The Bactericidal Activity of Temporin Analogues Against Methicillin Resistant Staphylococcus aureus

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
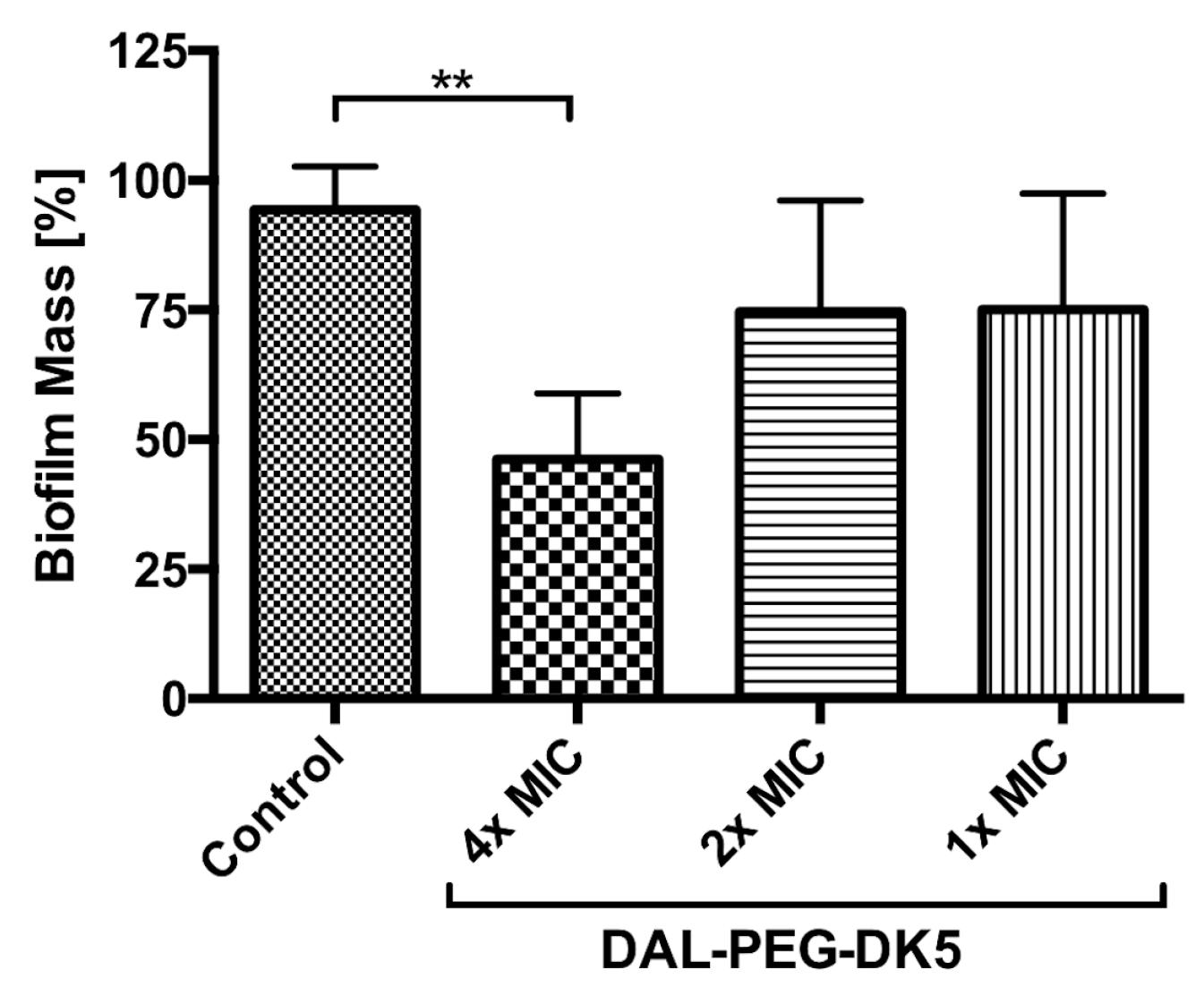
2.1. Antibacterial Activity of Temporin Conjugates Against Staphylococci
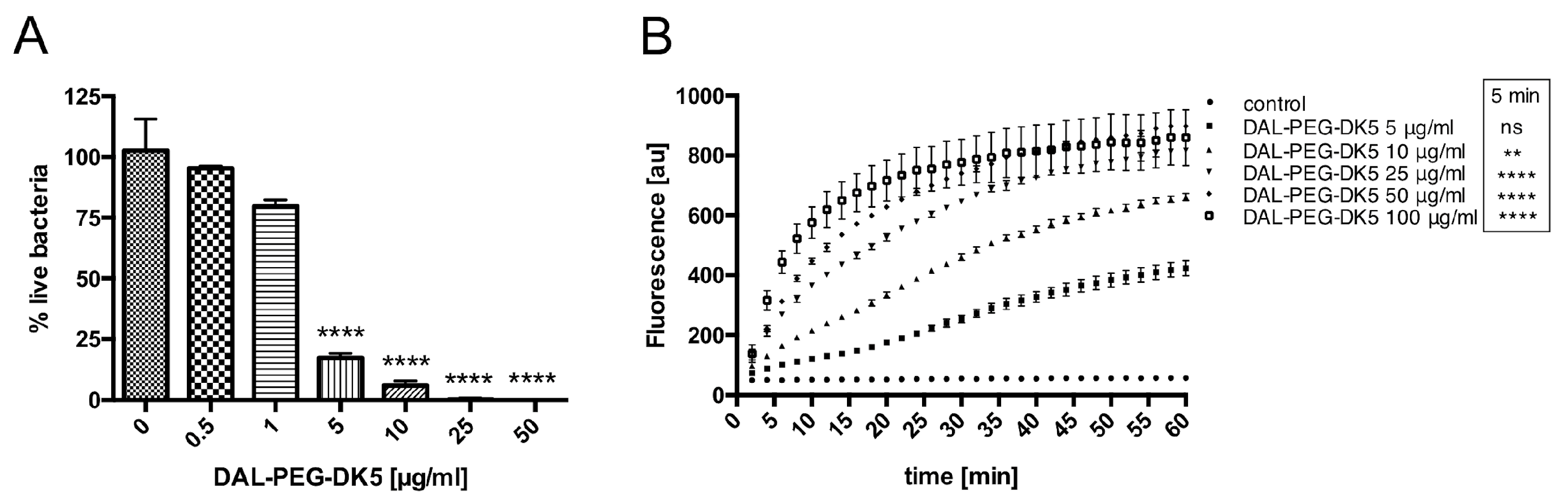
2.2. Mechanism Underlying the Bactericidal Activity of DAL-PEG-DK5
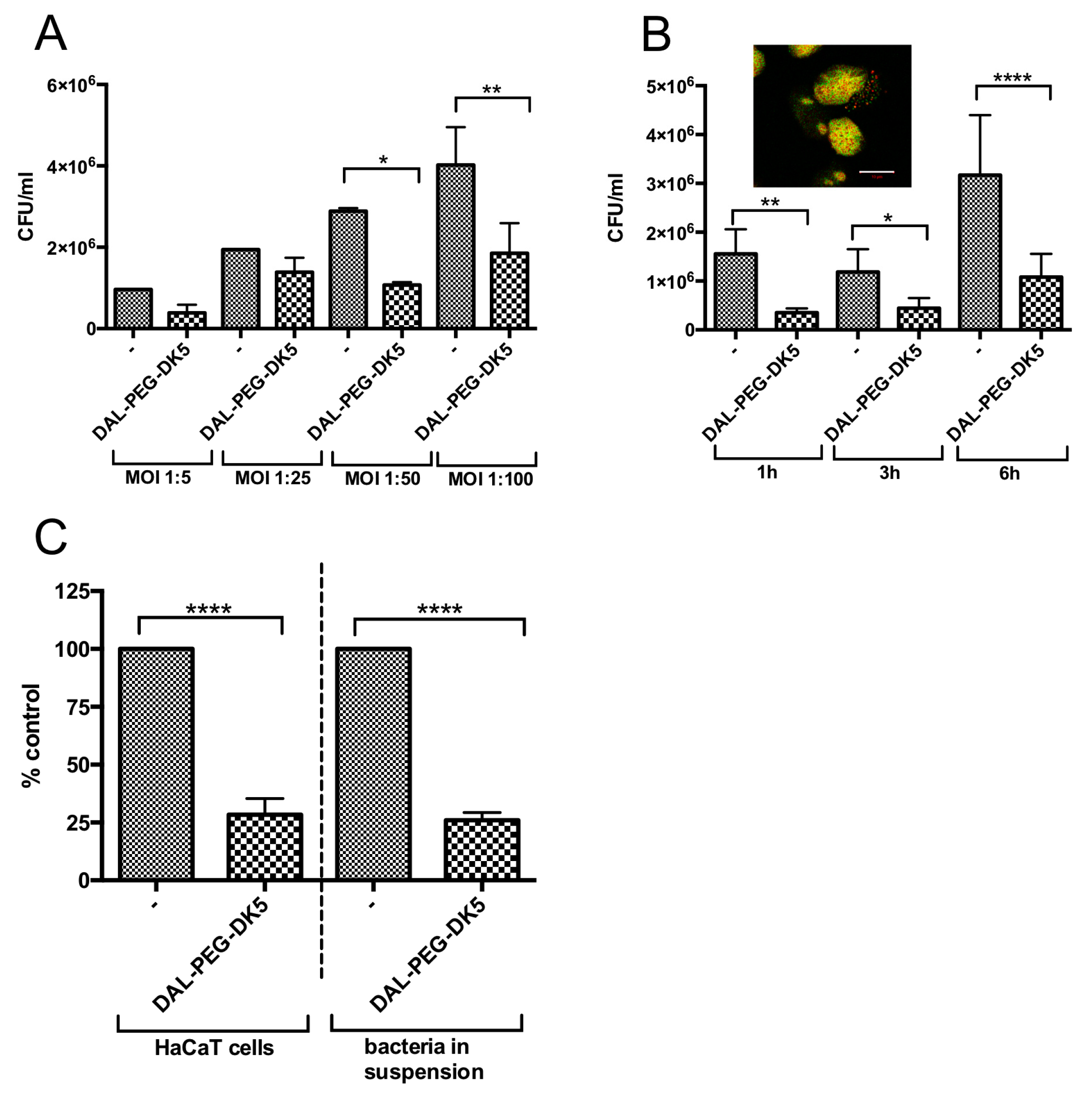
2.3. Activity of DAL-PEG-DK5 Against Intracellular S. aureus
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Peptides Synthesis and Purification
4.3. Cell Culture
4.4. Microorganisms
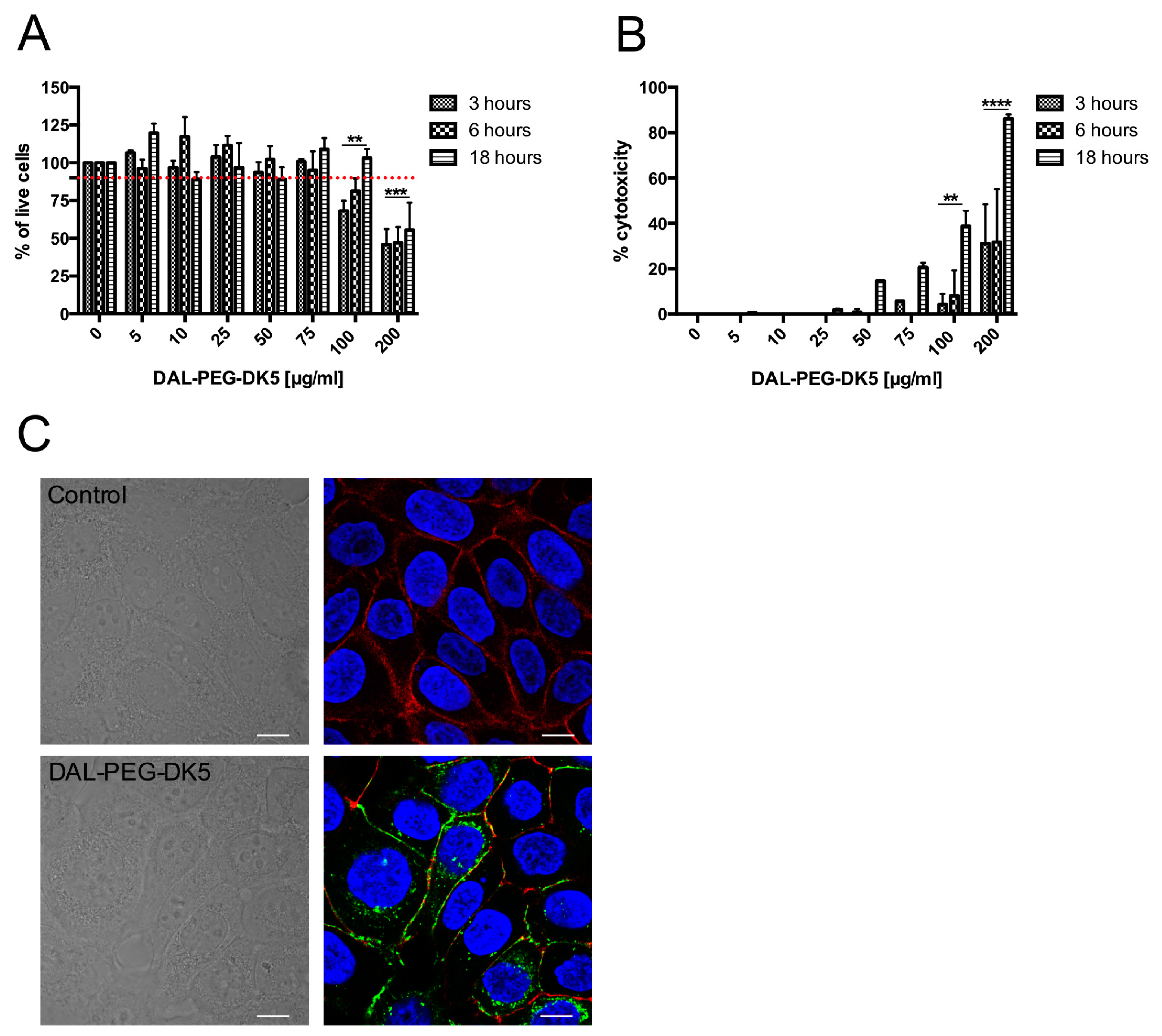
4.5. Cell Viability Test
4.6. Antimicrobial Activity
4.7. Efficacy of Peptides on S. aureus Biofilms
4.8. Sytox Green Uptake Analysis
4.9. Assessment of Bacterial Viability by Using the LIVE/ DEAD BacLight KIT
4.10. Antibacterial Efficacy of Peptides Against Intracellular S. aureus
4.11. Confocal Microscopy
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPs | Antimicrobial peptides |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| CFU | Colony forming units |
| FBS | Fetal bovine serum |
| MIC | Minimal inhibitory concentration |
| MOI | Multiplicity of infection |
| OD | Optical density |
| PBS | Phosphate buffered saline |
| PI | Propidium iodide |
| SCR | Scramble peptide |
References
- Peterson, E.; Kaur, P. Antibiotic Resistance Mechanisms in Bacteria: Relationships Between Resistance Determinants of Antibiotic Producers, Environmental Bacteria, and Clinical Pathogens. Front. Microbiol. 2018, 9, 2928. [Google Scholar] [CrossRef] [PubMed]
- Jevons, M.P. “Calbenin”-resistant staphylococci. BMJ 1961, 1, 124–125. [Google Scholar] [CrossRef]
- Zetola, N.; Francis, J.S.; Nuermberger, E.L.; Bishai, W.R. Community-acquired meticillin-resistant Staphylococcus aureus: An emerging threat. Lancet Infect. Dis. 2005, 5, 275–286. [Google Scholar] [CrossRef]
- Grundmann, H.; Aires-de-Sousa, M.; Boyce, J.; Tiemersma, E. Emergence and resurgence of meticillin-resistant Staphylococcus aureus as a public-health threat. Lancet 2006, 368, 874–885. [Google Scholar] [CrossRef] [Green Version]
- Diekema, D.J.; Pfaller, M.A.; Schmitz, F.J.; Smayevsky, J.; Bell, J.; Jones, R.N.; Beach, M.; SENTRY Partcipants Group. Survey of infections due to Staphylococcus species: Frequency of occurrence and antimicrobial susceptibility of isolates collected in the United States, Canada, Latin America, Europe, and the Western Pacific region for the SENTRY Antimicrobial Surveillance Program, 1997-1999. Clin. Infect Dis. 2001, 32, 114–132. [Google Scholar] [CrossRef]
- McCaig, L.F.; McDonald, L.C.; Mandal, S.; Jernigan, D.B. Staphylococcus aureus-associated skin and soft tissue infections in ambulatory care. Emerg. Infect. Dis. 2006, 12, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- Stryjewski, M.E.; Chambers, H.F. Skin and soft-tissue infections caused by community-acquired methicillin-resistant Staphylococcus aureus. Clin. Infect. Dis. 2008, 46, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Gorak, E.J.; Yamada, S.M.; Brown, J.D. Community-acquired methicillin-resistant Staphylococcus aureus in hospitalized adults and children without known risk factors. Clin. Infect. Dis. 1999, 29, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Haney, E.F.; Gill, E.E. The immunology of host defence peptides: Beyond antimicrobial activity. Nat. Rev. Immunol. 2016, 16, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; Chow, L.N.; Mookherjee, N. Cationic host defence peptides: Multifaceted role in immune modulation and inflammation. J. Innate Immun. 2012, 4, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.T.; Gellatly, S.L.; Hancock, R.E. Multifunctional cationic host defence peptides and their clinical applications. Cell. Mol. Life Sci. 2011, 68, 2161–2176. [Google Scholar] [CrossRef] [PubMed]
- Conlon, J.M.; Kolodziejek, J.; Nowotny, N. Antimicrobial peptides from ranid frogs: Taxonomic and phylogenetic markers and a potential source of new therapeutic agents. Biochim. Biophys. Acta 2004, 1696, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.L.; Grazia, A.D.; Cappiello, F.; Casciaro, B.; Luca, V. Naturally Occurring Peptides from Rana temporaria: Antimicrobial Properties and More. Curr. Top. Med. Chem. 2016, 16, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.L. Temporins, anti-infective peptides with expanding properties. Cell. Mol. Life Sci. 2006, 63, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Di Grazia, A.; Luca, V.; Segev-Zarko, L.A.; Shai, Y.; Mangoni, M.L. Temporins A and B stimulate migration of HaCaT keratinocytes and kill intracellular Staphylococcus aureus. Antimicrob. Agents Chemother. 2014, 58, 2520–2527. [Google Scholar] [CrossRef]
- Ciandrini, E.; Morroni, G.; Arzeni, D.; Kamysz, W.; Neubauer, D.; Kamysz, E.; Cirioni, O.; Brescini, L.; Baffone, W.; Campana, R. Antimicrobial Activity of Different Antimicrobial Peptides (AMPs) Against Clinical Methicillin-resistant Staphylococcus aureus (MRSA). Curr. Top. Med. Chem. 2018, 18, 2116–2126. [Google Scholar] [CrossRef]
- Shang, D.; Yu, F.; Li, J.; Zheng, J.; Zhang, L.; Li, Y. Molecular cloning of cDNAs encoding antimicrobial peptide precursors from the skin of the Chinese brown frog, Rana chensinensis. Zool. Sci. 2009, 26, 220–226. [Google Scholar] [CrossRef]
- Shang, D.; Li, X.; Sun, Y.; Wang, C.; Sun, L.; Wei, S.; Gou, M. Design of potent, non-toxic antimicrobial agents based upon the structure of the frog skin peptide, temporin-1CEb from Chinese brown frog, Rana chensinensis. Chem. Biol. Drug Des. 2012, 79, 653–662. [Google Scholar] [CrossRef]
- Golda, A.; Kosikowska-Adamus, P.; Babyak, O.; Lech, M.; Wysocka, M.; Lesner, A.; Potempa, J.; Koziel, J. Conjugate of Enkephalin and Temporin Peptides as a Novel Therapeutic Agent for Sepsis. Bioconjug. Chem. 2018, 29, 4127–4139. [Google Scholar] [CrossRef]
- Shekhter, A.B.; Solov’eva, A.I.; Spevak, S.E.; Titov, M.I. Effects of opioid peptide dalargin on reparative processes in wound healing. Biull. Eksp. Biol. Med. 1988, 106, 487–490. [Google Scholar] [CrossRef]
- Legeza, V.P.; Koshcheev, A.G.; Konovalova, L.N. Effect of dalargin on healing of a bullet wound of the soft tissues in rabbits. Patol. Fiziol. Eksp. Ter. 1995, 4, 45–48. [Google Scholar]
- Hipkiss, A.R. Aging, Proteotoxicity, Mitochondria, Glycation, NAD and Carnosine: Possible Inter-Relationships and Resolution of the Oxygen Paradox. Front. Aging Neurosci. 2010, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Cripps, M.J.; Hanna, K.; Lavilla, C., Jr.; Sayers, S.R.; Caton, P.W.; Sims, C.; De Girolamo, L.; Sale, C.; Turner, M.D. Carnosine scavenging of glucolipotoxic free radicals enhances insulin secretion and glucose uptake. Sci. Rep. 2017, 7, 13313. [Google Scholar] [CrossRef] [PubMed]
- Demidova-Rice, T.N.; Geevarghese, A.; Herman, I.M. Bioactive peptides derived from vascular endothelial cell extracellular matrices promote microvascular morphogenesis and wound healing in vitro. Wound Repair Regen. 2011, 19, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Kosikowska, P.; Pikula, M.; Langa, P.; Trzonkowski, P.; Obuchowski, M.; Lesner, A. Synthesis and Evaluation of Biological Activity of Antimicrobial--Pro-Proliferative Peptide Conjugates. PLoS ONE 2015, 10, e0140377. [Google Scholar] [CrossRef] [PubMed]
- Pletzer, D.; Hancock, R.E. Antibiofilm Peptides: Potential as Broad-Spectrum Agents. J. Bacteriol. 2016, 198, 2572–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayles, K.W.; Wesson, C.A.; Liou, L.E.; Fox, L.K.; Bohach, G.A.; Trumble, W.R. Intracellular Staphylococcus aureus escapes the endosome and induces apoptosis in epithelial cells. Infect. Immun. 1998, 66, 336–342. [Google Scholar]
- Kubica, M.; Guzik, K.; Koziel, J.; Zarebski, M.; Richter, W.; Gajkowska, B.; Golda, A.; Maciag-Gudowska, A.; Brix, K.; Shaw, L.; et al. A potential new pathway for Staphylococcus aureus dissemination: The silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS ONE 2008, 3, e1409. [Google Scholar] [CrossRef]
- Sayedyahossein, S.; Xu, S.X.; Rudkouskaya, A.; McGavin, M.J.; McCormick, J.K.; Dagnino, L. Staphylococcus aureus keratinocyte invasion is mediated by integrin linked kinase and Rac1. FASEB J. 2015, 29, 711–723. [Google Scholar] [CrossRef]
- Bitschar, K.; Wolz, C.; Krismer, B.; Peschel, A.; Schittek, B. Keratinocytes as sensors and central players in the immune defense against Staphylococcus aureus in the skin. J. Dermatol. Sci. 2017, 87, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Travers, J.B.; Norris, D.A.; Leung, D.Y. The keratinocyte as a target for staphylococcal bacterial toxins. J. Investig. Derm. Symp. Proc. 2001, 6, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Ulvatne, H. Antimicrobial peptides: Potential use in skin infections. Am. J. Clin. Derm. 2003, 4, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Wang, X.; Lushnikova, T.; Zhang, Y.; Golla, R.M.; Narayana, J.L.; Wang, C.; McGuire, T.R.; Wang, G. Antibacterial, antifungal, anticancer activities and structural bioinformatics analysis of six naturally occurring temporins. Peptides 2018, 106, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.L.; Maisetta, G.; Di Luca, M.; Gaddi, L.M.; Esin, S.; Florio, W.; Brancatisano, F.L.; Barra, D.; Campa, M.; Batoni, G. Comparative analysis of the bactericidal activities of amphibian peptide analogues against multidrug-resistant nosocomial bacterial strains. Antimicrob. Agents Chemother. 2008, 52, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Grassi, L.; Maisetta, G.; Maccari, G.; Esin, S.; Batoni, G. Analogs of the Frog-skin Antimicrobial Peptide Temporin 1Tb Exhibit a Wider Spectrum of Activity and a Stronger Antibiofilm Potential as Compared to the Parental Peptide. Front. Chem. 2017, 5, 24. [Google Scholar] [CrossRef] [Green Version]
- Capparelli, R.; Romanelli, A.; Iannaccone, M.; Nocerino, N.; Ripa, R.; Pensato, S.; Pedone, C.; Iannelli, D. Synergistic antibacterial and anti-inflammatory activity of temporin A and modified temporin B in vivo. PLoS ONE 2009, 4, e7191. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Wang, G. Ab initio design of potent anti-MRSA peptides based on database filtering technology. J. Am. Chem. Soc. 2012, 134, 12426–12429. [Google Scholar] [CrossRef]
- Boyle-Vavra, S.; Li, X.; Alam, M.T.; Read, T.D.; Sieth, J.; Cywes-Bentley, C.; Dobbins, G.; David, M.Z.; Kumar, N.; Eells, S.J.; et al. USA300 and USA500 clonal lineages of Staphylococcus aureus do not produce a capsular polysaccharide due to conserved mutations in the cap5 locus. MBio 2015, 6, e02585-14. [Google Scholar] [CrossRef]
- Kuipers, A.; Stapels, D.A.; Weerwind, L.T.; Ko, Y.P.; Ruyken, M.; Lee, J.C.; van Kessel, K.P.; Rooijakkers, S.H. The Staphylococcus aureus polysaccharide capsule and Efb-dependent fibrinogen shield act in concert to protect against phagocytosis. Microbiology 2016, 162, 1185–1194. [Google Scholar] [CrossRef]
- Herbert, S.; Newell, S.W.; Lee, C.; Wieland, K.P.; Dassy, B.; Fournier, J.M.; Wolz, C.; Döring, G. Regulation of Staphylococcus aureus type 5 and type 8 capsular polysaccharides by CO(2). J. Bacteriol. 2001, 183, 4609–4613. [Google Scholar] [CrossRef]
- Davis, S.C.; Ricotti, C.; Cazzaniga, A.; Welsh, E.; Eaglstein, W.H.; Mertz, P.M. Microscopic and physiologic evidence for biofilm-associated wound colonization in vivo. Wound Repair Regen. 2008, 16, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Schulin, T.; Voss, A. Coagulase-negative staphylococci as a cause of infections related to intravascular prosthetic devices: Limitations of present therapy. Clin. Microbiol. Infect. 2001, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Giormezis, N.; Kolonitsiou, F.; Foka, A.; Drougka, E.; Liakopoulos, A.; Makri, A.; Papanastasiou, A.D.; Vogiatzi, A.; Dimitriou, G.; Marangos, M.; et al. Coagulase-negative staphylococcal bloodstream and prosthetic-device-associated infections: The role of biofilm formation and distribution of adhesin and toxin genes. J. Med. Microbiol. 2014, 63, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, I.; Sanges, M.R.; Folgore, A.; Carratelli, C.R. Apoptosis of human keratinocytes after bacterial invasion. FEMS Immunol. Med. Microbiol. 2000, 27, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kong, L.; Liu, Y.; Fu, Q.; Cui, Z.; Wang, J.; Ma, J.; Wang, H.; Yan, Y.; Sun, J. A Phage Lysin Fused to a Cell-Penetrating Peptide Kills Intracellular Methicillin-Resistant Staphylococcus aureus in Keratinocytes and Has Potential as a Treatment for Skin Infections in Mice. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [PubMed]
- Pencheva, N.; Pospisek, J.; Hauzerova, L.; Barth, T.; Milanov, P. Activity profiles of dalargin and its analogues in mu-, delta- and kappa-opioid receptor selective bioassays. Br. J. Pharm. 1999, 128, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Koziel, J.; Chmiest, D.; Bryzek, D.; Kmiecik, K.; Mizgalska, D.; Maciag-Gudowska, A.; Shaw, L.N.; Potempa, J. The Janus face of α-toxin: A potent mediator of cytoprotection in staphylococci-infected macrophages. J. Innate Immun. 2015, 7, 187–198. [Google Scholar] [CrossRef]
- Mohamed, M.F.; Abdelkhalek, A.; Seleem, M.N. Evaluation of short synthetic antimicrobial peptides for treatment of drug-resistant and intracellular Staphylococcus aureus. Sci. Rep. 2016, 6, 29707. [Google Scholar] [CrossRef]
- Lee, J.K.; Seo, C.H.; Luchian, T.; Park, Y. Antimicrobial Peptide CMA3 Derived from the CA-MA Hybrid Peptide: Antibacterial and Anti-inflammatory Activities with Low Cytotoxicity and Mechanism of Action in Escherichia coli. Antimicrob. Agents Chemother. 2015, 60, 495–506. [Google Scholar] [CrossRef]
- Von Eiff, C.; Becker, K.; Metze, D.; Lubritz, G.; Hockmann, J.; Schwarz, T.; Peters, G. Intracellular persistence of Staphylococcus aureus small-colony variants within keratinocytes: A cause for antibiotic treatment failure in a patient with darier’s disease. Clin. Infect. Dis. 2001, 32, 1643–1647. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Staphylococcal strain | Strain ID | MIC [μg/mL] | |||
|---|---|---|---|---|---|
| DAL-PEG-DK5 | CAR-PEG-DK5 | CAR3-PEG-DK5 | COMB1-PEG-DK5 | ||
| S. aureus | USA300 | 40 | >190 | >190 | >190 |
| Newman | 140 | >190 | >190 | >190 | |
| ATCC 25923 | 40 | >190 | >190 | >190 | |
| S. epidermidis | ATCC 12228 | 60 | 40 | 110 | >190 |
| USA300 | MIC [μg/mL] |
|---|---|
| DK5 | >190 |
| DAL | >190 |
| DK5-PEG-DAL | 160 |
| DAL-PEG-DK5 | 40 |
| SCR | >190 |
| MRSA strain ID | MIC [μg/mL] | ||
|---|---|---|---|
| DAL-PEG-DK5 | Vancomycin | Linezolid | |
| 56A1 | 40 | - | - |
| 52B | 40 | 0.5 | 2 |
| 1694 | 70 | - | - |
| 2492 | 40 | - | - |
| 2706 | 40 | - | - |
| 2872cv | 40 | - | - |
| 3417 | 40 | - | - |
| 4187 | 40 | - | - |
| 6674 | 40 | - | - |
| 7219 | 30 | - | - |
| 7501 | 40 | - | - |
| 7569 | 40 | - | - |
| 7718 | 50 | - | - |
| USA300 | 40 | 1 | 1 |
| Staphylococcus Strains | Relevant Properties | Source |
|---|---|---|
| S. aureus | ||
| USA300 | Wilde type strain | L.N. Shaw [47] |
| ATCC 25923 | Clinical isolate | ATCC |
| Newman | Wilde type laboratory strain | T.J. Foster |
| 56A1 | Clinical isolate | * |
| 52B | Clinical isolate | * |
| 1694 | Clinical isolate | * |
| 2492 | Clinical isolate | * |
| 2706 | Clinical isolate | * |
| 2872cv | Clinical isolate | * |
| 3417 | Clinical isolate | * |
| 4187 | Clinical isolate | * |
| 6674 | Clinical isolate | * |
| 7219 | Clinical isolate | * |
| 7501 | Clinical isolate | * |
| 7569 | Clinical isolate | * |
| 7718 | Clinical isolate | * |
| S. epidermidis | ||
| ATCC 12228 | Wilde type strain | ATCC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golda, A.; Kosikowska-Adamus, P.; Kret, A.; Babyak, O.; Wójcik, K.; Dobosz, E.; Potempa, J.; Lesner, A.; Koziel, J. The Bactericidal Activity of Temporin Analogues Against Methicillin Resistant Staphylococcus aureus. Int. J. Mol. Sci. 2019, 20, 4761. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194761
Golda A, Kosikowska-Adamus P, Kret A, Babyak O, Wójcik K, Dobosz E, Potempa J, Lesner A, Koziel J. The Bactericidal Activity of Temporin Analogues Against Methicillin Resistant Staphylococcus aureus. International Journal of Molecular Sciences. 2019; 20(19):4761. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194761
Chicago/Turabian StyleGolda, Anna, Paulina Kosikowska-Adamus, Aleksandra Kret, Olena Babyak, Kinga Wójcik, Ewelina Dobosz, Jan Potempa, Adam Lesner, and Joanna Koziel. 2019. "The Bactericidal Activity of Temporin Analogues Against Methicillin Resistant Staphylococcus aureus" International Journal of Molecular Sciences 20, no. 19: 4761. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194761