliferation, cellular response to hypoxia, negative regulation of transcription, positive regulation of tyrosine phosphorylation of Stat3 protein, and negative regulation of apoptotic process were the most significant ones based on target enrichment and over-representation. All of these processes and terms are known players of tumorogenesis. The results of the enrichment analysis show that most miRNA targets are involved in signaling pathways and biological processes, which are critical for tumor formation, suggesting that circulating miRNAs could be potential regulatory factors in tumorogenesis. At the same time these data also show that the identified enriched pathways and GO terms are not specific for a given tumor type, our identified miRNA targets are associated with regulatory and signaling processes which are important in several different tumor types.
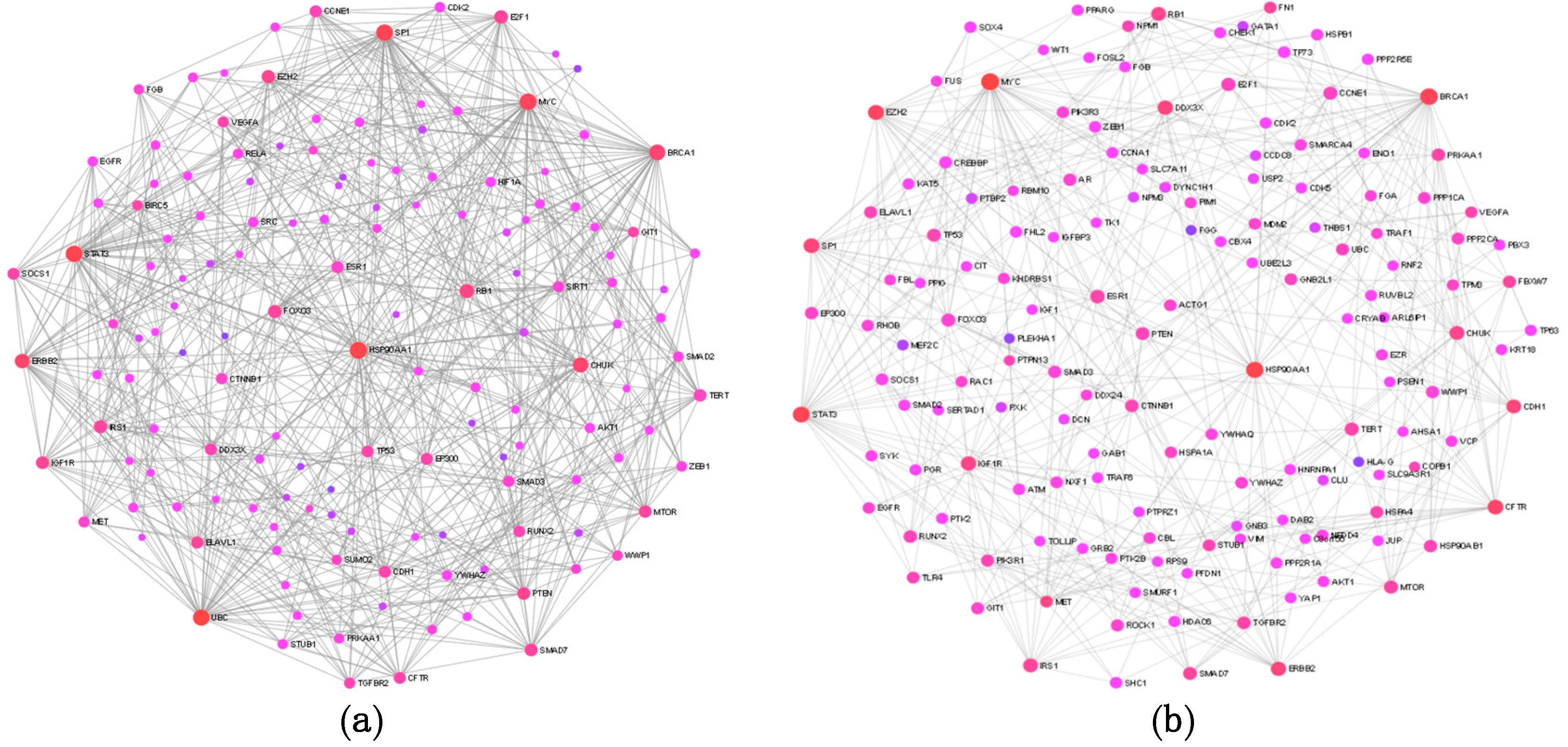
The target lists generated by the three prediction tools were merged into a single list and possible interactions between the target proteins and their functionally important interacting protein partners were analyzed by constructing general and ovary-specific PPI networks. The major hub proteins—HSP90AA1, MYC, SP1, BRCA1, RB1, CFTR, STAT3, E2F1, ERBB2, EZH2, and MET—were basically the same in the two networks, suggesting that our differentially expressed miRNAs regulate target genes which are involved in basic processes of tumor formation. This notion is strengthened by our network-based functional enrichment analysis, which provided a very similar enrichment pattern (both in KEGG and GO-BP terms) to those which were recognized by the DAVID tool for the different miRNA target groups.
In conclusion, our pilot study identified significantly differentially expressed circulating miRNAs in plasma samples of OC patients. Our functional annotation analysis showed that the experimentally validated targets of the differentially expressed miRNAs are key regulators of tumor formation, suggesting that circulating miRNAs might play an important pathophysiological role in the formation of different tumor types. On the other hand, these results also show that the differentially expressed miRNAs identified in our study have limited usefulness in the diagnosis of OC. A clear limitation of our study is the low sample size, however, we feel that our results would warrant validation in a large cohort of OC patients.
4. Materials and Methods
4.1. Patients and Samples
Twenty-four blood samples (six disease-free healthy controls, 18 serous ovarian cancer patients) were collected from the Department of Obstetrics and Gynecology, Faculty of Medicine, University of Debrecen. All patients that underwent surgery and tissue samples were histologically diagnosed. Pathological characterization of tumor stages was assessed according to the International Federation of Gynecology and Obstetrics (FIGO) criteria. None of the patients received chemotherapy or radiotherapy treatment prior to participation in the study. Each subject provided written informed consent. The study was approved by the Scientific and Research Ethics Committee of the Medical Research Council of the Ministry of Health, Budapest, Hungary (ETT TUKEB), (Project Identification Code: 30231-2/2016/EKU, date: 06 June 2016) and was conducted in accordance with the Declaration of Helsinki. Controls were followed up, none of them received gynecological treatment during the study period.
Peripheral blood (9 mL) was drawn into EDTA anticoagulated tubes (BD Vacutainer) from each patient and from healthy volunteers and kept at 4 °C until further processing (within two hours of collection). Plasma samples were subjected to a two-step centrifugation protocol (2500× g and 16,000× g; 10–10 min, 4 °C) to obtain plasma. After separation, the cell-free plasma samples were homogenized, aliquoted, and stored at −80 °C until further processing.
4.2. RNA Isolation and Purification for the NanoString Device
Prior to RNA isolation blood samples were thawed on ice, then circulating RNA was isolated from 500 µL plasma samples using the miRNeasy Serum/Plasma RNA isolation kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. The quality of the RNA was analyzed using the Nanodrop device (Thermo Scientific, Waltham, MA, USA).
4.3. RNA Expression Analysis
The miRNA content of all samples was analyzed using the nCounter Human v3 miRNA Panel of NanoString nCounter Analysis System (NanoString Technologies, Seattle, WA, USA), which contains 798 unique hsa-miRNA barcodes. 100 ng RNA/sample was used as input for the measurements, hybridization was carried out for 18 h, and miRNA counts were collected by scanning on the HIGH mode. The background correction of data was performed by subtracting the mean ± 2 standard deviation of the negative control set. Lane-by-lane technical variation was corrected by using the geometric median value of the positive code-set. The complete data set was normalized by calculating the geometric mean of 10 “housekeeping” miRNA counts for each sample to generate the normalization factor.
4.4. Prediction and Analysis of Experimentally Validated Target Genes
First, a miRNA–target gene network was constructed using the web based miRNet tool [
http://www.mirnet.ca]. Top miRNAs in the network were ranked by degree and betweenness centrality values. The prediction of experimentally validated target genes of miRNAs was carried out by using the web based miRNet, miRTarBase, and TargetScan software programs (
http://miRTarBase.mbc.nctu.edu;
www.targetscan.org). Target intersections were further validated by the miRWalk2 database (
www. http://zmf.umm.uni-heidelberg.deg). The general and ovary-specific protein–protein interaction (PPI) network of target genes was constructed using the NetworkAnalyst 3.0 tool [
www.networkanalyst.ca].
4.5. Functional Annotation and Pathway Enrichment Analysis
The lists of miRNA targets was used as input and the online Database for Annotation, Visualization, and Integrated Discovery (DAVID;
https://david.ncifcerf.gov) software tool was used to perform gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) based functional pathway enrichment analysis for the predicted target genes of prioritized differentially expressed hsa-miRNAs. The NetworkAnalyst tool was used to carry out ovary-specific enrichment analysis. A p-value of < 0.05 was considered statistically significant.
4.6. Validation of hsa-miRNA Expression by Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR) on Selected hsa-miRNAs
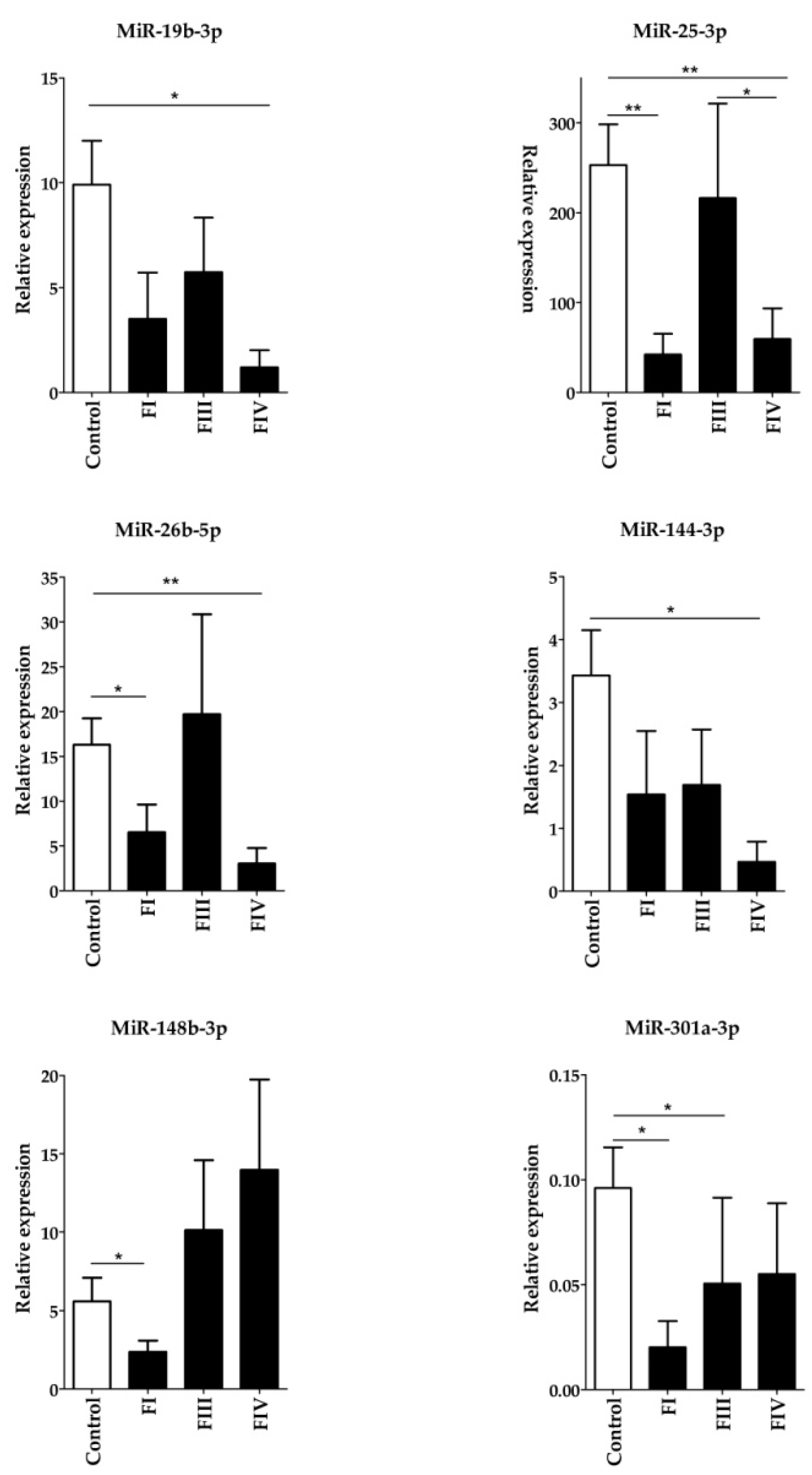
Circulating RNA was extracted from 200 µL plasma samples of 16 healthy control females and 18 OC patients by using the miRNeasy Serum/Plasma Kit (Qiagen, Hilden, Germany) including 3.5 µL miRNeasy Serum/Plasma Spike-In Control RNA, according to the manufacturer’s instructions. A miRNA-specific fluorometric assay on a Qubit® 2.0 Fluorimeter (Thermo Fischer Scientific, USA) was used to determine the concentration of RNA. To detect and measure the amounts of mature miRNAs the miScript PCR System (Qiagen, Hilden, Germany) was used. The miScript II RT Kit (Qiagen) was used for reverse transcription of miRNAs. The quantitative real-time PCR reaction was used (LightCycler®96; Roche Molecular Systems Inc., Pleasanton, CA, USA) to determine the level of hsa-miR-25-3p, hsa-miR-26b-5p, hsa-miR-144-3p, hsa-miR-19b-3p, hsa-miR-301a-3p, hsa-miR-148b-3p, hsa-miR-553, and hsa-miR-197 by using the miScript SYBR Green PCR Kit (Qiagen). The PCR reaction mixture contained 500 pg reverse transcription products. The reaction mixtures were first denatured at 95 °C for 15 min, followed by 50 amplification cycles of 94 °C for 15 s, 55 °C for 30 s and 70 °C for 30 s. Finally, a melting curve was generated by taking fluorescent measurements every 0.2 °C for 25 s from 50 °C until 95 °C to detect a single PCR product. Cycle threshold (Ct) values above the determinable range (up to 45) were assigned a Ct of 45. All measurements were performed in triplicate and the amounts of PCR products were normalized to an internal control (hsa-miR103-3p). Relative expression levels were calculated by the 2−∆Ct method.
4.7. Statistical Analysis
All data were analyzed using the GraphPad Prism statistical package (GraphPad Prism7, San Diego, CA, USA). Descriptive column statistics of each data set were performed and the distribution of data was analyzed by the Kolmogorov–Smirnov test. To assess the statistical significance of differences in miRNA counts between the control and patient groups the nonparametric one-way ANOVA Kruskal–Wallis test in combination with the post hoc Dunn’s test to adjust for multiple comparisons was applied. In all tests the difference was considered significant at p < 0.05 value. Where applicable, the Dunn’s p-values were indicated as: p < 0.05(*); p < 0.01(**). The fold change in the expression of a miRNA between the control data and a given FIGO stage data was calculated as: (FIGO stage mean count – Control mean count)/Control mean count.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}